0022-538X/07/$08.00
⫹
0
doi:10.1128/JVI.00968-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Precise Identification of a Human Immunodeficiency Virus Type 1
Antigen Processing Mutant
䌤
Peter Zimbwa,
1† Anita Milicic,
1†* John Frater,
1Thomas J. Scriba,
1Antony Willis,
2Philip J. R. Goulder,
3Tilly Pillay,
1Huldrych Gunthard,
4Jonathan N. Weber,
5Hua-Tang Zhang,
1and Rodney E. Phillips
1The James Martin 21st Century School at The Peter Medawar Building for Pathogen Research, Nuffield Department of
Clinical Medicine, University of Oxford, South Parks Road, Oxford OX1 3SY, United Kingdom
1; Department of
Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, United Kingdom
2; Department of Pediatrics,
University of Oxford, Oxford OX3 9DU, United Kingdom
3; University Hospital Zurich, Department of Medicine,
Division of Infectious Diseases and Hospital Epidemiology, Ramistrasse 100, CH-8091 Zurich,
Switzerland
4; and Jefferiss Research Laboratories, Wright-Fleming Institute, Imperial College,
St Mary’s Hospital, Norfolk Place, London W2 1PG, United Kingdom
5Received 11 May 2006/Accepted 9 November 2006
Human immunodeficiency virus type 1 (HIV-1) evokes a strong immune response, but the virus persists.
Polymorphisms within known antigenic sites result in loss of immune recognition and can be positively
selected. Amino acid variation outside known HLA class I restricted epitopes can also enable immune escape
by interfering with the processing of the optimal peptide antigen. However, the lack of precise rules dictating
epitope generation and the enormous genetic diversity of HIV make prediction of processing mutants very
difficult. Polymorphism E169D in HIV-1 reverse transcriptase (RT) is significantly associated with
HLA-B*0702 in HIV-1-infected individuals. This polymorphism does not map within a known HLA-HLA-B*0702 epitope;
instead, it is located five residues downstream of a HLA-B*0702-restricted epitope SPAIFQSSM (SM9). Here
we investigate the association between E169D and HLA-B*0702 for immune escape via the SM9 epitope. We
show that this single amino acid variation prevents the immune recognition of the flanked SM9 epitope by
cytotoxic T cells through lack of generation of the epitope, which is a result of aberrant proteasomal cleavage.
The E169D polymorphism also maps within and abrogates the recognition of an HLA-A*03-restricted RT
epitope MR9. This study highlights the potential for using known statistical associations as indicators for viral
escape but also the complexity involved in interpreting the immunological consequences of amino acid changes
in HIV sequences.
Cytotoxic T lymphocytes (CTL, CD8
⫹T cells) have T-cell
receptors which specifically recognize antigens presented by
HLA class I molecules. CTL have a key role in the immune
defense against viral infection and are crucial for the
contain-ment of human immunodeficiency virus type 1 (HIV-1)
repli-cation (4, 13, 26). In a single individual, HIV variation is
enormous. This results from a high proliferative capacity and
error-prone reverse transcriptase of HIV (33, 47) and so
en-ables the virus to evade HIV-1-specific CTLs (13, 16, 26).
Amino acid variation within HLA class I-restricted epitopes,
positively selected by host immune pressure, can lead to escape
from CTL recognition (13, 16, 26, 38). Evasion of the CTL
recognition and persistence in the face of a vigorous CTL
response typifies HIV-1 infection (26, 38, 40). HIV can escape
CTL responses when amino acid variation interferes with
pep-tide binding to the HLA class I (2, 10, 12, 15, 19, 27, 39) or
alters epitope recognition by the T-cell receptor (24, 41).
Emerging evidence shows that a mutation within a CTL
epitope can also affect its processing (49).
Antigen processing is often subverted by polymorphisms
outside epitopes (48) so that optimal epitope generation is
blocked (3, 9) or diminished (30, 42). Optimal epitope
gener-ation can be inhibited when proteolytic activity is redirected to
novel sites within the variant protein (34). Biochemical
de-scriptions of this phenomenon are emerging, but the
signifi-cance of this form of immune escape on the pathogenesis of
viral infection is very difficult to estimate.
Within an infected host, immune pressure exerted by the
HLA class I alleles leads to selection and the accumulation of
virions harboring escape variants (reviewed in reference 25).
Statistical approaches, where the frequencies of HLA class I
alleles in an HIV-infected population are analyzed for
associ-ation with viral amino acid polymorphisms, offer a means of
surveying a viral genome for escape mutants. Such studies have
revealed strong associations between amino acid
polymor-phisms within HIV-1 epitopes and their restricting HLA class
I alleles (21, 31, 50).
Here we show how the same statistical approach can expose
antigen-processing HIV mutants. We investigate the statistical
association between a polymorphism outside an
HLA-B*0702-restricted HIV reverse transcriptase (RT) epitope SPAIF
QSSM (SM9) and HLA-B*0702 (31). We demonstrate that
* Corresponding author. Mailing address: The James Martin 21st
Century School at The Peter Medawar Building for Pathogen
Re-search, Nuffield Department of Clinical Medicine, University of
Ox-ford, South Parks Road, Oxford OX1 3SY, United Kingdom. Phone:
44 794 1969 255. Fax: 44 1865 617028. E-mail: anita.milicic@clinpharm
.ox.ac.uk.
† P.Z. and A.M. contributed equally to this study.
䌤Published ahead of print on 15 November 2006.
2031
on November 8, 2019 by guest
http://jvi.asm.org/
to 172); however, no statistical association has been found
between E169D and HLA-A*03 (31).
MATERIALS AND METHODS
Patient samples.With the approval of the institutional review body, peripheral blood mononuclear cells (PBMCs) and/or plasma were obtained form 130 HIV-1-infected homosexual men recruited from St. Mary’s Hospital, London, England and the SSITT cohort (35). Fresh PBMCs were separated from whole blood by Ficoll-Hypaque (Axis Shield Diagnostics) density gradient centrifuga-tion. All patients were infected with clade B HIV-1 and were on structured treatment interruption therapy. RT was amplified from plasma as part of routine clinical care. RT was also amplified from proviral DNA in four patients, and clones were sequenced. Two molecular viral clones from one patient were iden-tified with 169E and 169D; these were used for HIV constructs. They were isogenic in the sequenced region of residues 2 to 262 other than at following loci: K30N, K49R, R83K K122E, I132V, I135T, E169D, and V245A. All patients were typed as HLA class I.
HLA class I typing.HLA class I typing was done by sequence-specific primer PCR on genomic DNA that was extracted from 3 ml whole blood using a Puregene DNA isolation kit (Gentra) per the manufacturer’s instructions (7).
Generation of B-lymphoblastoid cell lines.PBMCs were transformed with Epstein-Barr virus 95.8 stock for 2 h at 37°C in R-10 medium (RPMI 1640, 100
U/ml penicillin, 100g/ml streptomycin, 2 mML-glutamine, 10% fetal calf
serum) (28). Cyclosporine was then added at 0.5g/ml in a 24-well plate and
cultured at 37°C for several weeks, with regular changes of media.
Generation of CTL lines.CTL lines were generated from PBMCs of HLA-typed HIV-1-infected patients who responded to peptides RK9, SM9, and ER10. The lines were set up with corresponding synthetic peptides as described previ-ously (23). Briefly, around 3 million PBMCs were resuspended in 2 ml of R-10
medium (RPMI, 100 U/ml penicillin, 100g/ml streptomycin, 2 mML-glutamine,
10% fetal calf serum) containing 10M peptide and 25 ng/ml interleukin-7
(IL-7; Peprotech) and cultured in a 24-well plate. On day 3, the medium was replaced with R-10 medium containing 200 U/ml IL-2 (proleukin) and 5% T-Stim with phytohemagglutinin (PHA; BD Biosciences). The lines were subse-quently grown in the same medium (and restimulated using irradiated mixed
heterologous PBMCs and 4g/ml PHA) or maintained in R-10 containing 25
ng/ml IL-15 (Peprotech) (5, 32). CTL lines were enriched for specificity by
antigen stimulation and positive selection of gamma interferon (IFN-␥
)-produc-ing cells anti-IFN-␥microbeads per the manufacturer’s instructions (Miltenyi
Biotec).
We generated CTL lines specific for HLA-A*0301 p17 Gag RLRPGGKKK, HLA-B*0702 RT SPAIFQSSM, and a newly defined HLA-A*3301-restricted Integrase epitope ELKKIIGQVR (P. Zimbwa, A. Milicic, and R. Phillips, un-published data).
CD4ⴙT-cell lines.The SupT1 cell line used in making the HIV recombinants is a human lymphoma T-cell lymphoblast line obtained from J. A. Hoxie through the Centralized Facility for AIDS Reagents supported by EU Programme EVA/ MRC and the Medical Research Council, Hertfordshire, United Kingdom. Jurkat E6-1 is a human T-cell lymphoblast clone (HLA-A*0301/-B*0702/*3501 Cw*0401/*0701) that was obtained from R. A. Weiss through the same facility. The MT-2 cell line used for titration of HIV is an human T-cell leukemia virus type 1-infected human leukemic T-cell lymphoblast line obtained from G. Farrar through the European Collection of Cell Cultures, United Kingdom. These cell lines were maintained in R-10 medium.
ELISpot assay.Antigen-specific responses from PBMCs, CTL lines, or clones were measured using synthetic peptides corresponding to the relevant epitopes
and a standard enzyme-linked immunospot (ELISpot) assay for IFN-␥, as
de-scribed previously (22). Fresh or cryopreserved PBMCs (25,000 to 50,000 per well) or CTLs (500 to 2,500 per well) were used in overnight or 4-h assays. Jurkat E6-1 cells, either infected with HIV-1 or pulsed with exogenous peptide
(Bio-Synthesis, TX) at 2.5M were added as antigen-presenting targets. All assays
were performed in duplicate or triplicate, and positive (PHA) and appropriate negative controls were included in every assay. Spot quantification was auto-mated and standardized using an ELISpot plate reader (software version 3.2.3; Autoimmun Diagnostika, Germany).
(Sigma-Aldrich) was added at 10g/ml to prevent egress of IFN-␥from the
Golgi apparatus, and cells were incubated for a further 5 h at 37°C. Cells were then washed and stained for surface CD4, CD8, and HLA-A,B,C at 4°C for 20 min. After washing, cells were fixed and permeabilized with Cytofix/Cytoperm
(Becton Dickinson). Cells were washed again before staining for IFN-␥(30 min
at 4°C) or p24 (20 min at room temperature). After further washing, cells were resuspended in phosphate-buffered saline and analyzed on a FACScalibur flow cytometer (Becton Dickinson).
Peptide binding assay.The competitive fluorescent binding assay was per-formed as described previously (20, 29). Briefly, BCL expressing HLA-A*0301 were stripped of their naturally bound self-peptides by acid elution for 90 s
(pH⫽2.9). The B cells were then incubated for 24 h at 2 to 8°C with an
HLA-specific reference peptide conjugated to a fluorescein label and a test
peptide titrated between 200M and 20 nM. After the incubation, each sample
was stained with 7-amino-actinomycin D (Viaprobe; BD Biosciences) to exclude dead cells from subsequent analysis, fixed, and analyzed by flow cytometry. All assays were done three times. The inhibition of fluorescein-labeled reference peptide binding, through competition with the test peptide, is a measure of the binding affinity of the test peptides. Inhibition was determined as described previously (20).
Generation of HIV-1 RT fragments by PCR.RT fragments were amplified
from proviral DNA by nested PCR with outer primers RT18 (5⬘-GGA AAC
CAA AAA TGA TAG GGG GAA TTG GAG G-3⬘, nucleotides 2376 to 2406)
and RT21 (5⬘-CTG TAT TTC TGC TAT TAA GTC TTT TGA TGG G-3⬘,
nucleotides 3538 to 3508) and inner primers RT19 (5⬘-GGA CAT AAA GCT
ATA GGT ACA G-3⬘, nucleotides 2453 to 2474) and RT21. Initial denaturing at
95°C for 2 min was followed by 35 cycles of denaturing at 95°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 1 min. A final extension at 72°C was run for 5 min. PCR products were checked for size on a 1% agarose gel, and 2
l of the PCR product, purified using a DNA purification kit (QIAGEN), was
ligated into the TOPO TA plasmid using a TOPO TA cloning kit per the man-ufacturer’s instructions (Invitrogen). The RT insert and TOPO TA vector were
extracted from theEscherichia colicells using a DNA extraction kit (QIAGEN) per
the manufacturer’s instructions. After EcoRI digestion, the presence of the insert was confirmed using a 1% agarose gel.
Sequencing of proviral DNA.The cloned HIV-1 sequences were determined from both directions using primers M13F and M13R located in the TOPO TA plasmid on either side of the insert (Invitrogen). Cycling conditions were 96°C for 10 s, 50°C for 5 s, and 60°C for 4 min. Sequences were analyzed on an ABI 3700 automated analyzer.
Generation of recombinant HIV by electroporation.The recombinant HIVs were made as previously described (6). In short, RT sequences from proviral DNA were purified by gel extraction (QIAGEN) per the manufacturer’s instruc-tions and used to generate recombinant viruses with the RT-deleted
HXB2-based proviral molecular clone pHXB2⌬2-261RT. The amount of product was
determined by spectrophotometry. One microgram of the amplified RT was
mixed with 1g of SmaI-linearized plasmid pHXB2⌬2-261RT in 5⫻106
SupT1
cells in 250l of cold R-10. Cotransfection was achieved by applying a 40-ms
electric pulse with a Gene Pulser II set (Bio-Rad) at 250 V and 950F. After
electroporation, 500,000 more SupT1 cells were added and cells cultured in
25-cm3
and 75-cm3
flasks in R-10. The cultures were examined on days 5, 7, 10, 12, 14, 17, 19, and 21 for syncytium formation (45, 46). If most or all cells were forming syncytia, the supernatant was harvested by centrifugation and stored at
⫺80°C for subsequent titration. Recombinant viral titers (50% tissue culture
infectious dose) were determined in MT-2 cells using the Spearman-Karber formula (41a).
Infection of Jurkat E6-1 cells.Jurkat E6-1 (2⫻106) cells were pelleted in
15-ml conical tubes at 1,000⫻gfor 5 min and then resuspended in 10 ml R-10
containing 2g/ml hexadimethrine bromide (Polybrene; Sigma-Aldich). Cells
were then centrifuged at 1,000⫻gfor 5 min before resuspension in 1 ml R-10
with 2g/ml Polybrene-containing recombinant virus stock. Aliquots of culture
supernatant (250l) and resuspended cells (2 ml) were stored at⫺80°C for
subsequent viral sequencing and cellular assays every other day until day 13 when most cells had formed syncytia.
Proteasome digestion. Wild-type (QGWKGSPAIFQSSMTKILEPFRKQ
NPD) or mutant (QGWKGSPAIFQSSMTKILDPFRKQNPD) RT peptide (5
g) was added to 300l buffer (20 mM HEPES-KOH, pH 7.8, 2 mM MgAc2,2
on November 8, 2019 by guest
http://jvi.asm.org/
mM dithiothreitol [Sigma]). (The SM9 epitope is underlined.) The immuno or
constitutive 20S proteasome (Immatics) was then added at 2g per reaction
mixture and incubated at 37°C. Aliquots of the reaction mix were taken at times 0, 4, 6, 8, 12, 18, 24, and 48 h and added to acetic acid (10% final concentration) to terminate the reaction (43). Digestion experiments were set up in triplicate, and the assay was performed twice for each oligomer and proteasome. Digests were then analyzed by mass spectrometry (Ettan matrix-assisted laser desorption ionization–time of flight; Amersham Biosciences) and sequences inferred using PAWS (Protein Analysis Work Sheet).
RESULTS
Identification of a polymorphism associated with
HLA-B*0702 but outside a known epitope.
A study by Moore et al.
(31) reported a strong association between the E169D
poly-morphism within the RT protein of HIV-1 and HLA-B*0702
(odds ratio
⫽
12.57) in 473 patients infected with HIV-1. We
tested this finding in 130 HIV patients from St. Mary’s
Hospi-tal, London, and the Swiss-Spanish intermittent treatment trial
(SSITT) cohort (36). We sequenced the viral RT in these
patients and analyzed the statistical association between
HLA-B*0702 and E169D: 3 of 24 (12.5%) of HLA-HLA-B*0702
⫹patients
had E169D compared to 4 of 106 (3.8%) HLA-B*0702
⫺pa-tients (odds ratio
⫽
3.3,
P
⫽
0.016, Fisher’s exact test), thus
confirming the findings of Moore et al.
Position E169D lies within an HLA-A*0301 epitope MT
KILEPFR (amino acids [aa] 164 to 172) but not within any
known HLA-B*07 epitopes (http://www.hiv.lanl.gov/content
/immunology/). However, an HLA-B*0702-restricted epitope, SP
AIFQSSM (aa 156 to 164), maps five residues upstream of the
E169D polymorphism. Another HLA-A*03 epitope, AIFQS
SMTK (aa 158 to 166), also maps upstream of the E169D
poly-morphism (Fig. 1).
Alleles HLA-A*03 and HLA-B*0702 are in strong linkage
disequilibrium (8, 17). The original study by Moore et al. (31)
did not detect an association between HLA-A*0301 and
E169D independent of HLA-B*0702. In our patient group, all
of the HLA-B*0702
⫹patients were also HLA-A*03
⫹, so we
could not dissect this finding with respect to HLA-A*03.
Not-withstanding the lack of a statistical association between
E169D and HLA-A*03, we investigated the effect of this
vari-ant on the CTL recognition of the HLA-A*03-restricted
epitope MTKILEPFR (MR9).
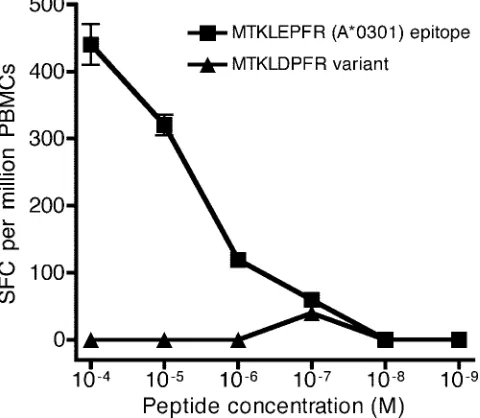
E169D mutation abolishes recognition of the
HLA-A*0301-restricted RT epitope MR9 (aa 164 to 172).
We compared
recognition of peptides MTKIL
E
PFR and MTKIL
D
PFR in
IFN-
␥
ELISpot assays directly ex vivo using PBMCs from an
HLA-A*0301-positive patient. In contrast to the wild-type MT
KIL
E
PFR peptide, the MTKIL
D
PFR variant did not activate
CTL (Fig. 2), indicating that the variant MTKIL
D
PFR is
un-likely to be recognized in HLA-A*0301 positive individuals
infected with HIV-1. We also tested the relative binding
affin-ities of these two peptides to HLA-A*0301, using a
competi-tive binding assay (20), and found a 44% reduction in the
binding of the variant peptide MTKIL
D
PFR in comparison
with wild-type peptide MTKIL
E
PFR (the E169D
polymor-phism is underlined).
Construction of recombinant pathogenic HIV-1 clones
bear-ing RT 169E and 169D.
The E169D change maps close to the
HLA-B*0702-restricted epitope SPAIFQSSM, and we wanted
to test the possibility that E169D might interfere with the
processing of this epitope. We have shown previously that a
single amino acid change can interfere with CTL recognition of
epitopes restricted by two distinct HLA alleles (29). To
inves-tigate whether E169D could also interfere with the generation
of the HLA-B*0702 peptide, we first screened 130 B-clade RT
sequences from HLA-typed patients. Of these, 123 had E (wild
type) and 7 had D (mutant) at position 169. We then generated
RT fragments (aa 2 to 261) which were inserted into an HIV-1
backbone to yield recombinant HIV-1 molecular clones HIV-1
(169E) (wild type) and HIV-1 (169D) (mutant), as described in
Materials and Methods.
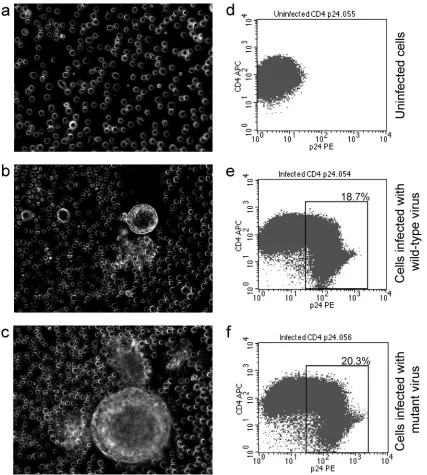
Both viruses were pathogenic, as demonstrated by the
for-mation of syncytia in infected SupT1 cell line by day 7 after
infection (Fig. 3a to c). When Jurkat E6-1 cells were infected
with an identical titer (multiplicity of infection [MOI]
⫽
10
⫺5)
of wild-type or mutant viruses, both recombinant forms
ex-pressed comparable levels of p24 Gag and down-regulated the
surface expression of CD4 on Jurkat cells (Fig. 3d to f).
To optimize the time course and viral titer for infection, the
recombinant wild-type HIV-1 (169E) virus was used to infect
FIG. 1. Position of three epitopes with different HLA restrictions
[image:3.585.302.542.72.281.2]within the HIV RT protein: HLA-B*0702-restricted SPAIFQSSM
(SM9, RT aa 156 to 164), HLA-A*0301-restricted AIFQSSMTK
(AK9, RT aa 158 to 166), and HLA-A*0301-restricted MTKILEPFR
(MR9, RT aa 164 to 172).
FIG. 2. MTKILDPFR variant evades recognition by CTL specific
for the MTKILEPFR epitope. The antigenicity of the peptide MTKI
LEPFR and its variant MTKIL
D
PFR were tested at different peptide
concentrations using PBMCs from an HLA-A*0301-positive patient
(50,000 PBMCs per well) directly ex vivo in a 16-h IFN-
␥
ELISpot
assay. The assay was performed in duplicate; data show means
⫾
standard errors of the means.
on November 8, 2019 by guest
http://jvi.asm.org/
Jurkat E6-1 cells (which are HLA-A*0301 and HLA-B*0702
positive) at an MOI of 1.6
⫻
10
⫺3. The infected and uninfected
cells were sampled on alternate days and stained for
intracel-lular p24 Gag. The proportion of p24 Gag-positive infected
cells increased from 4.6% on day 1 to 50.2% on day 5; the
proportion of p24 Gag-positive cells on day 5 also increased
with the titer of infecting virus from 2.3% at an MOI of 10
⫺5to 58.5% at an MOI of 3.2
⫻
10
⫺3(data not shown).
The successful presentation of HLA class I-restricted HIV-1
epitopes on the surface of the Jurkat cells infected with the
[image:4.585.80.507.67.542.2]wild-type HIV-1 construct was confirmed by IFN-
␥
ELISpot.
We used CTL specific for three epitopes:
HLA-A*0301-re-stricted p17 Gag RLRPGGKKK (RK9),
HLA-B*0702-re-stricted RT SPAIFQSSM (SM9), and HLA-A*3301-reHLA-B*0702-re-stricted
ELKKIIGQVR (ER9), as a negative control. Jurkat cell
clone E6-1 does not express HLA-A*3301, and the CTL
response to the ER9 epitope was absent, as expected (Fig.
4). CTL recognition of the RK9 and SM9 epitopes was
readily detected 5 days after infection of the target cells and
remained high (Fig. 4).
FIG. 3. Both wild-type and mutant recombinant HIV form syncytia in SupT1 cells and down-regulate CD4 expression on infected Jurkat cells.
SupT1 cells (a to c) or Jurkat E6-1 cells (d to f) were infected with either wild-type (E6-1.6.0) or mutant (E6-1.6.1) recombinant HIV. On day 5
of culture, the cells were investigated under the microscope for syncytium formation and stained for surface CD4 and intracellular HIV-1 p24. (a
and d) Uninfected cells; (b and e) cells infected with wild-type virus; (c and f) cells infected with mutant recombinant virus. Down-regulation of
the CD4 molecule as a result of HIV infection can be seen. The percentages represent the proportions of infected (p24 positive) cells.
on November 8, 2019 by guest
http://jvi.asm.org/
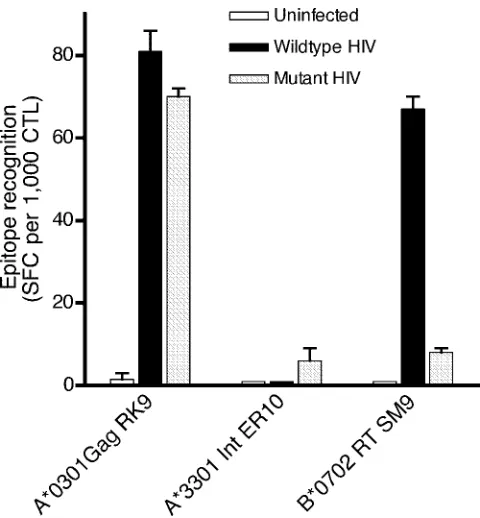
EI69D mutation abolishes the CTL recognition of the
neigh-boring HLA-B*0702-restricted RT epitope SM9 (aa 156 to
164).
To investigate whether E169D influenced the CTL
rec-ognition of SM9, we infected Jurkat E6-1 cells with either
wild-type (169E) or mutant (169D) virus and used them as
targets for the three CTL lines in ELISpot assays on day 5 of
infection. Both mutant and wild-type targets were recognized
equally well by the p17 Gag RLRPGGKKK-specific CTL line
(
P
⬎
0.5, Fisher’s exact test), and neither was recognized by
the HLA-A*3301-restricted ELKKIIGQVR-specific CTL
line (negative control). There was strong CTL recognition of
the HLA-B*0702-restricted SPAIFQSSM epitope in the
wild-type virus-infected cells, which was absent when tested
against mutant HIV-1-infected target cells (
P
⫽
9.3
⫻
10
⫺7,
Fisher’s exact test) (Fig. 5).
E169D impairs the generation of the SM9 epitope by the
proteasome.
These results show that D at position 169, five
residues downstream from the SPAIFQSSM epitope (aa 156 to
164), specifically interferes with SM9-specific CTL recognition.
In line with our previous study (30), we hoped to determine
whether the SM9 epitope is proteasome dependent, but the
SM9-specific T-cell clone did not survive long enough to
com-plete these experiments. Instead, we performed in vitro assays
of proteasome processing. We investigated the effect of the
E169D substitution on digestion of oligopeptides
correspond-ing to a part of the HIV-1 RT protein, similar to our earlier
study (30).
Two 27-mer synthetic oligopeptides corresponding to the aa
residues 151 to 177 of the wild-type (QGWKGSPAIFQSSMT
KILEPFRKQNPD) and mutant (QGWKGSPAIFQSSMTKIL
DPFRKQNPD) RT protein were digested in vitro using the
constitutive and immuno 20S proteasome (see Materials and
Methods). (The E169D polymorphism is underlined.) Aliquots
were taken at 0, 4, 6, 8, 12, 18, 24, and 48 h of digestion, and
products were analyzed using mass spectrometry. The
diges-tion with the constitutive 20S proteasome resulted in very few
fragments whereas the 20S immunoproteasome produced a
wide array of digestion products. A similar observation has
been reported previously for HIV-1 RT (44). Lactacystin, an
inhibitor of the 20S proteasome, completely blocked the
sub-strate-specific proteasomal activity.
The products of the in vitro cleavage of the wild-type and
mutant oligopeptides and the position of the three epitopes in
this region of RT are illustrated in Fig. 6. Digestion of the
wild-type (169E) oligopeptide within 6 h released an
interme-diate peptide QGWKGSPAIFQSSM, which has the
appropri-ate carboxyl terminus (M) for the HLA-B*0702-restricted
epitope SM9. Importantly, this fragment was absent following
the digestion of the mutant oligopeptide (169D) even after
48 h, by which time nonspecific proteasomal degradation of the
oligomers starts to occur. The correct carboxyl termini for the
HLA-A*0301-restricted AIFQSSMTK and MTKILEPFR
epitopes were detected after 6 h of digestion of both the
wild-type and mutant synthetic oligopeptides (Fig. 6).
[image:5.585.301.541.66.325.2]These results provide an explanation for the lack of
recog-nition of the mutant recombinant HIV-1 virus by SPAIFQSS
M-specific CTL (Fig. 5). They strongly suggest that the E169D
mutation abolishes CTL recognition of SM9 by blocking the
correct proteasomal cleavage.
FIG. 4. Infected Jurkat E6-1 cells are recognized by CTL after day
5 of infection. Jurkat E6-1 cells infected with wild-type virus were used
as targets in 16-h IFN-
␥
ELISpot assays after the indicated number of
days postinfection; each assay contained 250,000 infected cells and
2,500 CTL. The HLA-A*3301 ER10-specific CTL line represents the
negative control, as Jurkat E6-1 cells do not express HLA-A*3301.
RK9, p17 Gag RLRPGGKKK (HLA-A*0301 restricted); SM9, RT
SPAIFQSSM (HLA-B*0702 restricted); ER10, integrase ELKKII
GQVR (HLA-A*3301 restricted).
FIG. 5. Flanking mutation E169D specifically abolishes recognition
of the RT epitope SM9 (RT aa 156 to 164). Jurkat E6-1 cells,
unin-fected or inunin-fected with wild-type or mutant recombinant viruses (day
5), were used in IFN-
␥
ELISpot assays with CTL lines specific for three
epitopes. RK9, HLA-A*0301-restricted p17 Gag RLRPGGKKK
(pos-itive control); ER10, HLA-A*3301-restricted integrase ELKKII
GQVR (negative control); SM9, HLA-B*0702-restricted RT SPAIFQ
SSM. Each assay was carried out using 100,000 targets and 1,000 CTL
per well.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.54.276.68.247.2]DISCUSSION
CTL escape due to intraepitope amino acid alteration can
occur through various mechanisms. It can affect peptide
bind-ing to the HLA class I or recognition of the peptide/HLA class
I complex by the T-cell receptor. Variation within a CTL
epitope can also affect antigen processing (12, 34, 49). Studies
which evaluated the effect of artificially altered
epitope-flank-ing residues on antigen processepitope-flank-ing predicted that naturally
occurring flanking variants could also interfere with epitope
presentation. Recent reports have confirmed this prediction (3,
9, 30, 42).
[image:6.585.116.473.68.535.2]Processing mutants can be detected by searching for
evi-dence that amino acid polymorphisms in the vicinity of a
known epitope are positively selected during an infection.
Our group has detected this phenomenon in one case (30).
In this study, we show that escape mutants can also be
FIG. 6. In vitro proteasome digestion of RT peptides. (a) Products detected after 6 h of in vitro 20S immunoproteasome digestion of 27-mer
oligopeptides corresponding to wild-type (169E) and mutant (169D) RT sequence. The correct precursor for the SPAIFQSSM (SM9) epitope is
shown in boldface type; this fragment was absent when the mutant 169D sequence was digested. Sequences in blue represent digestion products
detected only with the mutant oligomer. The fragments shown were identified in triplicate in two independent experiments. (b) A diagram showing
the three RT epitopes: red, HLA-B*0702-restricted SPAIFQSSM (SM9, RT aa 156 to 164); green, HLA-A*0301-restricted AIFQSSMTK (AK9,
RT aa 158 to 166); yellow, HLA-A*0301-restricted MTKILEPFR (ER9, RT aa 164 to 172). Arrows indicate digestion products detected after 24 h
of incubation with the 20S immunoproteasome. Colored arrows correspond to the correct carboxyl termini for the appropriate epitopes. The
correct restriction site for epitope SM9 is marked with a red arrow. The variant position E169D is shown within the blue box.
on November 8, 2019 by guest
http://jvi.asm.org/
predicted from statistical surveys which associate HLA class
I molecules and amino acid polymorphisms adjacent to CTL
epitopes (21, 31, 50).
In HIV-1 RT, HLA-B*0702 was found to be strongly
asso-ciated with E169D (31). There have been no reports to indicate
that the E169D variant is associated with antiretroviral drug
treatment. Although no HLA-B*0702-restricted epitope maps
to this polymorphism, position 169 lies 5 amino acids
down-stream of an HLA-B*0702-restricted epitope, SM9 (aa 156 to
164). Position 169 also maps within an overlapping
HLA-A*0301-restricted epitope, MR9 (aa 164 to 172). Despite the
strong linkage disequilibrium between HLA-A*0301 and
B*0702 and the high frequency of HLA A*03 in the population
(8, 17), statistical association between E169D and
HLA-A*0301, independent of HLA-B*0702, has not been found.
Using ELISpot IFN-
␥
assay, we showed that E169D confers
escape from pressure mediated through
HLA-A*0301-re-stricted MR9-specific CTL. The statistical association with
HLA-B*0702 suggested that the same polymorphism might
also confer viral escape in HLA-B*0702
⫹individuals infected
with HIV. In HLA-B*0702
⫹patients, the E169D mutation
could allow viral escape in one of two ways: position 169 might
lie within an as yet unidentified HLA-B*0702-restricted
epitope or, alternatively, E169D might confer escape from
SM9-mediated CTL pressure by interfering with antigen
pro-cessing.
Previous studies on HIV-1 antigen processing have utilized
systems in which HIV-1 antigens were expressed in BCLs by
recombinant vaccinia virus (30) or HIV-1 mRNA inserts (1, 9).
We have established a novel HIV-1 CD4
⫹target assay to
investigate the effect of E169D on the processing of the SM9
epitope. We reconstituted HIV-1 with an RT sequence derived
from a patient. This clone bore the polymorphism under
scru-tiny in its natural context, which would ensure viral viability
(11, 14, 19, 37). We infected CD4
⫹Jurkat E6-1 cells, a natural
HIV-1 host, allowing HIV-1 protein to be expressed and
pro-cessed in a more physiological manner. Initial in vitro studies
showed both recombinant viruses, HIV-1 (169E) and HIV-1
(169D), to be equally viable and infectious.
When the HIV recombinant virus-infected CD4
⫹cells were
presented to SM9-specific CTLs, the targets expressing HIV-1
(169E) were recognized significantly better than those
express-ing the HIV-1 (169D) mutant virus (
P
⫽
6.7
⫻
10
⫺10). Yet
there was no disparity in recognition by a control line specific
for the p17 Gag epitope RK9 which is isogenic in both 169E
and 169D HIV constructs (
P
⬍
0.51). This suggested that
epitope SM9 was not being presented at the surface of the
Jurkat E6-1 cell. We investigated whether E169D mutation
might inhibit proteasome cleavage of the SM9 epitope and
subjected 27-mer synthetic polypeptides with either E or D at
position 169 to in vitro digestion by the 20S
immunoprotea-some. Digestion of the 169E polypeptide liberated the correct
carboxyl terminals for the HLA-B*0702-restricted epitope
SM9, as well as HLA-A*0301-restricted epitopes MR9 and
AK9. However, digestion of the 169D polypeptide liberated
the appropriate fragments for the HLA-A*0301-restricted
epitopes only; the potential precursor of the SM9 epitope was
extended at the carboxyl end.
This study shows that statistical approaches can be a way of
identifying antigen-processing escape mutants. A single amino
acid mutation, E169D, can confer escape from dual attack by
CTL, governed by either HLA-A*0301 and B*0702, though by
different mechanisms. However, it also highlights how difficult
it would be to predict the effect of HIV variation on CTL
recognition on purely statistical evidence, without a detailed
functional analysis of individual variants.
ACKNOWLEDGMENTS
This work was funded by the Wellcome Trust, United Kingdom.
We thank Andy Sewell for help and support during the preparation
of the manuscript.
REFERENCES
1.Allen, T. M., M. Altfeld, X. G. Yu, K. M. O’Sullivan, M. Lichterfeld, S. Le Gall, M. John, B. R. Mothe, P. K. Lee, E. T. Kalife, D. E. Cohen, K. A. Freedberg, D. A. Strick, M. N. Johnston, A. Sette, E. S. Rosenberg, S. A. Mallal, P. J. Goulder, C. Brander, and B. D. Walker.2004. Selection, trans-mission, and reversion of an antigen-processing cytotoxic T-lymphocyte es-cape mutation in human immunodeficiency virus type 1 infection. J. Virol.
78:7069–7078.
2.Ammaranond, P., J. Zaunders, C. Satchell, D. van Bockel, D. A. Cooper, and A. D. Kelleher.2005. A new variant cytotoxic T lymphocyte escape mutation in HLA-B27-positive individuals infected with HIV type 1. AIDS Res. Hum.
Retrovir.21:395–397.
3.Beekman, N. J., P. A. van Veelen, T. van Hall, A. Neisig, A. Sijts, M. Camps, P. M. Kloetzel, J. J. Neefjes, C. J. Melief, and F. Ossendorp.2000. Abroga-tion of CTL epitope processing by single amino acid substituAbroga-tion flanking the
C-terminal proteasome cleavage site. J. Immunol.164:1898–1905.
4.Benito, J. M., M. Lopez, and V. Soriano.2004. The role of CD8⫹T-cell
response in HIV infection. AIDS Rev.6:79–88.
5.Berard, M., K. Brandt, S. Bulfone-Paus, and D. F. Tough.2003. IL-15
promotes the survival of naive and memory phenotype CD8⫹T cells. J.
Im-munol.170:5018–5026.
6.Boucher, C. A., W. Keulen, T. van Bommel, M. Nijhuis, D. de Jong, M. D. de Jong, P. Schipper, and N. K. Back.1996. Human immunodeficiency virus type 1 drug susceptibility determination by using recombinant viruses gen-erated from patient sera tested in a cell-killing assay. Antimicrob. Agents
Chemother.40:2404–2409.
7.Bunce, M.2003. PCR-sequence-specific primer typing of HLA class I and
class II alleles. Methods Mol. Biol.210:143–171.
8.Doran, T. J., H. V. Bashir, J. Trejaut, M. L. Bassett, J. W. Halliday, and L. W. Powell.1981. Idiopathic haemochromatosis in the Australian
popula-tion: HLA linkage and recessivity. Hum. Immunol.2:191–200.
9.Draenert, R., S. Le Gall, K. J. Pfafferott, A. J. Leslie, P. Chetty, C. Brander, E. C. Holmes, S. C. Chang, M. E. Feeney, M. M. Addo, L. Ruiz, D. Ramduth, P. Jeena, M. Altfeld, S. Thomas, Y. Tang, C. L. Verrill, C. Dixon, J. G. Prado, P. Kiepiela, J. Martinez-Picado, B. D. Walker, and P. J. Goulder.2004. Immune selection for altered antigen processing leads to cytotoxic T
lym-phocyte escape in chronic HIV-1 infection. J. Exp. Med.199:905–915.
10.Feeney, M. E., Y. Tang, K. A. Roosevelt, A. J. Leslie, K. McIntosh, N. Karthas, B. D. Walker, and P. J. Goulder.2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive
long-term-nonprogressing child. J. Virol.78:8927–8930.
11.Friedrich, T. C., C. A. Frye, L. J. Yant, D. H. O’Connor, N. A. Kriewaldt, M. Benson, L. Vojnov, E. J. Dodds, C. Cullen, R. Rudersdorf, A. L. Hughes, N. Wilson, and D. I. Watkins.2004. Extraepitopic compensatory substitutions partially restore fitness to simian immunodeficiency virus variants that es-cape from an immunodominant cytotoxic-T-lymphocyte response. J. Virol.
78:2581–2585.
12.Furutsuki, T., N. Hosoya, A. Kawana-Tachikawa, M. Tomizawa, T. Odawara, M. Goto, Y. Kitamura, T. Nakamura, A. D. Kelleher, D. A. Cooper, and A. Iwamoto.2004. Frequent transmission of cytotoxic-T-lymphocyte escape mu-tants of human immunodeficiency virus type 1 in the highly HLA-A24-positive
Japanese population. J. Virol.78:8437–8445.
13.Gandhi, R. T., and B. D. Walker.2002. Immunologic control of HIV-1.
Annu. Rev. Med.53:149–172.
14.Garcı´a-Lerma, J. G., H. MacInnes, D. Bennett, H. Weinstock, and W. Heneine.2004. Transmitted human immunodeficiency virus type 1 carrying the D67N or K219Q/E mutation evolves rapidly to zidovudine resistance in vitro and shows a high replicative fitness in the presence of zidovudine.
J. Virol.78:7545–7552.
15.Goulder, P. J., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland-Jones.1997. Late escape from an immunodominant cyto-toxic T-lymphocyte response associated with progression to AIDS. Nat. Med.
3:212–217.
16.Goulder, P. J., and D. I. Watkins.2004. HIV and SIV CTL escape:
impli-cations for vaccine design. Nat. Rev. Immunol.4:630–640.
on November 8, 2019 by guest
http://jvi.asm.org/
Phillips.2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J.
Exp. Med.193:375–386.
20.Kessler, J. H., B. Mommaas, T. Mutis, I. Huijbers, D. Vissers, W. E. Benckhuijsen, G. M. Schreuder, R. Offringa, E. Goulmy, C. J. Melief, S. H. van der Burg, and J. W. Drijfhout.2003. Competition-based cellular peptide binding assays for 13 prevalent HLA class I alleles using
fluores-cein-labeled synthetic peptides. Hum. Immunol.64:245–255.
21.Kiepiela, P., A. J. Leslie, I. Honeyborne, D. Ramduth, C. Thobakgale, S. Chetty, P. Rathnavalu, C. Moore, K. J. Pfafferott, L. Hilton, P. Zimbwa, S. Moore, T. Allen, C. Brander, M. M. Addo, M. Altfeld, I. James, S. Mallal, M. Bunce, L. D. Barber, J. Szinger, C. Day, P. Klenerman, J. Mullins, B. Korber, H. M. Coovadia, B. D. Walker, and P. J. Goulder.2004. Dominant influence of HLA-B in mediating the potential co-evolution of HIV and
HLA. Nature432:769–775.
22.Lalvani, A., R. Brookes, S. Hambleton, W. J. Britton, A. V. Hill, and A. J. McMichael.1997. Rapid effector function in CD8⫹memory T cells. J. Exp.
Med.186:859–865.
23.Lalvani, A., T. Dong, G. Ogg, A. A. Patham, H. Newell, A. V. Hill, A. J. McMichael, and S. Rowland-Jones.1997. Optimization of a peptide-based protocol employing IL-7 for in vitro restimulation of human cytotoxic T
lymphocyte precursors. J. Immunol. Methods210:65–77.
24.McAdam, S., P. Klenerman, L. Tussey, S. Rowland-Jones, D. Lalloo, R. Phillips, A. Edwards, P. Giangrande, A. L. Brown, F. Gotch, et al.1995. Immunogenic HIV variant peptides that bind to HLA-B8 can fail to
stimu-late cytotoxic T lymphocyte responses. J. Immunol.155:2729–2736.
25.McMichael, A., and P. Klenerman.2002. HIV/AIDS. HLA leaves its
foot-prints on HIV. Science296:1410–1411.
26.McMichael, A. J., and S. L. Rowland-Jones.2001. Cellular immune
re-sponses to HIV. Nature410:980–987.
27.Meier, U. C., P. Klenerman, P. Griffin, W. James, B. Koppe, B. Larder, A. McMichael, and R. Phillips.1995. Cytotoxic T lymphocyte lysis inhibited by
viable HIV mutants. Science270:1360–1362.
28.Meinl, E., and R. Hohlfeld.2000. T cell transformation with herpesvirus
saimiri: a tool for neuroimmunological research. J. Neuroimmunol.103:1–7.
29.Milicic, A., C. T. Edwards, S. Hue, J. Fox, H. Brown, T. Pillay, J. W. Drijfhout, J. N. Weber, E. C. Holmes, S. J. Fidler, H. T. Zhang, and R. E. Phillips.2005. Sexual transmission of single human immunodeficiency virus type 1 virions encoding highly polymorphic multisite cytotoxic T-lymphocyte
escape variants. J. Virol.79:13953–13962.
30.Milicic, A., D. A. Price, P. Zimbwa, B. L. Booth, H. L. Brown, P. J. Easterbrook, K. Olsen, N. Robinson, U. Gileadi, A. K. Sewell, V. Cerundolo, and R. E. Phillips.2005. CD8⫹T cell epitope-flanking mutations disrupt
proteasomal processing of HIV-1 Nef. J. Immunol.175:4618–4626.
31.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal.2002. Evidence of HIV-1 adaptation to HLA-restricted immune
responses at a population level. Science296:1439–1443.
32.Mueller, Y. M., P. M. Bojczuk, E. S. Halstead, A. H. Kim, J. Witek, J. D. Altman, and P. D. Katsikis.2003. IL-15 enhances survival and function of
HIV-specific CD8⫹T cells. Blood101:1024–1029.
33.O’Neil, P. K., G. Sun, H. Yu, Y. Ron, J. P. Dougherty, and B. D. Preston.
2002. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral
mutagenesis. J. Biol. Chem.277:38053–38061.
34.Ossendorp, F., M. Eggers, A. Neisig, T. Ruppert, M. Groettrup, A. Sijts, E. Mengede, P. M. Kloetzel, J. Neefjes, U. Koszinowski, and C. Melief.1996. A single residue exchange within a viral CTL epitope alters
proteasome-medi-ated degradation resulting in lack of antigen presentation. Immunity5:115–
124.
35.Oxenius, A., D. A. Price, S. J. Dawson, H. F. Gunthard, M. Fischer, L. Perrin, E. Ramirez, C. Fagard, B. Hirschel, G. Scullard, J. N. Weber, A. R.
infection. Proc. Natl. Acad. Sci. USA99:13747–13752.
37.Peyerl, F. W., D. H. Barouch, W. W. Yeh, H. S. Bazick, J. Kunstman, K. J. Kunstman, S. M. Wolinsky, and N. L. Letvin.2003. Simian-human immu-nodeficiency virus escape from cytotoxic T-lymphocyte recognition at a
struc-turally constrained epitope. J. Virol.77:12572–12578.
38.Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards, A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. Bangham, and C. R. Rizza.
1991. Human immunodeficiency virus genetic variation that can escape
cy-totoxic T cell recognition. Nature354:453–459.
39.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips.1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl.
Acad. Sci. USA94:1890–1895.
40.Price, D. A., U. C. Meier, P. Klenerman, M. A. Purbhoo, R. E. Phillips, and A. K. Sewell. 1998. The influence of antigenic variation on cytotoxic T
lymphocyte responses in HIV-1 infection. J. Mol. Med.76:699–708.
41.Reid, S. W., S. McAdam, K. J. Smith, P. Klenerman, C. A. O’Callaghan, K. Harlos, B. K. Jakobsen, A. J. McMichael, J. I. Bell, D. I. Stuart, and E. Y. Jones.1996. Antagonist HIV-1 Gag peptides induce structural changes in
HLA B8. J. Exp. Med.184:2279–2286.
41a.Schmidt, N. J.1989. Cell culture procedures for diagnostic virology, p. 78–79.
InN. J. Schmidt and R. W. Emmons (ed.), Diagnostic procedures for viral,
rickettsial, and chlamydial infections. American Public Health Association, Washington, DC.
42.Seifert, U., H. Liermann, V. Racanelli, A. Halenius, M. Wiese, H. Wedemeyer, T. Ruppert, K. Rispeter, P. Henklein, A. Sijts, H. Hengel, P. M. Kloetzel, and B. Rehermann.2004. Hepatitis C virus mutation affects proteasomal epitope
pro-cessing. J. Clin. Investig.114:250–259.
43.Seifert, U., C. Maranon, A. Shmueli, J. F. Desoutter, L. Wesoloski, K. Janek, P. Henklein, S. Diescher, M. Andrieu, H. de la Salle, T. Weinschenk, H. Schild, D. Laderach, A. Galy, G. Haas, P. M. Kloetzel, Y. Reiss, and A. Hosmalin.2003. An essential role for tripeptidyl peptidase in the generation
of an MHC class I epitope. Nat. Immunol.4:375–379.
44.Sewell, A. K., D. A. Price, H. Teisserenc, B. L. Booth, Jr., U. Gileadi, F. M. Flavin, J. Trowsdale, R. E. Phillips, and V. Cerundolo.1999. IFN-gamma exposes a cryptic cytotoxic T lymphocyte epitope in HIV-1 reverse
transcrip-tase. J. Immunol.162:7075–7079.
45.Sylwester, A., D. Wessels, S. A. Anderson, R. Q. Warren, Shutt, R. C. Kennedy, and D. R. Soll.1993. HIV-induced syncytia of a T cell line form
single giant pseudopods and are motile. J. Cell Sci.106(Pt 3):941–953.
46.Tersmette, M., R. E. de Goede, B. J. Al, I. N. Winkel, R. A. Gruters, H. T. Cuypers, H. G. Huisman, and F. Miedema.1988. Differential syncytium-inducing capacity of human immunodeficiency virus isolates: frequent detection of syncytium-inducing isolates in patients with acquired
immuno-deficiency syndrome (AIDS) and AIDS-related complex. J. Virol.62:2026–
2032.
47.Walker, B. D., and B. T. Korber.2001. Immune control of HIV: the obstacles
of HLA and viral diversity. Nat. Immunol.2:473–475.
48.Yellen-Shaw, A., and L. C. Eisenlohr.1997. Regulation of class I-restricted
epitope processing by local or distal flanking sequence. J. Immunol.158:
1727–1733.
49.Yokomaku, Y., H. Miura, H. Tomiyama, A. Kawana-Tachikawa, M. Takiguchi, A. Kojima, Y. Nagai, A. Iwamoto, Z. Matsuda, and K. Ariyoshi.2004. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are ma-jor escape mechanisms from CTL immune pressure in human
immunodefi-ciency virus type 1 infection. J. Virol.78:1324–1332.
50.Yusim, K., C. Kesmir, B. Gaschen, M. M. Addo, M. Altfeld, S. Brunak, A. Chigaev, V. Detours, and B. T. Korber.2002. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J.
Vi-rol.76:8757–8768.
on November 8, 2019 by guest
http://jvi.asm.org/