Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Latent Adeno-Associated Virus Infection Elicits Humoral but
Not Cell-Mediated Immune Responses in a Nonhuman
Primate Model
YOSBANI J. HERNANDEZ,
1,2,3JIANMING WANG,
1,2,3WILLIAM G. KEARNS,
4SCOTT LOILER,
1,2,3AMY POIRIER,
1,2,3ANDTERENCE R. FLOTTE
1,2,3*
Gene Therapy Center
1and Departments of Pediatrics
2and Molecular Genetics and Microbiology,
3University of Florida
College of Medicine, Gainesville, Florida 32610, and Department of Pediatrics, Johns Hopkins University
School of Medicine, Baltimore, Maryland 21287
4Received 6 April 1999/Accepted 18 June 1999
Latent infection with wild-type (wt) adeno-associated virus (AAV) was studied in rhesus macaques, a species
that is a natural host for AAV and that has some homology to humans with respect to the preferred locus for
wt AAV integration. Each of eight animals was infected with an inoculum of 10
10IU of wt AAV, administered
by either the intranasal, intramuscular, or intravenous route. Two additional animals were infected
intrana-sally with wt AAV and a helper adenovirus (Ad), while one additional animal was inoculated with saline
intranasally as a control. There were no detectable clinical or histopathologic responses to wt AAV
adminis-tration. Molecular analyses, including Southern blot, PCR, and fluorescence in situ hybridization, were
performed 21 days after infection. These studies indicated that AAV DNA sequences persisted at the sites of
administration, albeit at low copy number, and in peripheral blood mononuclear cells. Site-specific integration
into the AAVS1-like locus was observed in a subset of animals. All animals, except those infected by the
intranasal route with wt AAV alone, developed a humoral immune response to wt AAV capsid proteins, as
evidenced by a
>
fourfold rise in anti-AAV neutralizing titers. However, only animals infected with both wt AAV
and Ad developed cell-mediated immune responses to AAV capsid proteins. These findings provide some
insights into the nature of anti-AAV immune responses that may be useful in interpreting results of future
AAV-based gene transfer studies.
Adeno-associated virus type 2 (AAV) is a nonpathogenic
parvovirus that generally requires coinfection with a helper
virus, such as an adenovirus (Ad) or herpesvirus, to undergo
productive infection (3, 4, 7). In cultured cells infected with
wild-type (wt) AAV in the absence of helper virus, AAV
es-tablishes a latent infection (5, 11, 22) and often integrates site
specifically into a sequence located on the q arm of human
chromosome 19, termed the AAVS1 site (19, 20, 29–31, 35).
AAV proviral DNA remains in this latent state until rescued by
a helper virus. Studies on the mode of viral integration suggest
that tandem copies of the viral DNA are inserted into the host
cell chromosome in a head-to-tail orientation via the inverted
terminal repeats of the virus (28).
Due to its lack of pathogenicity and its ability to establish
persistent infections in human cells, AAV has gained
accep-tance as a potential vector for human gene therapy (18).
Re-combinant AAV (rAAV) vectors mediate stable in vivo
expres-sion in a wide range of different tissues including the lungs
(16), muscles (12, 13, 26, 40), brain (2, 24, 27, 39), spinal cord
(34), retinas (14, 32, 43), and liver (36). The use of AAV
vectors has not been associated with significant toxicity in
an-imal models. In addition, two phase I trials of recombinant
AAV vectors have been undertaken in patients with cystic
fibrosis (15, 38).
Despite the growing body of data regarding the biology of
AAV latency in vitro, very few studies have examined this
phenomenon in vivo. Epidemiologic studies have shown a high
prevalence of AAV2 seropositivity (6, 8, 9). However, it is not
known whether seropositivity is indicative of previous
produc-tive infection or of latent infection. Although infectious wt
AAV has been cultured from samples from the respiratory and
gastrointestinal tract in association with productive helper
vi-rus infection (9), latent AAV DNA has been found only in
peripheral blood mononuclear cells (PBMCs) (21). Since
hu-mans and monkeys are the only species known to possess the
AAVS1 integration sequence (35), they are the only two in vivo
models which would be expected to faithfully reflect the wt
AAV latency pathway. One previous study examined the
in-teractions between rAAV vectors, wt-AAV and Ad in a rhesus
macaque model and demonstrated that the productive phase
of the wt AAV life cycle could be reproduced in that model (1).
In this study, we have examined latent wt AAV infection in
that same rhesus model. Additional studies investigated the
cell specificity of persistence, the integration state of latent
viral genomes, and the immune responses to viral infection. In
this setting, persistence was observed, although site-specific
integration was infrequent. wt AAV genomes were detectable
both at the site of administration and in circulating PBMCs.
Neutralizing antibodies directed against wt AAV were
ob-served in serum 21 days postinfection in animals infected
in-tramuscularly or intravenously with wt AAV alone, but
cell-mediated immune responses were elicited only in the presence
of helper Ad.
MATERIALS AND METHODS
Preparation of wt AAV.wt AAV was prepared with 293 cells grown as mono-layers in Dulbecco modified Eagle medium, supplemented with 10% fetal bovine serum and 100 U of penicillin-streptomycin per ml, at 37°C under humidified air containing 5% CO2. The cells were grown to confluency on Cell Factories (Nunc)
* Corresponding author. Mailing address: University of Florida
Gene Therapy Center, Academic Research Bldg. Rm. R1-191, 1600
SW Archer Rd. (JHMHSC Box 100266), Gainesville, FL 32610-0266.
Phone: (352) 846-2739. Fax: (352) 846-2738. E-mail: flotttr@peds.ufl
.edu.
8549
on November 9, 2019 by guest
http://jvi.asm.org/
and infected with wt AAV seed stock at a multiplicity of infection (MOI) of 1. The cells were coinfected with Ad5 at an MOI of 3. After 48 h, the cultures were harvested, resuspended in phosphate-buffered saline (PBS), and frozen and thawed three times. The crude lysate was heated at 56°C for 15 min to inactivate Ad and then centrifuged for 5 min at 9,000⫻gto remove cellular debris. The supernatant’s volume was raised to 10 ml with PBS and loaded onto a HiTrap heparin affinity column (Pharmacia Biotech) at a rate of 1 ml per min with a peristaltic pump (Bio-Rad). The column was then washed with 20 ml of 0.01 M sodium phosphate buffer (pH 8) at a rate of 1 ml per minute. The flowthrough was retained and later analyzed. Virus was eluted at a rate of 1 ml per min (15 ml total volume) with a salt gradient (0 to 1 M NaCl) in 0.01 M sodium phosphate buffer (pH 8). Fifteen fractions were collected and analyzed by PCR and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). PCR-positive fractions were pooled, and CsCl was added to the pooled sample to a density of 1.41 g per ml (refractive index, 1.3710). The sample was then loaded onto an ultracentrifuge tube (Beckman) and centrifuged in an SW50 swinging-bucket rotor (Beckman) at 35,0000 rpm at 4°C for 24 h. The contents of the tube were then fractionated (500-l fractions) and dialyzed against PBS by using an ultraconcentrating tube with a molecular mass cutoff of 50 kDa (Ami-con). The dialyzed fractions were then analyzed for the wt AAV genome by PCR. The positive fractions were pooled and analyzed by SDS-PAGE.
SDS-PAGE.Viral capsid proteins were analyzed by PAGE. A 5-l volume from each fraction was denatured, mixed with 1⫻loading dye (final concentra-tions, 12.5 mM NaH2PO4, 35 mM Na2HPO4, 0.5% SDS, 0.5% -mercaptoetha-nol, 75g of bromophenol blue per ml, and 3 M urea), and loaded onto a 10% polyacrylamide gel (Bio-Rad). Samples were electrophoresed at 100 V for 2 h at room temperature with a MiniProtein electrophoresis apparatus (Bio-Rad). The gel slab was then fixed, stained with Coomassie blue (2.5 mg/ml in 45% metha-nol–10% acetic acid) at room temperature overnight, and destained (in 30% methanol–6% acetic acid) for several minutes until the background cleared, to visualize protein bands.
QC-PCR to determine the physical titer of AAV.The physical titer was as-sessed by a PCR-based protocol. A 1-l sample was taken from the purified stock and treated with 10 U of DNase (Boehringer Mannheim) in 10 mM MgCl2–50 mM Tris-HCl (pH 7.5) (total volume, 100l) for 1 h at 37°C. The sample was then treated with proteinase K (Boehringer Mannheim) at a final concentration of 0.2g/ml for 1 h at 37°C, using the manufacturer’s recommended buffer conditions. Viral DNA was then purified by two phenol-chloroform extractions and one phenol extraction. The sample was precipitated with ethanol and then centrifuged at 10,000⫻gfor 15 min. The supernatant was carefully discarded. The DNA pellet was resuspended in 10l of distilled H2O. The sample was then serially diluted. A PCR cocktail containing 1l of serially diluted viral sample and different amounts of the internally deleted competitor template was pre-pared. The PCR mixture consisted of 50 mM KCl, 10 mM Tris-HCl (pH 9.0), 1.5 mM MgCl2, 200 mM each dATP, dGTP, dCTP, and dTTP, 0.5 U ofTaq polymerase (Promega), and 5 pmol of each amplification primer. The primers used for quantitative competitive (QC)-PCR were as follows: the 5⬘ primer sequence was 5⬘-TGGCCCACCACCACCAAAGCCCGCA-3⬘hybridizing to wt AAV nucleotides (nt) 2283 to 2308, and the 3⬘primer sequence was 5⬘-TGGC CCGCCTTTCCGGTTCCCGAGG-3⬘hybridizing to wt AAV nt 2668 to 2693. Thirty-five cycles of PCR were performed with the following program: 96°C for 1 min, 72°C for 1 min, and 60°C for 1 min. The products were analyzed on a 1.5%
were visualized by autoradiography and counted manually.
Vector construction and production.The recombinant AAV, rAAV-UF5, expressing the humanized green fluorescent protein (hGFP) transgene driven by a cytomegalovirus promoter was packaged as previously described by Zolotukhin et al. (43) with Ad5-infected 293 cells cotransfected with a helper plasmid (to providerepandcapfrom an ori construct) and a vector plasmid containing the cDNA flanked by AAV inverted terminal repeats. The vector was purified by two successive CsCl ultracentrifugation steps.
Animal experiments.Eleven female rhesus macaques ranging from 2 to 3 years of age and weighing between 2.8 and 4.2 kg were obtained (Covance Research). Sera from the animals were assayed for AAV antibodies prior to purchasing. The animals were housed at the University of Florida Animal Facility. The animals were sedated during all procedures by administration of 10 mg of Ketamine per kg intramuscularly. wt AAV (1010IU) was administered intravenously (into the right femoral vein) to two animals in a 3-ml suspension of bacteriostatic 0.9% sodium chloride. Three animals received 1010 IU of wt AAV into the left quadriceps muscle (6.5 cm from the patella) in a 500-l suspension of bacteri-ostatic 0.9% sodium chloride. Three other animals received 5⫻109IU of wt AAV in a 250-l suspension of bacteriostatic 0.9% sodium chloride into each nostril (for a total dosage of 1010IU). Two animals received coadministrations of wt AAV and a mutant form of Ad. These two animals received 5⫻109IU of wt AAV in a 250-l suspension of bacteriostatic 0.9% sodium chloride into each nostril (for a total wt AAV dosage of 1010IU) plus 5⫻107PFU of AdHR405 per nostril (for a total AdHR405 dosage of 108PFU). AdHR405 is a host range mutant form of Ad selected for growth on monkey cells (1, 10). The control animal was given 250l of bacteriostatic 0.9% sodium chloride into each nostril and was sedated regularly along with the other animals. The animals were bled every 7 days throughout the study.
[image:2.612.59.287.72.231.2]Genomic DNA analysis.High-molecular-weight DNA was extracted from an-imal tissue by using QIAamp tissue kits (Qiagen). The DNA concentration was determined by spectrophotometric analysis of the optical density at 260 nm. The DNA (30g) was digested for 24 h withKpnI (New England BioLabs) under conditions recommended by the manufacturer. The DNA was then separated by agarose gel electrophoreses (1% agarose) in TBE buffer (10 mM Tris borate, 2 mM EDTA [pH 8]). The agarose gel was acid treated for 20 min with 0.2 N HCl and denatured for 15 min with 1.5 M NaCl–0.5 M NaOH. The agarose gel was then neutralized with 3 M NaCl–0.5 M Tris and then blotted via capillary forces FIG. 1. Analysis of heparin-Sepharose affinity column fractions. (A)
AgCl-stained polyacrylamide gel of fractions eluted from a heparin-Sepharose affinity column. The lanes represent aliquots of each of 15 successive 1-ml fractions eluted with a continuous NaCl gradient at concentrations ranging from 0 to 1 M. (B) Ethidium bromide-stained agarose gel of wt AAV-specific PCR amplifica-tion products from each of the same 15 fracamplifica-tions.
FIG. 2. Analysis of CsCl gradient fractions. (A) Ethidium bromide-stained agarose gel of wt AAV-specific PCR amplification products from each of 10 successive fractions of a continuous CsCl density gradient. (B) Silver-stained polyacrylamide gel of the PCR-positive fractions.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.331.524.516.692.2]by using 20⫻SSC (1⫻SCC is 0.15 M NaCl plus 0.015 M sodium citrate) (Sigma) onto nylon membranes. The nylon membrane was then baked for 2 h at 80°C under vacuum. The membranes were hybridized at 60°C with an AAV [32P]DNA probe specific for wt AAV (AAV2) radiolabeled by random priming (Boehringer Mannheim). The hybridization solution contained 6⫻SSC, 0.5% SDS, 1⫻ Den-hardt’s solution, 20g of herring sperm DNA per ml, and 0.01 M EDTA. The membrane was washed in large volumes of 2⫻SSC–0.1% SDS at 60°C, dried, placed in an X-ray cassette (Kodak), and exposed to X-ray film (Kodak) for several days.
DNA PCR for detection of viral genomes and site-specific integration.
Genomic DNA samples from peripheral blood mononuclear cells (PBMCs) and from the sites of virus administration were purified with QIAamp blood kits (Qiagen). High-molecular-weight DNA was extracted from animal tissue by using QIAamp tissue kits. PCR was carried out with 100 ng of genomic DNA (or 1l of fractioned material) added to 50l of total PCR cocktail (ingredients and primers are given above). Thirty-five cycles of PCR were performed with the following program: 96°C for 1 min, 72°C for 1 min, and 56°C for 1 min. The products were analyzed on a 1% agarose gel, stained with ethidium bromide, transferred to nitrocellulose, and hybridized with a wt AAV-specific probe ra-diolabeled by random priming (Boehringer Mannheim).
To detect site-specific integration, DNA samples that were positive for AAV sequences by the internal PCR described above were also analyzed by the PCR dot blot methods described by Yang et al. (41), in which the 5⬘PCR primer was chosen from within the 3⬘end of the wt AAV sequence (5⬘-ATAAGTAGCAT GGCGGGTTA-3⬘) and was directed outward from the proviral insert while the 3⬘primer was chosen from within the AAVS1 site (5⬘-GCATAAGCCAGTAG AGCTCA-3⬘). Homology between the primer sequence and the rhesus sequence was confirmed by sequence alignment. PCR products were immobilized on nylon membranes by using a Schleicher and Schuell Minifold II dot blot manifold, and a random-primed32P-labeled probe from the end of the AAV genome [the 180-bpPvuII-XbaI fragment from pSub201(⫹)] was used for the hybridization. The specificity of this signal for AAV-chromosomal junctions was confirmed by comparison with a control in which only the internal AAV probe was used.
Fluorescence in situ hybridization (FISH) analysis.Metaphase chromosomes and interphase nucleus preparations were prepared by a mitotic shakeoff method (25). Hypotonic fixation and slide preparation were performed by standard cytogenetic methods. A wild-type AAV probe was labeled and hybridized to each preparation as previously described (25). Photomicrographic images of nuclear signals were acquired by using a cooled charge-coupled device camera under the control of the Metamorph software package.
Neutralizing-antibody assay.293 cells were plated in a 96-well plate at 50 to 75% confluency (5⫻103cells per well). The cells were cultured overnight at 37°C in humidified air containing 5% CO2. The following day, serial dilutions of animal serum (day 0 and day 21) were incubated with 105IU (equivalent to an MOI of 10) of recombinant AAV expressing the hGFP transgene (rAAV-UF5). The dilutions were performed in Hanks balanced salt solution a 100-l total volume. The sample was then incubated at 37°C for 1 h. After 1 h, the medium from the previously plated cells was removed and 100l of Dulbecco modified Eagle medium supplemented with 20% heat-inactivated fetal bovine serum and 200 U of penicillin-streptomycin per ml containing 2⫻105PFU of Ad was added. Additionally, 100l of the serially diluted serum plus rAAV-UF5 solu-tion was added to the wells. The cells were cultured for 24 h at 37°C in humidified air containing 5% CO2. After 24 h, the transgene product was visualized under a fluorescence microscope. The end point was defined as the dilution of serum which inhibited the transgene efficiency by at least 10-fold.
Antigen-specific lymphocyte proliferation assay to assess cell-mediated im-munity to AAV.Heparinized whole blood (5 ml) was collected and diluted 1:1 in
Hanks buffered salt solution in a conical centrifuge tube. Ficoll-Hypaque (5 ml; Pharmacia) was slowly layered at the bottom of the conical tube. The tube was then centrifuged for 30 min at 500⫻gat room temperature. The layer above the clear layer was carefully removed with a sterile transfer pipette. The removed material was transferred to a centrifuge tube containing 10 ml of Hanks buffered salt solution and centrifuged for 10 min at 500⫻gat room temperature. The supernatant was removed, and the cell pellet was washed again with 10 ml of Hanks buffered salt solution and recentrifuged for 10 min at 500⫻gat room temperature. The supernatant was discarded, and the cell pellet was resuspended in 2 ml of RPMIC⫹medium (CellGrow). The cells were counted by a Trypan blue exclusion method.-Mercaptoethanol was added at 2l per ml of cell suspension (adjusted to account for 106cells per ml). Two 96-well plates were set up for every animal, one for the antigen (VP3 capsid proteins) and a second for the mitogen phytohemagglutinin as a positive control. Cells were plated on the wells, and the respective agent was added and incubated for 3 days at 37°C in humidified air containing 5% CO2. On day 3, 20l of a 1:20 [3H]thymidine dilution was added to the mitogen-treated plate. On day 4, the mitogen-treated cells were removed and the level of radioactivity was determined. On day 5, 20 l of a 1:20 [3H]thymidine dilution was added to the antigen-treated plate. After 24 h, the plate was harvested and the level of radioactivity was determined by liquid scintillation counting.
Tissue preparation and histology.Tissues from the lungs, nasal passages, trachea, thymus, bronchial lymph nodes, heart, liver, spleen, pancreas, kidney, jejunum, mesenteric lymph nodes, gonads, brain, and muscles were isolated aseptically and placed in 4% paraformaldehyde for 24 h at 4°C. The tissues were then embedded in paraffin, and 10-m sections were made. The sections were then stained with hematoxylin and eosin, coverslipped, and photographed with a Zeiss Axioskop upright microscope.
RESULTS
Preparation of wt AAV.
Since one of the primary goals of this
study was to characterize immune responses to AAV in both
FIG. 3. Quantitation of biological and physical titers of wt AAV. (A) Autoradiographic images of nylon membranes onto which had been immobilized Ad-infected 293 cells infected with serial dilutions of the wt AAV stock used in later rhesus experiments. A32P-labeled wt AAV-specific probe was used to detect “infectious centers”, each of which appears as a discrete dot on the autoradiograph. (B) Ethidium bromide-stained gel of amplification products from the quantitative competitive PCR used to quantitate wt AAV genomes.FIG. 4. Southern blot and PCR analysis for wt AAV DNA at the site of administration. (A) Southern blot of unamplified,KpnI-digested genomic DNA isolated from the site of wt AAV administration. Liver tissue from animals infected intravenously (lanes 1 and 2), muscle tissue from animals infected intramuscularly (lanes 4 to 6), and nasal tissue from animals infected intranasally either without (lanes 8 and 9) or with (lanes 10 and 11) helper Ad were exam-ined. Liver, muscle, and nasal epithelial DNAs from a saline-injected control animal were also examined (lanes 3, 7, and 12, respectively). Plasmid DNAs equating to 0.1, 1, 10, and 100 copies of wt AAV genomes per cell were included as standards (lanes 13 to 16, respectively). (B) PCR analysis for internal AAV-Rep gene sequences was performed for each of the same samples.
on November 9, 2019 by guest
http://jvi.asm.org/
latent and productive AAV infections, it was essential to
elim-inate contaminating proteins that could serve as adjuvants to
an anti-AAV host response. To purify wild-type AAV free of
Ad proteins and other contaminants, an affinity purification
method based on the recently described discovery of heparan
sulfate proteoglycan as an attachment receptor for AAV2 was
developed (37). A cleared lysate of Ad5- and AAV2-infected
293 cells was loaded directly onto a heparin affinity column,
washed in low-salt buffer, and eluted with a continuous NaCl
gradient, ranging in concentration from 0 to 1 M. Fifteen
successive fractions were analyzed for viral proteins by
SDS-PAGE with silver staining (Fig. 1A) and for viral genomes by
PCR (Fig. 1B). AAV genomes were detected in fractions 1
through 10, while the majority of cellular proteins were eluted
in fractions 9 through 15. While this is not quantitative, more
intense PCR signals were observed in fractions 6 to 9. These
fractions corresponded to a concentration of 0.4 to 0.6 M NaCl
in the elution buffer, indicating that this would be the optimal
salt concentration for eluting the bound viral particles. The
positive fractions were further purified by CsCl density
gradi-ent ultracgradi-entrifugation, and the resultant CsCl gradigradi-ent
frac-tions were analyzed for wt AAV DNA by PCR. PCR-positive
fractions (Fig. 2A) were observed at a refractive index of
1.3715 (
⫽
1.41 g/cm
3). Peak fractions were pooled and
ex-amined by PAGE with silver staining (Fig. 2B). In the final
material, only three protein bands were detectable, and these
migrated at apparent molecular masses identical to those
pre-dicted for AAV capsid proteins (62, 73, and 87 kDa). No
contaminating proteins were detectable.
To determine the yield and infectivity of virus, we
indepen-dently assessed the physical and biological titers of wt AAV in
the preparation. The physical titer was determined by genome
quantitation via QC-PCR. By using this technique, it was
esti-sites within this locus, the Rep binding element and terminal
resolution site (
trs
), while the intervening sequence was
iden-tical. Based on these findings, an in vivo model of latent AAV
infection was established. Each of eight rhesus macaques was
infected with 10
10IU by one of several routes (intranasal,
intravenous, or intramuscular). Two additional animals had a
productive infection with wt AAV established by intranasal
coadministration of wt AAV and Ad2HR405, a host range
mutant Ad selected for growth on monkey cells (10). One
additional animal was inoculated with isotonic saline
intrana-sally as a control. All the animals were analyzed for antibodies
to AAV capsid proteins prior to experiments.
Analysis of DNA persistence at the site of virus entry.
To
determine whether infection with wt AAV resulted in
persis-tence of AAV DNA at the site of inoculation, DNA was
iso-lated from each site and examined by Southern blot analysis.
Based on a previous study with recombinant AAV indicating
that systemically delivered vector is distributed mostly to the
liver (17), liver tissue was taken from animals infected
venously. Muscle tissue was used from animals infected
intra-muscularly, and nasal tissue was used from animals infected
intranasally with and without helper viruses. The abundance of
viral DNA was below the level of detection by Southern
blot-ting at each of the sites of infection (Fig. 4A), despite having a
sensitivity of
⬍
0.1 copy of AAV DNA per cell. This was
un-expected, given that we delivered at least 10
3to 10
4viral
genome copies per cell at the site of administration in the
muscle, assuming that between 10
7and 10
8nuclei would be
present in the region of muscle subtended by a 500-
l
injec-tion. The same samples were then analyzed by a PCR assay for
internal wt AAV sequences (sensitive to 0.001 copy per cell),
and several were found to be positive for AAV DNA (Fig. 4B).
PBMC DNA.
One previous study (21) had shown evidence of
[image:4.612.77.267.73.302.2]AAV DNA persistence in peripheral blood cells after naturally
FIG. 5. PCR detection of wt AAV sequences in PBMC DNA. GenomicDNAs isolated from purified PBMCs from intravenously infected animals (lanes 1 and 2), intramuscularly infected animals (lanes 3 to 5), animals infected intra-nasally without helper Ad (lanes 6 and 7), and animals infected intraintra-nasally with both AAV and Ad2HR405 (lanes 8 and 9) were amplified by using wt AAV primers and probed with AAV sequences. DNA from a saline-injected control animal was also examined (lane 10). (A) Ethidium bromide-stained agarose gel of the PCR products. (B) Southern blot hybridization of that same gel.
FIG. 6. PCR dot blot assay for site-specific integration into the rhesus AAVS1 site. A PCR dot blot assay specific for AAV-AAVS1 junctions was performed with both primers (bottom row of dots) or with the single internal AAV primer (top row), which serves as a control to distinguish signals from AAV-AAV junctions. Lanes: 1, positive control DNA from AAV-infected IB3-1 cells; 2, negative control DNA from uninfected IB3-1 cells; 3, negative control DNA from a saline-injected monkey; 4 and 5, cell DNA from the nose of AAV-and Ad2HR405-infected monkeys; 6 AAV-and 7, cell DNA from the nose of animals infected with AAV alone; 8 to 10, muscle DNA from animals infected intramus-cularly; 11 and 12, liver cell DNA from animals receiving intravenous doses of AAV.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.324.537.571.637.2]occurring infections. To determine whether this could have
occurred in our animals, genomic DNA was isolated from
lymphocytes and amplified by PCR with wt AAV primers.
DNA was isolated from lymphocytes 21 days after viral
infec-tion and amplified with wt AAV primers (see Materials and
Methods) under optimized conditions. Lymphocytes isolated
from both of the intravenously infected animals were positive
for AAV DNA (Fig. 5, lanes 1 and 2). Of the intramuscularly
infected animals, only one (95B005) was positive for AAV
DNA (lane 5). Additionally, one of the intranasally infected
animals was positive for AAV DNA (95B032) (lane 7). Neither
of the intranasally infected animals given helper virus was
positive for AAV DNA (lanes 8 and 9).
Site-specific integration.
To determine whether site-specific
integration had occurred within the rhesus AAVS1-like locus,
all organ DNA samples that had scored positive by PCR for
internal AAV sequences were also assayed by a junction PCR
dot blot assay described by Yang et al. (41). Junction
se-quences were amplified with a 5
⬘
primer within the AAV “tail”
(3
⬘
end) directed outward from the genome sequence (5
⬘
-AT
AAGTAGCATGGC-GGGTTA-3
⬘
) and a 3
⬘
primer from the
AAVS1 site (5
⬘
-GCATAAGCCAGTAGAGCTCA-3
⬘
). A
dot-blot hybridization was used to detect amplified products, since
previously published data indicated that this amplification
pro-duces a heterogeneous mix of products that are not easily
distinguished by agarose gel electrophoresis with ethidium
staining. To distinguish tail-to-tail junctions between two viral
genomes from bona fide AAV-cell DNA junctions, the
reac-tions were also performed with a single internal AAV primer
in the reaction. This primer alone would be expected to
am-plify inverted tandem (tail-to-tail) forms whether or not they
are integrated.
[image:5.612.108.496.72.207.2]The positive control for this assay was genomic DNA
ex-tracted from a culture of IB3-1 cells (CF bronchial epithelial
cell line) infected with wt AAV2 at an MOI of 5. This cell line
showed both a strong signal with the double primer pair (Fig.
6, bottom row) and a weaker signal with the single internal
AAV primer, consistent with a tail-to-tail junction between two
viral genomes. The negative control was genomic DNA from
uninfected IB3-1 cells. Evidence of site-specific integration in
both the nasal epithelium and the PBMCs from one of the
animals that had received vector intranasally was observed by
this assay (Fig. 6). Two of the animals that had received
intra-venous injections of AAV showed very faint site-specific
inte-gration signals in hepatocyte DNA. The specificity of this
sig-nal for AAV-cell DNA junctions as opposed to AAV-AAV
junctions was confirmed by the lack of amplification with the
single AAV PCR primer. DNA sequencing of such junctions is
a subject for future studies.
[image:5.612.313.547.492.681.2]FIG. 7. FISH for detection of wt AAV DNA in interphase nuclei of PBMCs. PBMC cultures taken from animals 21 days after wt AAV infection were examined for viral DNA. wt AAV DNA was visualized in the interphase nuclei of lymphocytes isolated from animals coinfected with wt AAV and Ad (A), an intramuscularly injected animal (B), and a control animal (C).
[image:5.612.53.293.606.695.2]FIG. 8. Frequency of FISH positivity. A total of 100 interphase nuclei were counted, and the number of cells displaying at least one signal was counted and considered a positive FISH result. No signals were observed in interphase nuclei from myoblast, nasal epithelial, or skin fibroblast cell cultures. IM, intramuscu-lar.
TABLE 1. FISH signals on interphase nuclei of PBMCs
Treatment
No. of nuclei showing following no. of FISH signals/nucleusa
0 1 2 3 4
Saline control
99
1
0
0
0
Nasal (AAV alone)
97
2
1
0
0
Nasal (w/helper)/95B039
95
3
2
0
0
Nasal (w/helper)/96C082
91
3
5
1
0
Intramuscular
85
12
1
0
2
aOne hundred interphase nuclei were examined from each individual animal.
Anywhere from 0 to 4 FISH signals per nucleus were observed. The values in the table indicate the number of nuclei showing each of the indicated numbers of signals per nucleus.
on November 9, 2019 by guest
http://jvi.asm.org/
FISH analysis.
FISH was performed on cell cultures isolated
either from solid organs at the site of administration (muscle
or nose), from the skin fibroblasts from the intravenously
in-fected animals, or from PBMCs from wt AAV-inin-fected
ani-mals 21 days postinfection. Of particular note, none of the
muscle, nose, or skin samples were positive by FISH, while
numerous PBMCs were positive. These FISH data were
con-sistent with the Southern blot data indicating a low copy
num-ber of AAV DNA at the site of administration. FISH
prepa-rations of lymphocyte interphase nuclei showed wt AAV DNA
signals in animals infected intranasally with helper virus (Fig.
7A) and animals infected intramuscularly (Fig. 7B). No signals
were observed in lymphocyte interphase nuclei of the control
animal (Fig. 7C). Additionally, the signal numbers were
greater in the lymphocytes isolated from the animal infected
intranasally with wt AAV and helper virus (Table 1).
Meta-phase nuclei were examined by FISH for wt AAV DNA in all
the groups of animals, and none were positive. However, due
to the low mitotic index of these primary cultures, there were
only one to five metaphase nuclei available for scoring on most
culture samples. This very small number of metaphase spreads
present would make it difficult to detect integration by this
method.
The overall incidence of FISH-positive PBMCs was also
analyzed. Of 100 PBMC interphase nuclei examined from each
study group, 3% of the PBMCs from animals infected with
AAV alone intranasally were positive, compared with 15%
from the intramuscularly infected animals and 6% from
ani-mals coinfected with AAV and Ad (Fig. 8). The 1% of
lym-phocytes that apparently show positive FISH signals represent
the background on this assay.
Neutralizing-antibody responses to wt AAV infection.
To
assess whether rhesus monkeys infected with wt AAV
devel-oped humoral immune responses, sera were collected prior to
and 21 days after infection and assayed for in vitro anti-AAV
neutralizing activity. Neutralizing activity was found in most of
the preinfection sera. A significant increase in
neutralizing-antibody response was defined as a fourfold increase in
neu-tralizing titer between the pre- and postinfection samples.
Most of the infected animals did develop a significant
neutral-izing-antibody response (Fig. 9). This included animals that
were infected with AAV alone by the intravenous or
intramus-cular route and animals infected intranasally with both AAV
and Ad. In contrast, animals infected nasally with wt AAV
alone did not develop a significant increase in anti-AAV
neu-tralizing antibodies (Table 2). To determine whether 21 days
was sufficient to detect a response, one of these animals
(96B026) was monitored for over 7 weeks and bled on two
additional occasions, and it still did not have a detectable
response. This animal was later immunized with 100
g of
purified VP3 as a positive control and was then found to
develop neutralizing antibodies within 21 days after
adminis-tration (data not shown).
Cell-mediated immunity to wt AAV.
To determine whether
[image:6.612.76.533.74.409.2]cell-mediated immune responses to AAV had occurred,
lym-phocytes from wt AAV-infected animals were exposed ex vivo
FIG. 9. Anti-AAV neutralizing antibody activity. Shown are fluorescence micrographs of Ad-infected 293 cells coinfected with a rAAV-GFP vector preincubated with monkey serum from either day 0 (day of infection) or day 21 after in vivo coinfection with wt AAV and Ad2HR405.on November 9, 2019 by guest
http://jvi.asm.org/
to AAV capsid antigen and the extent of antigen-specific
stim-ulation of lymphocyte proliferation was assessed. The
stimula-tion index was calculated by dividing the number of cpm of
[
3H]thymidine incorporated into lymphocyte cultures in the
presence of the specific antigen (with 1, 5, or 10
g of the
antigen) by the number of cpm incorporated into parallel
cul-tures grown in the absence of antigen (28). Based on previous
norms for this assay, a positive response was defined as a
stimulation index of 3. The viability of each of these PBMC
cultures was confirmed by PHA stimulation of parallel wells,
and each showed a stimulation index of
ⱖ
3.0.
At baseline, all animals were negative in this assay. On day
26, animals infected with wt AAV intravenously (95B002 and
95B003) were negative in this assay (stimulation index,
⬍
1), as
were the animals infected intramuscularly (stimulation indices,
1.8 and 2.7) and intranasally (stimulation indices, 2.1 and 1.9)
(Fig. 10). In contrast, animal 95B039, which was infected
in-tranasally with both wt AAV and Ad, had a stimulation index
of 3.8. Thus, only the animal with productive helper virus
infection developed a specific cell-mediated immune response
to AAV capsid proteins.
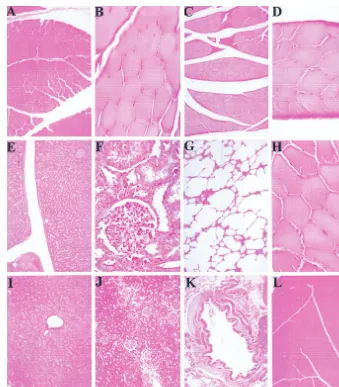
Tissue examination.
Histological examination was
per-formed to determine if inflammation or cellular infiltration
had occurred in response to wt AAV infection at the site of
infection. Tissues isolated from muscle, liver, kidney, and lungs
from animals infected intramuscularly and intravenously were
histologically examined. No morphological abnormalities were
observed in any case compared to matched tissue samples from
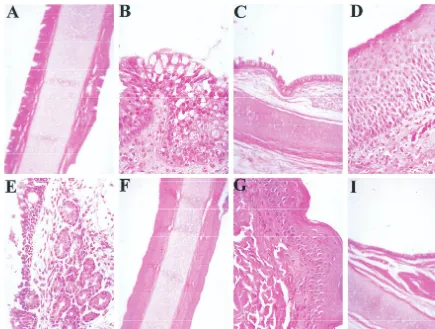
the control animal (Fig. 11). Tissues from the nasal cavity and
trachea of intranasally infected animals were also examined
(Fig. 12). No abnormalities were observed in the animals
in-fected intranasally with wt AAV alone. However, animal
95B039, which was coinfected with wt AAV and Ad
demon-strated goblet cell hyperplasia (Fig. 12B) in the nasal
epithe-lium. The control animal had no apparent increase in goblet
cell number.
DISCUSSION
Despite the high prevalence of AAV infection in humans,
relatively little data is available with regard to the latency of wt
AAV in vivo. Current models of AAV latency are derived from
experiments performed with immortalized and primary cell
lines. Our findings indicate that viral DNA persists both at the
site of administration and in peripheral blood cells. Notably,
the only published data about isolation of latent AAV DNA
from humans was from peripheral blood cells in a pattern
consistent with our findings (21). The data presented here did
not allow a precise determination of the in vivo integration
frequency. Although site-specific integration was apparently
present in some instances, the frequency of this process
ap-pears to be rather low.
Previous findings with recombinant AAV in the respiratory
tract also indicated infrequent integration (1). However,
pre-vious studies with recombinant AAV vectors in muscle have
indicated that vector genomes persist in muscle either as
inte-grated proviral genomes or as high-molecular-weight
concate-mers (12, 13, 40). There are several potential reasons why our
studies might have underestimated the integration frequency
somewhat. One possibility is that sampling of the sites of
ad-ministration may have been imprecise. It is also possible that
despite having integrated into host cells, these cells were
elim-inated by the immune response. This seems very unlikely,
how-ever, since elimination of infected cells by cytotoxic T
lympho-cytes would typically be associated with positive findings on the
antigen-specific lymphocyte proliferation assay and with
histo-logical changes at the site of delivery. Finally, it is possible that
rhesus monkeys simply do not represent a suitable host for
AAV. The fact that productive infections have been achieved
in this model (1) and that it possesses an AAVS1-like site with
moderate homology to the human sequence argue against that
possibility. However, there are significant limitations to the
rhesus model, both as a model of productive infection and as a
model of latency. The host range mutant Ad strains are less
efficient for replication in culture than are wt Ad strains and
thus may not faithfully reproduce Ad infection in vivo.
Fur-thermore, the AAVS1-like sequences we identified have not
been proven to be functional for AAV integration.
[image:7.612.54.293.91.235.2]FISH analysis of PBMCs also suggested that wt AAV
se-quences persist in these cells, as evidenced by signals on
inter-phase nuclei. However, no signals were detectable on
meta-phase chromosomes. Since less than 15% of cells were positive
FIG. 10. Antigen-specific lymphocyte proliferation. Peripheral blood [image:7.612.54.290.462.652.2]lym-phocytes from animals infected with wt AAV (intravenous [i.v.], intramuscular [i.m.], intranasal, and intranasal coinfection with Ad) were incubated in the presence of wt AAV capsid proteins (VP3) and assayed for proliferative re-sponses. The stimulation index represents the ratio of the amount (cpm) of [3H]thymidine incorporated in the presence of the specific antigen to the amount incorporated by parallel cultures grown in the absence of the specific antigen. A positive response to the AAV capsid antigen is indicated by a stimulation index ofⱖ3.
TABLE 2. Anti-AAV neutralization titer 21 days after
AAV inoculation
Mode of administration/ animal no.
Titera
ⱖ4-fold increase in titer
Preadminis-tration Postadministration(21 days)
Intravenous/95B002
0
2,560
⫹
Intravenous/95B003
0
320
⫹
Intramuscular/95B005
40
320
⫹
Intramuscular/95B025
320
2,560
⫹
Intramuscular/95B041
320
2,560
⫹
Nasal/96B026
0
0
⫺
Nasal/95B032
160
320
⫺
Nasal/96C120
40
40
⫺
Nasal (w/helper)/95B039
0
40
⫹
Nasal (w/helper)/96C082
320
1,280
⫹
Saline control/96C153
40
40
⫺
aTiters are expressed as the reciprocal dilution of serum required for a 10-fold
neutralization in vitro.
on November 9, 2019 by guest
http://jvi.asm.org/
in the interphase nuclei in every case, however, one would not
expect the examination of such a small number of metaphase
spreads (five or fewer per sample) to show integration even if
it occurred in every case where AAV DNA was persistent. The
accessibility of these rather short target sequences to
hybrid-ization with the FISH probes would also be expected to be
greater with the unwound chromatin of interphase nuclei than
with that observed in condensed metaphase chromosomes.
There was also a notable lack of FISH signals on nuclei from
primary cells isolated from the site of vector administration.
While our group has found positive FISH signals from
bron-chial epithelial cells harvested from the site of rAAV-CFTR
administration (1), the possibility remains that the process of
establishing primary culture somehow selects for a population
of cells that are less likely to be latently infected. Recent in vivo
data with rAAV indicate that terminally differentiated cells
may be more permissive for AAV infection. If this is the case,
the process of establishing a primary culture, which favors the
growth of less differentiated cells, could substantially
underes-timate the actual frequency of integration in vivo.
Previous studies indicated that 60% of adults are
seroposi-tive for AAV and that this seroconversion occurs early in life
(6). It has never been known whether this humoral immune
response to AAV capsid was elicited primarily by productive
infection or by latent infection. The evidence presented here
suggests that nonhuman primates can develop a
neutralizing-antibody response to wt AAV in the absence of helper virus if
infected parenterally but that mucosal exposure without helper
virus does not elicit such a response. These results are
com-patible with data generated from experiments with rAAV, in
which administration to the maxillary sinus did not elicit an
anti-capsid antibody response, while intramuscular
administra-tion did so in several cases (12, 33, 40).
[image:8.612.133.472.70.457.2]It is also notable that cell-mediated immune responses to a
single dose of rAAV were observed only in the presence of
active AAV replication, i.e., in the presence of helper virus.
This is also consistent with previous results with rAAV. These
data are consistent with the findings of Joos et al. (23), which
indicated that antigen-presenting cells are relatively resistant
to AAV infection. Alternatively, the helper virus may simply be
providing an adjuvant effect. It is important to point out that
these studies represent a single-exposure paradigm and that
FIG. 11. Absence of cellular infiltration at site of injection after wt AAV infection. Paraformaldehyde-fixed tissue sections were prepared from animals 21 days after infection with wt AAV. Muscle tissue from intramuscularly infected animals 96C041 (A), 95B005 (B), 95B025 (C), and 95B025 (D) was examined for infiltrative cellular responses at the site of injection. Liver (E), kidney (F), lung (G), and muscle (H) tissue from intravenously injected animal was also examined. No histological abnormalities were observed compared to liver (I), kidney (J), lung (K), and muscle (L) tissues from the control animal.on November 9, 2019 by guest
http://jvi.asm.org/
repeated dosing might have resulted in more vigorous
re-sponses.
To limit any adjuvant effects from contaminants in our wt
AAV preparations, careful purification and quality control
as-says were used. We used properties of the recently described
AAV receptor to purify large quantities of wt AAV. The use of
a heparin affinity column prior to isopycnic centrifugation
yield-ed a large viral particle number. Additionally, the viral stock
appeared to be free of Ad proteins and other contaminating
proteins. Viral genomes were quantified by QC-PCR, and the
infectivity of the virus was quantified by the infectious-center
assay. Thus, the physical titer was 10
13particles per ml,
com-pared to the biological titer of 10
11IU per ml, for a
particle-to-IU ratio of 100, which is nearly optimal for a DNA virus.
The method described herein is similar to one recently
de-scribed by Zolotukhin et al. (42).
In summary, the rhesus macaque was used as a model of
latent wt AAV infection. In this model, wt AAV persisted both
at the site of administration and in peripheral blood cells and
in some instances integrated within an AAVS1-like site.
Fur-thermore, latent AAV infection was capable of eliciting
hu-moral immune responses but not cell-mediated immunity.
While the immune response data is quite consistent with the
results of previous reports with wt AAV, the lack of
site-specific integration calls into question the relevance of such
findings in cell cultures ex vivo. Additional studies by methods
more sensitive for detecting low-frequency integration are
re-quired before a definite conclusion can be drawn about the
capacity of AAV to integrate site specifically in vivo.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institute for
Diabetes, Digestive, and Kidney Diseases (DK51809).
Many thanks to David Muir for assistance with column
chromatog-raphy techniques, to Kye Chesnut, Barry Byrne, and Nick Muzyczka
for advice on this work, and to Mark Atkinson for assistance with
immune response assays.
REFERENCES
1.Afione, S. A., C. K. Conrad, W. G. Kearns, S. Chunduru, R. Adams, T. C. Reynolds, W. B. Guggino, G. R. Cutting, B. J. Carter, and T. R. Flotte.1996. In vivo model of adeno-associated virus vector persistence and rescue. J. Vi-rol.70:3235–3241.
2.Alexander, I. E., D. W. Russell, A. M. Spence, and A. D. Miller.1996. Effects of gamma irradiation on the transduction of dividing and nondividing cells in brain and muscle of rats by adeno-associated virus vectors. Hum. Gene Ther.
7:841–850.
3.Berns, K. I.1990. Parvovirus replication. Microbiol. Rev.54:316–329. 4.Berns, K. I., and R. M. Linden.1995. The cryptic life style of
adeno-associated virus. Bioessays17:237–245.
5.Berns, K. I., T. C. Pinkerton, G. F. Thomas, and M. D. Hoggan.1975. Detection of adeno-associated virus (AAV)-specific nucleotide sequences in DNA isolated from latently infected Detroit 6 cells. Virology68:556–560. 6.Blacklow, N. R.1988. Adeno-associated viruses of humans, p. 165–174.In
J. R. Pattison (ed.), Parvoviruses and human disease. CRC Press, Inc., Boca Raton, Fla.
7.Blacklow, N. R., M. D. Hoggan, A. Z. Kapikian, J. B. Austin, and W. P. Rowe.
[image:9.612.85.520.71.400.2]1968. Epidemiology of adenovirus-associated virus infection in a nursery FIG. 12. Lack of cellular infiltration after intranasal wt AAV infection. Paraformaldehyde-fixed tissue sections from the nose and trachea were prepared from animals 21 days after infection with wt AAV. Infection with helper virus (AdHR405) gave no cellular response in tracheal tissue (A and C) compared to the control (F and I). There was, however, an enlargement of goblet cells in intranasally infected animals (B). No infiltrative cellular response was observed in animals infected intranasally with wt AAV alone (D and E) compared to the control (G).
on November 9, 2019 by guest
http://jvi.asm.org/
Hum. Gene Ther.8:659–669.
13.Fisher, K. J., K. Jooss, J. Alston, Y. Yang, S. E. Haecker, K. High, R. Pathak, S. E. Raper, and J. M. Wilson.1997. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med.3:306–312.
14.Flannery, J. G., S. Zolotukhin, M. I. Vaquero, M. M. LaVail, N. Muzyczka, and W. W. Hauswirth.1997. Efficient photoreceptor-targeted gene expres-sion in vivo by recombinant adeno-associated virus. Proc. Natl. Acad. Sci. USA94:6916–6921.
15. Flotte, T., B. Carter, C. Conrad, W. Guggino, T. Reynolds, B. Rosenstein, G. Taylor, S. Walden, and R. Wetzel.1996. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Hum. Gene Ther.7:1145–1159.
16. Flotte, T. R., S. A. Afione, C. Conrad, S. A. McGrath, R. Solow, H. Oka, P. L. Zeitlin, W. B. Guggino, and B. J. Carter.1993. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc. Natl. Acad. Sci. USA90:10613–10617. 17. Flotte, T. R., X. Barraza-Ortiz, R. Solow, S. A. Afione, B. J. Carter, and W. B.
Guggino.1995. An improved system for packaging recombinant adeno-associated virus vectors capable of in vivo transduction. Gene Ther.2:29–37. 18. Flotte, T. R., and B. J. Carter.1995. Adeno-associated virus vectors for gene
therapy. Gene Ther.2:357–362.
19. Giraud, C., E. Winocour, and K. I. Berns.1995. Recombinant junctions formed by site-specific integration of adeno-associated virus into an episome. J. Virol.69:6917–6924.
20. Giraud, C., E. Winocour, and K. I. Berns.1994. Site-specific integration by adeno-associated virus is directed by a cellular DNA sequence. Proc. Natl. Acad. Sci. USA91:10039–10043.
21. Grossman, Z., E. Mendelson, F. Brok-Simoni, F. Mileguir, Y. Leitner, G. Rechavi, and B. Ramot.1992. Detection of adeno-associated virus type 2 in human peripheral blood cells. J. Gen. Virol.73:961–966.
22. Hoggan, M., G. Thomas, F. Thomas, and F. Johnson.1972. Continuous carriage of adeno-associated virus genome in cell culture in the absence of helper adenoviruses, p. 243–253.InProceedings of the 4th Lepetit Collo-quium, Cocoyac, Mexico. North-Holland, Amsterdam, The Netherlands. 23. Jooss, K., Y. Yang, K. J. Fisher, and J. M. Wilson.1998. Transduction of
dendritic cells by DNA viral vectors directs the immune response to trans-gene products in muscle fibers. J. Virol.72:4212–4223.
24. Kaplitt, M. G., P. Leone, R. J. Samulski, X. Xiao, D. W. Pfaff, K. L. O’Malley, and M. J. During.1994. Long-term gene expression and phenotypic correc-tion using adeno-associated virus vectors in the mammalian brain. Nat. Genet.8:148–154.
25. Kearns, W. G., S. A. Afione, S. B. Fulmer, M. C. Pang, D. Erikson, M. Egan, M. J. Landrum, T. R. Flotte, and G. R. Cutting.1996. Recombinant adeno-associated virus (AAV-CFTR) vectors do not integrate in a site-specific fashion in an immortalized epithelial cell line. Gene Ther.3:748–755. 26. Kessler, P. D., G. M. Podsakoff, X. Chen, S. A. McQuiston, P. C. Colosi, L. A.
Laughlin, S. McLaughlin, N. Muzyczka, M. Rocchi, and K. I. Berns.1990. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA87:2211–2215.
32. Lewin, A. S., K. A. Drenser, W. W. Hauswirth, S. Nishikawa, D. Yasumura, J. G. Flannery, and M. M. LaVail.1998. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat. Med.4:967–971. (Erratum,4:1081.)
33. Manning, W. C., S. Zhou, M. P. Bland, J. A. Escobedo, and V. Dwarki.1998. Transient immunosuppression allows transgene expression following read-ministration of adeno-associated viral vectors. Hum. Gene Ther.9:477–485. 34. Peel, A. L., S. Zolotukhin, G. W. Schrimsher, N. Muzyczka, and P. J. Reier.
1997. Efficient transduction of green fluorescent protein in spinal cord neu-rons using adeno-associated virus vectors containing cell type-specific pro-moters. Gene Ther.4:16–24.
35. Samulski, R. J., X. Zhu, X. Xiao, J. D. Brook, D. E. Housman, N. Epstein, and L. A. Hunter.1991. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J.10:3941–3950. (Erratum,
11:1228, 1992.)
36. Snyder, R. O., C. H. Miao, G. A. Patijn, S. K. Spratt, O. Danos, D. Nagy, A. M. Gown, B. Winther, L. Meuse, L. K. Cohen, A. R. Thompson, and M. A. Kay.1997. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat. Genet.
16:270–276.
37. Summerford, C., and R. J. Samulski.1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol.72:1438–1445.
38. Wagner, J. A., T. Reynolds, M. L. Moran, R. B. Moss, J. J. Wine, T. R. Flotte, and P. Gardner.1998. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet351:1702–1703. (Letter.)
39. Xiao, X., J. Li, T. J. McCown, and R. J. Samulski.1997. Gene transfer by adeno-associated virus vectors into the central nervous system. Exp. Neurol.
144:113–124.
40. Xiao, X., J. Li, and R. J. Samulski.1996. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol.70:8098–8108.
41. Yang, C. C., X. Xiao, X. Zhu, D. C. Ansardi, N. D. Epstein, M. R. Frey, A. G. Matera, and R. J. Samulski.1997. Cellular recombination pathways and viral terminal repeat hairpin structures are sufficient for adeno-associated virus integration in vivo and in vitro. J. Virol.71:9231–9247.
42. Zolotukhin, S., B. J. Byrne, E. Mason, I. Zolotukhin, M. Potter, K. Chesnut, C. Summerford, R. J. Samulski, and N. Muzyczka.Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Submitted for publication.
43. Zolotukhin, S., M. Potter, W. W. Hauswirth, J. Guy, and N. Muzyczka.1996. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol.70:4646–4654.
on November 9, 2019 by guest
http://jvi.asm.org/