0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
Poliovirus Protein 3AB Forms a Complex with and Stimulates
the Activity of the Viral RNA Polymerase, 3D
polSTEPHEN J. PLOTCH*ANDOLGA PALANT
Molecular Biology Research Section, Lederle Laboratories, Medical Research Division, American Cyanamid Company, Pearl River, New York 10965
Received 16 March 1995/Accepted 16 August 1995
Poliovirus protein 3B (also known as VPg) is covalently linked to the 5* ends of both genomic and antigenomic viral RNA. Genetic and biochemical studies have implicated protein 3AB, the membrane-bound precursor to VPg, in the initiation of genomic RNA synthesis. We have purified 3AB to near homogeneity following thrombin cleavage of purified glutathioneS-transferase-3AB. When added to transcription reaction mixtures catalyzed by poliovirus RNA polymerase (3Dpol
), 3AB stimulated RNA synthesis up to 75-fold with oligo(U)-primed virion RNA, globin mRNA, and unprimed synthetic, full-length minus-strand viral RNA as the templates. Synthetic VPg also stimulated RNA synthesis but was only 1 to 2% as effective as 3AB on a molar basis. The increased level of transcription was not the result of enhancing the elongation rate of the poly-merase. No evidence was found for uridylylation of 3AB or for covalent linkage to RNA transcription products. 3AB sedimented as a multimer in glycerol gradients. In the presence of the polymerase, the sedimentation rate of both proteins increased, suggesting the formation of a complex. Detergent prevented both multimerization and complex formation. The polymerase also bound to immobilized glutathione S-transferase-3AB; this procedure was used to purify the polymerase to near homogeneity. These results suggest a mechanism for bringing together 3AB, 3Dpol
(or its precursor 3CD), and viral RNA in host cell membranous vesicles in which all viral RNA synthesis occurs.
A detailed understanding of the steps involved in the repli-cation of poliovirus RNA has not yet been achieved. Following infection, the 7.5-kb polyadenylated viral genomic RNA (36, 67, 76, 91) is translated into an incipient polyprotein that is autocatalytically cleaved by encoded proteases into structural and nonstructural proteins (28, 57, 59, 75, 85). Biochemical and genetic evidence has identified proteins 2A, 2B, 2C, 3AB, 3B, 3CD, and 3Dpolas participants in the process of viral RNA replication (4, 5, 9, 13, 16, 24, 30, 32, 33, 35, 37, 38, 40, 43, 48, 51, 53, 63, 64, 66, 68, 69, 83, 84, 86, 89).
Most of these proteins are associated with newly synthesized membranous vesicles in the infected cell, which are the sites where viral RNA synthesis occurs (14, 15, 17, 78, 80, 81). Protein 3B (also known as VPg) and its membrane-bound precursor 3AB are required for the initiation of both plus- and minus-strand RNA synthesis; 3B is covalently linked to uridine residues located at the 59end of both plus and minus strands, including nascent strands in replicative intermediate RNA (1, 3, 9, 22, 24, 41, 42, 58, 61, 73). Protein 3CD, the precursor to the mature protease 3C and the RNA-dependent RNA poly-merase 3Dpol, is itself a protease, responsible for maturation of the viral structural proteins (34, 93). 3CD is also involved in binding to structures at both the 59and 39ends of the viral plus strand; mutations of the 59structure block plus-strand (but not minus-strand) RNA synthesis in vivo (4, 5, 30). Protein 3Dpol catalyzes the elongation of both plus and minus strands; how-ever, it is incapable of initiating RNA synthesis in the absence of a primer (10, 21, 23, 45, 56, 87, 90, 92). In vitro the purified enzyme can elongate a variety of RNAs, both viral and nonvi-ral, from hairpin structures at their 39ends, generating
tem-plate-primed, double-length molecules (18, 31, 44). Double-stranded snap-back viral RNA species have also been detected in vivo (70). The polymerase can also unwind preformed RNA duplexes during the course of elongation (18); the role of this activity during viral RNA replication is not known. Protein 2C binds RNA, has NTPase activity, and has sequence elements similar to those found in known RNA helicases (25, 26, 49, 72). It is also the target of guanidine, a specific inhibitor of polio-virus RNA replication (8, 63, 64, 82). In addition, it (and protein 2BC) induces membranous vacuole formation similar to that seen during poliovirus replication (2). The function of protein 2B is unknown; however, mutants defective in RNA synthesis have been generated which were not complement-able in trans (13, 33). Protein 2BC, the precursor to proteins 2B and 2C, and proteins P2-3AB and 2C3AB, precursors to pro-tein 3AB, have also been found in the membrane-bound rep-lication complex (15, 74, 81). The function of protein 2A pro-tease in replication is unknown, but its deletion from a dicistronic virus prevents RNA replication (51).
In addition to the viral proteins, experiments in vivo and in vitro have suggested that host proteins play a role in the initi-ation of RNA synthesis. One or more 67- to 68-kDa host proteins were shown to provide 3Dpolwith the ability to tran-scribe genomic RNA in vitro in the absence of a primer. This protein(s) variously bound to 3Dpolimmobilized on a column of CNBr-agarose (11), coimmunoprecipitated with 3Dpol(19), had protein kinase activity with specificity for eukaryotic initi-ation factor 2 (54), or had terminal uridylyl transferase (TUT-ase) activity. The latter was proposed to act by generating a poly(A)-oligo(U) hairpin at the 39 end of the viral RNA, thereby providing a self-priming template for the polymerase to elongate to a double-length molecule (6, 7, 92). Whether this protein(s) is involved in RNA replication in vivo is not known. At least one other host cell protein may also participate in RNA replication. Gel mobility shift assays detected a pro-tein of 36 kDa that binds to the stem-loop structure at the 59
* Corresponding author. Mailing address: Molecular Biology Research Section, Lederle Laboratories, Medical Research Division, American Cy-anamid Company, 401 N. Middletown Rd., Pearl River, NY 10965. Phone: (914) 732-4378. Fax: (914) 732-2480. Electronic mail address: Steve_Plotch_at_USPRMG41@internetmail.pr.cyanamid.com.
7169
on November 9, 2019 by guest
http://jvi.asm.org/
terminus of viral plus-strand RNA, in association with 3CD (4, 30).
The mechanism of initiation of plus- and minus-strand syn-thesis has not been determined. Models for strand-specific initiation have been proposed on the basis of experiments in vitro with either a membranous crude replication complex (CRC) or with purified polymerase. CRCs from infected cells were shown to synthesize uridylylated derivatives of VPg, mol-ecules also detected in infected cells, and these species could be chased into longer VPg-bound polynucleotides with a se-quence identical to the 59 end of plus-strand RNA (78, 80). The CRC could also synthesize full-length viral RNA. Other studies suggested that the uridylylation of VPg was catalyzed by the viral polymerase (86) and that this reaction was also dependent on template RNA (79). Studies with a temperature-sensitive viral mutant in protein 3A demonstrated inhibition of the initiation and synthesis of plus-strand RNA in vivo and the uridylylation of VPg in vitro at the nonpermissive temperature (24). These results suggested a model in which protein 3AB, the most abundant membrane-bound precursor of VPg (81) and an apparent glycoprotein (20), is the source of uridylylated VPg, which in turn acts as primer for the initiation of plus-strand RNA synthesis (24, 69). Whether the uridylylation re-action occurs before or after the cleavage of VPg from 3AB by the viral protease is not known. The most recent models pro-pose that an initiation complex consisting of 3CD and either a 36-kDa cellular protein or 3AB bound to the 59 end of the plus-strand RNA, together with membrane-bound 3AB, cata-lyzes in trans the synthesis of new plus-strand RNA (4, 30). Whether proteins 2C and 2B play a role in any of these pro-cesses is not known. Two models, based entirely on in vitro studies, have been proposed for the synthesis of minus strands. In one model the hairpin-primed double-length RNA gener-ated by the concerted action of TUTase and the RNA poly-merase on the genomic RNA is cleaved in a transesterification reaction at the poly(A)-oligo(U) junction by either VPg or 3AB, resulting in the linkage of the protein to the newly gen-erated 59 end of the minus strand (84). In a second model, membrane-bound 3AB acts as primer for minus-strand initia-tion and elongainitia-tion is catalyzed by 3Dpol generated by the autoproteolysis of 3CD, which is bound to the 39 end of genomic RNA in a complex with 3AB (30).
The crucial contribution of the membrane component to the proper functioning of the replication complex has been clearly demonstrated by studies showing the disruption of nascent RNA synthesis in the CRC by detergent treatment (78, 80) and by the potent inhibition of viral RNA synthesis in vivo by brefeldin A or cerulenin, inhibitors of membranous vesicle formation (27, 47). Because the architectural integrity of the replication complex is apparently necessary for its ability to replicate viral RNA, disruption of the complex to assess the biochemical role of its components has not succeeded. An alternative approach to elucidate the individual functions of the viral and host factors involves reconstruction experiments, using purified viral (and host) proteins. In this report, we describe the purification of protein 3AB and its interactions with 3Dpol. Purified 3AB sedimented as a multimer in glycerol gradients and could bind to the polymerase. 3AB stimulated the activity of the polymerase up to 75-fold on a variety of RNA templates, including synthetic full-length minus-strand viral RNA. VPg also stimulated the activity of the polymerase but at a 50- to 100-fold-higher molar excess than 3AB. The stimulatory effect of 3AB appeared to be the result of an enhanced binding of the polymerase to the RNA template. Neither uridylylation of 3AB nor covalent linkage of the pro-tein into product RNA was detected. These results are
dis-cussed with reference to models proposed for the initiation of viral RNA synthesis in vivo. In reports published after the work presented below was completed, purified 3AB was shown to strongly stimulate the oligo(U)-primed poly(U) polymerase activity of 3Dpol(40, 60), to bind to both 3CD and 3Dpol(50), and to promote the binding of 3CD to both the 59 and 39 termini of the poliovirus genome (30).
MATERIALS AND METHODS
Preparation of poliovirus RNA.HeLa S3 cells at 107cells per ml were infected
with poliovirus type 1 (Mahoney strain) at a multiplicity of infection of 20 PFU/ml in S-minimal essential medium (Gibco BRL, Life Technologies Inc.) without serum. After 30 min at room temperature, the suspension was diluted twofold with S-minimal essential medium plus 5% calf serum and maintained at 378C for 7 h. Cells were harvested, and virus and virion RNA were purified exactly as previously described (52).
Construction of plasmids.In order to prepare full-length poliovirus minus-strand RNA terminating at its 39end with a sequence exactly complementary to the nucleotides at the 59end of plus-strand virion RNA, the sequences flanking the poliovirus sequences in plasmid pT7-PV1-5 (which generates plus-strand RNA upon transcription by T7 RNA polymerase [65, 77, 88]) were modified. Briefly, the T7 promoter at the 59end was removed and replaced with a MluI site. At the 39end, a new T7 promoter was inserted, allowing transcription in the opposite direction. First, pT7PV1-5 was digested with SalI and Tth111I, filled in with Klenow DNA polymerase, and religated. This removed a 1.6-kb vector fragment whose presence reduced the plasmid copy number. The resulting plas-mid was digested with BglI and SalI, and the fragment containing the T7 pro-moter and nucleotides 1 to 35 of the poliovirus genome was replaced with an oligonucleotide pair that rebuilt the poliovirus sequences and incorporated a MluI site between nucleotide 1 and the SalI site. The resulting plasmid was digested with EcoRI and then with mung bean nuclease (New England Biolabs, Inc.) to remove the 59overhanging sequences. The blunt ends were ligated to an oligonucleotide pair composed of the T7 promoter adjacent to a XhoI site to produce the final plasmid, pT7-PV1(2). The digestion of this plasmid with MluI and the removal of the protruding nucleotides with mung bean nuclease results in a template that directs the synthesis of a 7.5-kb minus-strand RNA with five nonviral nucleotides, GGCCG, preceding the poly(U) tract at its 59end and an authentic viral 39end. To construct plasmid pGEX-3AB, the source of protein glutathione S-transferase (GST)-3AB, plasmid pT7-2C3AB (12) was digested with AvaII and BamHI, releasing a 345-nucleotide fragment containing 3AB sequences which, after filling in with Klenow polymerase, was ligated to plasmid pGEX-2T (Pharmacia Biotech Inc.), which had been digested with BamHI, blunted with mung bean nuclease, and dephosphorylated with calf intestine phosphatase. The cleavage of GST-3AB protein with thrombin generates au-thentic protein 3AB, the NH2and COOH-termini of which are identical to the
protein expressed in poliovirus-infected cells. To construct plasmid pGEX-2C, the source of protein GST-2C, plasmid pGEX-2T was digested with BamHI and then treated with mung bean nuclease. The DNA was ligated to an oligonucle-otide pair extending from the second nucleoligonucle-otide at the 59end of the sequence coding for protein 2C to the SphI site. After digestion with SphI and SmaI, the DNA was ligated to a;950-nucleotide fragment containing the remainder of the gene coding for 2C. This fragment, containing sequences from the SphI site to the 39end of the coding region followed by two stop codons, a SalI, site and a blunted NotI site, was obtained from plasmid pT7-2C, which in turn was derived from plasmid pT7-2C3AB. All plasmids were confirmed by restriction digests and sequencing.
Synthesis of poliovirus minus-strand RNA.Plasmid pT7-PV1(2) was digested with MluI, and then the 59overhanging nucleotides were removed with mung bean nuclease. RNA (without radiolabel) was synthesized with the MEGAscript T7 RNA polymerase kit (Ambion, Inc.). After being treated with DNase, the RNA was extracted with phenol-chloroform (1:1), and unincorporated nucleoti-des were removed by passing the RNA through two successive Sephadex G-50 spin columns (Pharmacia Biotech Inc.). After ethanol precipitation, the RNA was redissolved in H2O.
Expression and purification of proteins GST-3AB and GST-2C.Escherichia coli harboring pGEX-3AB was induced with 1.0 mM isopropyl-b-D -thiogalacto-pyranoside (IPTG) as previously described (65), except that growth was contin-ued overnight at 258C. The cells (1 liter) were harvested by centrifugation, and the pellet was suspended in 20 ml of phosphate-buffered saline (PBS) containing 1 mM dithiothreitol (DTT) and 1% Triton X-100 (buffer A). The suspension was sonicated at 08C until it was no longer viscous and then centrifuged at 15,0003 g for 20 min. The pellet was resuspended in 10 ml of buffer A plus 1.0 M NaCl and sonicated again, and the suspension was centrifuged. The supernatant was diluted fivefold with buffer A. The two supernatants were pooled, filtered through a 0.45-mm-pore-size filter apparatus (Falcon; Becton Dickinson and Co.), and mixed with 2.5 ml of glutathione-agarose gel (Sigma Chemical Co.) equilibrated in buffer A. After stirring for 1 h at room temperature, the suspen-sion was loaded onto a 0.45-mm-pore-size filter apparatus and suction was ap-plied until the gel was dry. The gel was washed with 50 volumes of buffer A and
on November 9, 2019 by guest
http://jvi.asm.org/
then with 50 volumes of 50 mM Tris (pH 8.0)–1 mM DTT. The GST-3AB was eluted with 20 ml of a solution containing 50 mM Tris (pH 8.0), 10 mM gluta-thione, 10 mM DTT, and 0.1% Triton X-100 (or, alternatively, 0.5% octyl glucoside). Protein GST-2C, expressed in cells transformed with pGEX-2C, was purified by the same procedure. Protein GST was obtained from cells trans-formed with the parent vector pGEX-2T, except that no detergent was present in the final elution buffer.
Purification of protein 3AB.Purified GST-3AB (4 mg) was dialyzed against 50 mM Tris (pH 8.0)–1 mM DTT–0.1% Triton X-100 and incubated with 10mg of thrombin (Sigma Chemical Co.) at 308C for 2 h. The solution was passed through a 0.5-ml column of Q-Sepharose (Pharmacia Biotech Inc.) equilibrated in the same buffer but without the Triton X-100. The flowthrough, diluted to about two times the volume loaded on the column and containing nearly homogeneous 3AB, was adjusted to 10% in glycerol and stored at2708C. The working con-centration of the preparation was ;60 mg/ml, as determined by amino acid analysis.
Radiolabeling of poliovirus RNA polymerase.[35
S]methionine labeling of E. coli-expressing poliovirus polymerase was done as described previously (65). After labeling, the bacteria were pelleted and then lysed in 50 mM Tris (pH 8.0)–0.5 mM EDTA–1 mM DTT–0.1% Nonidet P-40 by passage several times through a narrow-gauge syringe. After centrifugation, the supernatant was ad-justed to 10% in glycerol and used for binding to GST fusion proteins immobi-lized on glutathione-agarose. Extracts were stored at2708C.
Binding of radiolabeled poliovirus polymerase to glutathione-agarose-immo-bilized GST proteins.Columns (0.2 ml) of glutathione-agarose were equilibrated in PBS–1 mM DTT. Two hundred micrograms of purified GST, GST-2C, or GST-3AB was dialyzed against this buffer and bound to individual columns. After washing the columns with loading buffer (50 mM Tris, pH 8.0, 1 mM DTT), [35S]methionine-labeled bacterial extracts containing poliovirus polymerase were
diluted 1:10 and loaded onto the columns. The columns were washed with loading buffer and then with loading buffer containing 1% Triton X-100. The final elution was with loading buffer containing 1% Triton X-100 and 1.0 M NaCl. The remaining enzyme bound to the column was released by boiling the glutathione-agarose resin in Laemmli buffer (39).
Purification of poliovirus RNA polymerase.Polymerase was purified as de-scribed previously (65). To remove trace amounts of poly(U) leeched from the poly(U)-Sepharose (Pharmacia Biotech Inc.) column used in the final purifica-tion step, the polymerase was dialyzed against 50 mM Tris (pH 8.0)–2.0 mM DTT–10% glycerol (buffer B) and loaded onto a phosphocellulose (Whatman P-11; Whatman International Ltd.) column equilibrated in this buffer. After washing with two column volumes of buffer B, the enzyme was eluted with buffer B plus 400 mM KCl. The polymerase was dialyzed against buffer B plus 50 mM KCl and stored at2708C at a concentration of;60mg/ml. Polymerase was also purified by affinity chromatography with GST-3AB bound to glutathione-agar-ose. All operations were performed at 48C. Bacterial extract from a 200-ml culture containing the polymerase was applied to phosphocellulose in a low-salt buffer as previously described (65), and after washing the column with buffer B plus 100 mM NaCl, partially purified enzyme was batch eluted with buffer B plus 400 mM NaCl (or KCl). After dialysis against buffer B, the enzyme was applied to a glutathione-agarose column (1.0 ml) to which was previously bound 4 mg of purified GST-3AB. The column was washed successively with buffer B, buffer B plus 0.5% Nonidet P-40, and then again against buffer B. The nearly homoge-neous polymerase was eluted with buffer B plus 1.5 M NaCl plus 40% added glycerol (50% total). The enzyme was dialyzed against buffer B plus 50 mM KCl and stored at2708C.
Enzymatic assays.RNA polymerase assays were performed in 25-ml volumes unless otherwise indicated. Reaction mixtures contained 60 ng of purified RNA polymerase, 50 mM HEPES (N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid, pH 8.0), 5 mM DTT, 0.5 mM (each) ATP, CTP, and GTP, 10mM UTP, 3.5
mM magnesium acetate, 50 U of RNasin (Promega Corporation), 2.5mCi of
[a-32P]UTP, and either 0.1mg of virion RNA, 0.1mg of globin mRNA (Gibco
BRL, Life Technologies Inc.), or varying amounts, as indicated in the text, of
minus-strand RNA synthesized from plasmid pT7-PV1(2). The RNA
concen-tration was estimated by ethidium bromide staining of test and standard marker RNAs on neutral agarose gels. When virion RNA or globin mRNA was added, 5 ng of synthetic oligo(U)20(made on an Applied Biosystems DNA 380B
syn-thesizer) was included, where indicated. Purified protein 3AB or synthetic VPg (made on an Applied Biosystems 430A peptide synthesizer) was added as indi-cated in the text. Incubations were at 308C for 60 min unless indicated otherwise. Aliquots (10ml) were removed and spotted on DE-81 (Whatman) filters, which were then washed in 0.5 M Na2HPO4, and the bound radioactivity was measured
in scintillation fluid. For analysis on gels, reactions were terminated with the addition of 10 mM EDTA and extracted with phenol-chloroform (1:1), and the RNA was recovered by ethanol precipitation in the presence of 10mg of carrier RNA.
Gel electrophoresis and Western gel analysis.RNA was analyzed by electro-phoresis on 0.8% agarose gels after glyoxal denaturation (46). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (15% acrylamide; ac-rylamide/bisacrylamide ratio, 125:1) was performed as previously described (39). Western (immunoblot) analysis of blotted gels was done by the ECL detection system (Amersham Corporation). The preparation of polyclonal antibody to poliovirus polymerase was described previously (65). Polyclonal antibody to 3AB was prepared by injecting rabbits with 100mg of synthetic VPg three times at 2-week intervals. Serum was collected after 6 weeks and stored at2208C.
Glycerol-gradient centrifugation.Samples were loaded onto 15 to 30% glyc-erol gradients prepared in 50 mM HEPES (pH 8.0)–2 mM DTT either lacking or containing 0.1% Triton X-100. Gradients were centrifuged at 40,000 rpm for 23 h at 48C in a Beckman SW 55 rotor. Fractions were collected from the top and analyzed by SDS-PAGE and then by Western analysis of blotted gels.
RESULTS
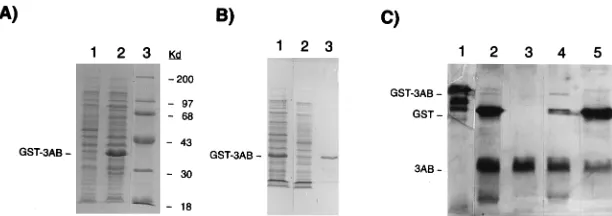
Purification of GST-3AB and 3AB.GST-3AB expressed in bacteria (Fig. 1A) was purified by glutathione-agarose chro-matography (Fig. 1B). The yield of the fusion protein was approximately 10 to 20 mg/liter of bacterial culture. Elution of the column with buffer lacking detergent yielded little or no GST-3AB (data not shown). The treatment of the GST-3AB with low levels of thrombin resulted in the quantitative cleav-age of the fusion protein and the release of native protein 3AB (Fig. 1C, lane 2). Most of the 3AB flowed through a Q-Sepha-rose column equilibrated in a low-ionic-strength buffer (Fig. 1C, lane 3); GST and thrombin were retained on the column under these conditions, resulting in a preparation of 3AB that was nearly homogeneous.
[image:3.612.155.461.71.179.2]Binding of poliovirus RNA polymerase to immobilized GST-3AB and affinity purification.3Dpolhas been implicated in the uridylylation of VPg (86), and genetic and biochemical evi-dence points to a role for 3AB in the initiation of viral RNA synthesis (24). These results suggested that 3AB and 3Dpolmay interact with each other. To test this, a [35S]methionine-labeled crude bacterial extract containing 3Dpol was loaded onto a glutathione-agarose column to which purified GST-3AB was previously bound. Most of the labeled 3Dpol bound to the
FIG. 1. Purification of proteins GST-3AB and 3AB. (A) SDS-PAGE of lysate from bacteria harboring pGEX-3AB following induction for 3 h with 1.0 mM IPTG, lane 2; no IPTG, lane 1; molecular weight markers, lane 3. (B) Elution of GST-3AB from glutathione-agarose column with glutathione plus Triton X-100, lane 3; sample applied to column, lane 1; column flowthrough, lane 2. (C) Purification of 3AB. GST-3AB (purified as in panel B, lane 3), lane 1; GST-3AB digested with thrombin, lane 2; thrombin-digested GST-3AB applied to a Q-Sepharose column, low-ionic-strength buffer flowthrough, lane 3; 0.1 M NaCl eluant, lane 4; 0.5 M NaCl eluant, lane 5. Proteins were stained with Coomassie blue (A) and (B) or silver (C).
on November 9, 2019 by guest
http://jvi.asm.org/
column, whereas the remaining bacterial proteins did not (Fig. 2A, compare lanes 9 and 13). The release of the polymerase from the column was resistant to a low-salt buffer wash con-taining nonionic detergent (Fig. 2A, lane 10), and only a por-tion of the polymerase could be eluted with buffer containing 1 M NaCl (lane 11). Much of the remainder, however, was released after boiling the glutathione-agarose with SDS (Fig. 2A, lane 12). The binding is specific since little or no poly-merase bound to glutathione-agarose columns to which had been previously bound either a fusion protein consisting of GST and poliovirus protein 2C (GST-2C) (Fig. 2A, lanes 5 to 8) or GST alone (lanes 1 to 4). We have also failed to detect any significant binding of the polymerase to a glutathione-agarose column to which was bound a fusion protein consisting of GST and poliovirus protein 2B (data not shown). To elim-inate the possibility that 3AB is a nonspecifically sticky protein, we tested whether poliovirus protein 3C protease could bind to GST-3AB. No binding was detected (data not shown).
These results suggested that a column containing immobi-lized GST-3AB could be utiimmobi-lized for the rapid purification of
milligram quantities of the polymerase. To reduce the back-ground of bacterial proteins applied to the column, an unla-beled extract from bacteria expressing 3Dpol was first passed through a phosphocellulose column. The polymerase binds to phosphocellulose and, after step elution with a buffer contain-ing 400 mM NaCl or KCl, is recovered quantitatively with a 10-to 20-fold increase in specific activity (data not shown [65]). Dialyzed eluant was applied to a column of glutathione-agar-ose to which was bound GST-3AB (Fig. 2B). Virtually all the bacterial proteins passed unbound through the column. As in the case with [35S]methionine-labeled protein, washing the column with detergent (Fig. 2B, lane 4) or low-concentration salt (lane 5) caused little or no polymerase to be eluted, and only a fraction of the enzyme was eluted with 1 M NaCl plus 10% glycerol (lane 6). Elution with 1 M NaCl plus 50% glyc-erol (Fig. 2B, lane 7), however, resulted in a significant im-provement in the recovery of the enzyme, with little additional recovery occurring when this was followed by elution with 1 M NaCl plus 50% ethylene glycol (lane 8). No GST-3AB was released from the column. The apparent purity of the enzyme rivaled that obtained by conventional ion exchange chroma-tography (65). A 10-fold scaleup of the column shown here yielded about 3 mg of purified polymerase from 2 liters of bacterial culture. Although nearly homogeneous, the specific activity of the enzyme purified on the GST-3AB column was less than half that of enzyme purified by conventional ion exchange chromatography with Mono Q (Pharmacia) and poly(U)-Sepharose columns (data not shown). When poly-merase purified by GST-3AB chromatography was applied to a poly(U)-Sepharose column or a GTP-agarose column (71), only a portion of the protein bound to these columns and the unbound portion was nearly devoid of enzymatic activity (data not shown). Thus, immobilized poly(U) and GTP appear able to discriminate between more active or less active forms of the polymerase, whereas 3AB does not. These results are consis-tent with poly(U) and GTP binding to the polymerase at its catalytic site and with 3AB binding elsewhere on the enzyme. Inactive enzyme may not present an accessible site for poly(U) and GTP binding, whereas 3AB may be able to bind both active and inactive enzymes.
3AB stimulates 3Dpol
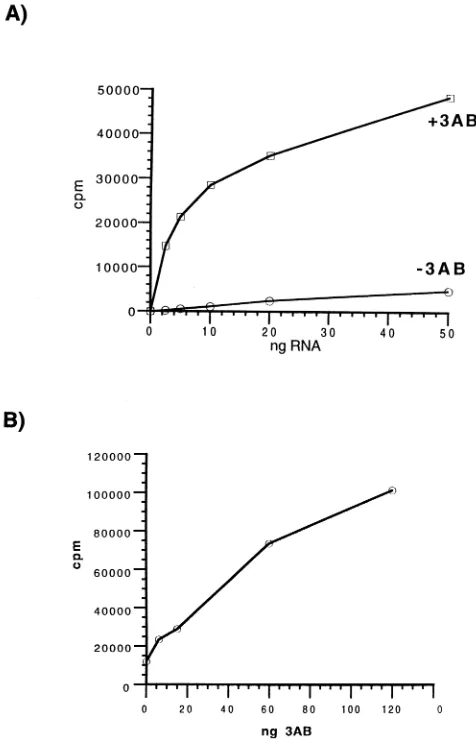
-catalyzed transcription of viral and nonviral RNA templates. The ability of immobilized protein 3AB to specifically bind the polymerase suggested that, if this interaction occurred in solution, it might result in the modu-lation of the enzymatic activity of the polymerase. To test this, purified 3AB was added to reaction mixtures containing poly-merase and synthetic minus-strand RNA as the template (Fig. 3). This RNA had five extra nonviral nucleotides at its 59end and was exactly complementary at its 39end to the nucleotides at the 59end of plus-strand RNA. Assays were performed with either a fixed concentration of 3AB and varying amounts of minus-strand RNA template or a fixed concentration of RNA and increasing amounts of added 3AB. In the absence of 3AB, levels of RNA synthesis increased with increasing amounts of template RNA (Fig. 3A). At a fixed level of template RNA, the addition of increasing amounts of 3AB resulted in a linear stimulation of RNA synthesis (Fig. 3B). At the highest level of 3AB tested, the molar ratio of 3AB to 3Dpol to RNA was
[image:4.612.83.274.74.371.2];10:1.2:0.04. The degree of stimulation was dependent on the amount of RNA template; at the lowest level of RNA tested, stimulation by 3AB approached 75-fold (Fig. 3A). Under these conditions, the molar ratio was;10:1.2:0.001. Similar template concentration-dependent stimulation of 3Dpol by 3AB was seen when poly(A)zoligo(dT) was used as the template-primer (60). GST-3AB could also stimulate RNA transcription (data not shown).
FIG. 2. Binding of 3Dpol
to immobilized GST-3AB and affinity purification. (A) [35
S]methionine-labeled bacterial extract containing 3Dpol
(lane 13) was applied to glutathione-agarose columns to which were previously bound purified GST (lanes 1 to 4), GST-2C (lanes 5 to 8), or GST-3AB (lanes 9 to 12). Low-salt buffer (see Materials and Methods) flowthrough, lanes 1, 5, and 9; 1% Triton X-100 eluant, lanes 2, 6, and 10; 1% Triton X-100 plus 1.0 M NaCl eluant, lanes 3, 7, and 11; protein solubilized after boiling column beads in Laemmli buffer, lanes 4, 8, and 12. (B) Affinity purification of 3Dpol
on glutathione-agarose-immobilized GST-3AB. 3Dpol, partially purified by phosphocellulose
chromatog-raphy (lane 1) was applied to immobilized GST-3AB equilibrated in buffer B (containing 10% glycerol). Flowthrough, lane 2; buffer B wash, lane 3; buffer B plus 0.5% Nonidet P-40, lane 4; buffer B plus 0.3 M NaCl, lane 5; buffer B plus 1.0 M NaCl, lane 6; buffer B containing 50% glycerol plus 1.0 M NaCl, lane 7; buffer B containing 50% ethylene glycol plus 1.0 M NaCl, lane 8. Proteins were stained with silver.
on November 9, 2019 by guest
http://jvi.asm.org/
The RNA synthesized in the absence of 3AB migrated as a heterogeneous mixture of products when examined by dena-turing agarose gel electrophoresis (Fig. 4A, lane 1). In the presence of 3AB, a substantial increase in this heterogeneous mixture was seen (Fig. 4A, lane 2), with the largest species migrating more slowly than the oligo(U)-primed RNA made from a 7.5-kb viral RNA template (lane 3). Computer models and nuclease digestion studies of the 39end of poliovirus mi-nus-strand RNA predict a hairpin structure which could the-oretically serve as a template-primer for the elongation of minus-strand RNA by the polymerase to a double-length prod-uct (62). The size of the largest RNA prodprod-uct seen here is consistent with a template-priming mechanism, suggesting that 3AB greatly stimulates the ability of the polymerase to utilize hairpin structures for self-priming. The stimulation of template priming by 3AB was also observed for poly(A) templates con-taining 39terminal heterogeneous nucleotides, presumably be-cause of the formation of hairpin structures (60). Bebe-cause of their heterogeneous nature, it is likely that the majority of the RNA products are generated not from the putative hairpin at the 39end of the full-length template but rather by hairpins at
the 39end of subgenomic fragments either already present or generated by the nicking of the template RNA during the reaction.
Transcription of the plus-strand poliovirion RNA by the purified polymerase requires the presence of a complementary oligonucleotide to act as the primer, presumably because the polyadenylated 39end cannot form a hairpin structure capable of self-priming. We previously showed that synthetic, polyade-nylated plus-strand poliovirus RNA prepared from a plasmid could self-prime, generating a double-length product in the presence of the polymerase (65). These results were subse-quently shown to be an artifact caused by the EcoRI-derived nucleotides 39 to the poly(A) sequence, because when these nucleotides were removed, no self-priming occurred (unpub-lished experiments and reference 18). With oligo(U) as the primer, the transcription of viral RNA generated RNA prod-ucts, the largest of which was 7.5 kb (Fig. 4B, lane 3). The addition of 3AB greatly increased the amount of these RNA species (Fig. 4B, lane 4), indicating that, as with hairpin struc-tures, 3AB also stimulates the ability of the polymerase to utilize noncovalently linked complementary oligonucleotides as primers (see also reference 60). In the absence of 3AB and oligo(U), no transcription was detected (Fig. 4B, lane 1). How-ever, in the presence of 3AB and the absence of oligo(U), a low level of transcription products was seen, almost all of which were smaller than 7.5 kb (Fig. 4B, lane 2). The nature of these products is not known, but they might be due to random priming by fragments of RNA present in the viral RNA prep-aration, whose ability to prime transcription can only be de-tected when the polymerase is stimulated by 3AB.
[image:5.612.319.553.71.264.2]With globin mRNA as the template, 3AB also strongly stim-ulated transcription primed by oligo(U) (Fig. 4A, compare lanes 4 and 5). In the absence of oligo(U), a barely detectable level of RNA product which migrated more slowly than the
FIG. 3. Stimulation of 3Dpol-catalyzed poliovirus minus-strand RNA
tran-scription by 3AB. (A) Counts per minute of [32P]UMP incorporated into product
RNA as a function of increasing amounts of PV1 minus-strand RNA template with or without 3AB (120 ng). (B) Counts per minute incorporated as a function of increasing amounts of 3AB in the presence of 100 ng of PV1 minus-strand RNA template.
FIG. 4. RNA synthesized in the presence or absence of 3AB. (A) [32
P]UMP-labeled RNA products transcribed from PV1 minus-strand RNA template (100 ng) with 3Dpolin the absence (lane 1) or presence (lane 2) of 120 ng of 3AB.
Lane 3, RNA product transcribed from virion plus-strand RNA template (100 ng) in the presence of oligo(U)20primer (5 ng). Lanes 4 to 6: RNA products
transcribed from globin mRNA (100 ng) in the presence of oligo(U)20primer
(lane 4), oligo(U)20primer plus 120 ng of 3AB (lane 5), and 120 ng of 3AB (lane
6). Lane 7, 32
P-labeled HindIII digest of lambda DNA. (B) RNA products transcribed from virion plus-strand RNA template. Lane 1, no additions; lane 2, 120 ng of 3AB; lane 3, oligo(U)20primer; lane 4, oligo(U)20primer plus 120 ng
of 3AB; lane 5,32
P-labeled HindIII digest of lambda DNA. RNA and DNA were analyzed by electrophoresis on 0.8% agarose gels after glyoxal denaturation (42).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.57.295.72.442.2]oligo(U)-primed product was transcribed in the presence of 3AB (Fig. 4A, lane 6). The nature of this RNA is not known but may be related to the apparent double-length RNA syn-thesized in the absence of oligo(U) described previously (65). VPg also stimulates the activity of the polymerase. In an initial attempt to determine which amino acid residues in 3AB mediate the stimulatory effect on the catalytic activity of the polymerase, synthetic peptide VPg was added to reaction mix-tures utilizing minus-strand RNA as the template (Fig. 5). RNA synthesis was stimulated up to eightfold with increasing amounts of added VPg, showing a degree of stimulation sim-ilar to that seen with 3AB and the same amount of RNA template (Fig. 3B). In addition, as with 3AB, heterogeneous RNA products up to double the length of the template in size were seen (data not shown). VPg also stimulated the
transcrip-tion of viral RNA in the presence of an oligo(U) primer, whereas other unrelated peptides of similar sizes had no effect (data not shown). The molar ratio of VPg to 3AB required to reach equivalent levels of stimulation was, however, 50- to 100-fold greater. Thus, the effect of 3AB appears to be medi-ated in part by VPg residues, but residues in 3A probably play a role in strengthening the interaction with the polymerase.
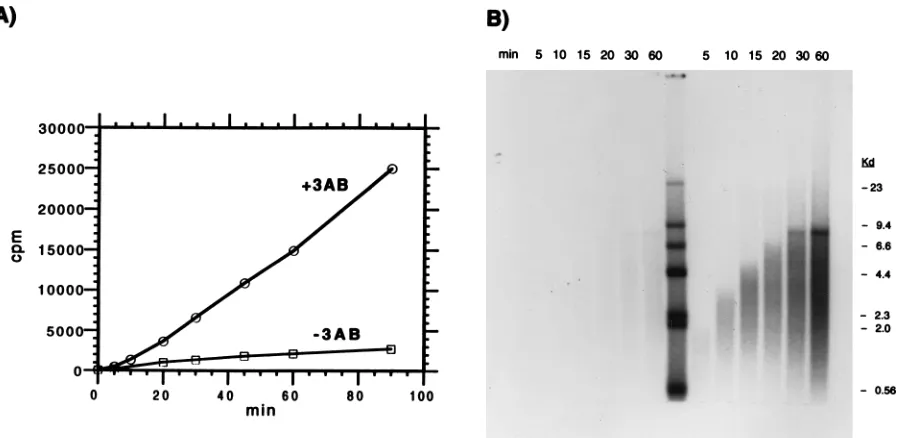
[image:6.612.71.286.70.236.2]3AB does not affect the rate of elongation.Several mecha-nisms can be proposed to explain how the binding of 3AB enhances the activity of the polymerase. The polymerase is a fairly unstable enzyme, with a half-life of about 1 min at 428C (71). Binding of 3AB may stabilize the enzyme even at 308C, allowing more extensive transcription to occur over longer times. 3AB may enhance the binding of polymerase to the template-primer, generating increased amounts of productive transcription complexes. Finally, 3AB may stimulate the rate of transcription, generating longer transcripts per unit of time. To test the latter hypothesis, aliquots were removed at various times and analyzed for the amount of radioactive label incor-porated and also by denaturing agarose gel electrophoresis. When added at the start of the reaction, stimulation by 3AB was observed even at the earliest time point taken (5 min) and increased with time (Fig. 6A). If 3AB was added 5 min after the start of the reaction, the degree of stimulation was signif-icantly reduced, suggesting that once transcription initiates, the ability of 3AB to bind to the polymerase is inhibited (data not shown). These results are in contrast to those of Paul et al. (60), who showed that the later addition of 3AB to reactions already initiated by 3Dpol with poly(A)zoligo(dT) template-primers resulted in stimulation equal to that observed when 3AB was present from the start. The rate of transcription, however, was not affected by 3AB, even if added at the start of the reaction (Fig. 6B). The observed elongation rate (;300 bp/min) was the same in the presence or absence of 3AB; only the total amount of product was enhanced by the presence of 3AB. Thus, 3AB appears to act by increasing the absolute
FIG. 5. Stimulation of 3Dpol-catalyzed poliovirus minus-strand RNA
tran-scription by VPg. Counts per minute of [32P]UMP incorporated into product
RNA as a function of increasing amounts of VPg in the presence of 100 ng of PV1 minus-strand RNA template.
FIG. 6. Effect of 3AB on the rate of elongation. (A) Virion RNA (100 ng) and oligo(U) primer were incubated with 3Dpol
either with or without 120 ng of 3AB in the transcription reaction buffer. At the times indicated, aliquots were removed and the counts per minute of [32
P]UMP incorporated into product RNA were determined. (B) Aliquots from panel A were removed at the times indicated and, after glyoxal denaturation, were analyzed by agarose gel electrophoresis and autoradiography.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.84.534.469.688.2]amount of productive transcription complexes without affect-ing the rate of elongation.
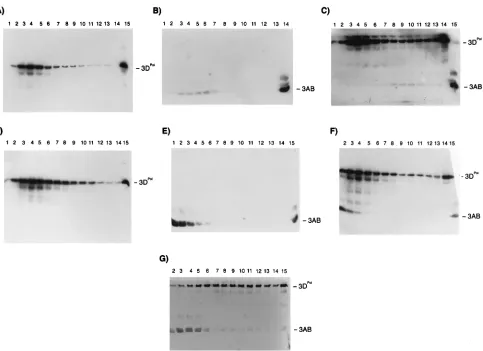
Effects of detergents.3AB is a hydrophobic protein and is found associated with membranes in the infected cell. The addition of detergent disrupts the membranous CRCs isolated from infected cells and blocks uridylylation of VPg and nascent RNA synthesis (71, 78). The elution by glutathione of GST-3AB immobilized on glutathione-agarose is inefficient unless detergent is included in the buffer. These results suggest that detergent may affect both the physical state of 3AB as well its ability to interact with and stimulate the activity of the poly-merase. To determine the effect of detergent on 3AB, increas-ing amounts of either Triton X-100, CHAPS {3-[(3-cholami-dopropyl)-dimethyl-ammonio]-1-propanesulfonate}, or octyl glucoside were added to oligo(U)-primed viral RNA transcrip-tion reactranscrip-tion mixtures catalyzed by the polymerase (Fig. 7A to C). In the absence of 3AB, none of the detergents had any significant effect on the activity of the polymerase. In the pres-ence of 3AB, each of the detergents, when present at levels at or above their respective critical micelle concentrations, com-pletely blocked the stimulatory effect of 3AB. Similar results were seen by Paul et al. (60) who used Nonidet P-40 to block 3Dpolstimulation by 3AB. Because 3AB presumably stimulates the polymerase by the formation of a complex between the two proteins, the effect of detergents is probably due to the disrup-tion of this interacdisrup-tion. To determine whether this is the case, 3AB, polymerase, and mixtures of 3AB and polymerase were analyzed by glycerol-gradient centrifugation in the presence or absence of 0.1% Triton X-100 (Fig. 8A to G). In the absence of detergent, the polymerase (52 kDa) sedimented in these gradients at about the same rate as albumin (68 kDa) (Fig. 8A). 3AB (12 kDa) sedimented at about the same rate or slightly higher, indicating that the protein behaves as a mul-timer under these conditions (Fig. 8B). The recovery of 3AB in these gradients was not quantitative; significant amounts pre-cipitated during centrifugation in the absence of detergent and were pelleted (the pelleted fractions were not analyzed by SDS-PAGE). When 3AB was present at a 1.75-molar excess over the polymerase, virtually all of the 3AB and some of the polymerase sedimented at a higher rate, suggesting that some of the polymerase formed a complex with most of the 3AB (Fig. 8C). When 3AB was present at a 4.5-molar excess over the polymerase, some of the 3AB and most of the polymerase sedimented at higher rates, again suggesting complex forma-tion (Fig. 8G). Because the total amount of 3AB is greater, it would be expected that the relative band intensity of the faster-sedimenting 3AB species should be greater in the latter gra-dient than in the former. This is not readily apparent from the autoradiogram. However, because of the variable levels of precipitation, comparing the absolute amounts of the faster-and more slowly sedimenting forms of 3AB between different gradients is probably not informative. In the presence of de-tergent, the sedimentation rate of the polymerase was the same as that in the absence of detergent (compare Fig. 8A and D). 3AB however, sedimented much more slowly, suggesting that detergent blocked the multimerization of 3AB (Fig. 8E). When 3AB was mixed with the polymerase and sedimented in the presence of detergent, no change in the sedimentation rates of either protein was detected, indicating that no complex forma-tion occurred (Fig. 8F). These results suggest that the interac-tion between the polymerase and 3AB may depend on the multimeric state of 3AB. This may be the case only in solution, however, because detergent treatment (in the absence of glu-tathione) of polymerase bound to GST-3AB immobilized on glutathione-agarose did not result in the elution of significant amounts of the polymerase (Fig. 2).
3AB is not uridylylated or covalently linked to RNA tran-scription products.In the infected cell, VPg is incorporated at the 59ends of nascent plus- and minus-strand RNA. In addi-tion, uridylylated forms of VPg have also been detected in vivo and in reactions catalyzed by CRCs, leading to the proposal that these molecules act as primers for further RNA synthesis. It was therefore of interest to determine whether 3AB, the presumed precursor of VPg, could be uridylylated or incorpo-rated into product RNA in vitro. 3AB was incubated in reac-tion mixtures containing polymerase, minus-strand RNA as the template, and either radiolabeled UTP alone, UTP plus ATP, or all four nucleoside triphosphates. After being digested with RNase A and T1, reaction products were analyzed by
SDS-FIG. 7. Effects of detergents on the stimulation of transcription with 3AB. Virion plus-strand RNA (100 ng) was incubated with oligo(U)20(5 ng) and 3D
pol
(60 ng) either with or without added 3AB (120 ng, 2ml) in the presence of increasing amounts of (A) octyl glucoside, (B) CHAPS, or (C) Triton X-100. Counts per minute of [32
P]UMP incorporated into product RNA were deter-mined. Critical micelle concentrations (CMC) of each detergent are indicated. The concentrations of detergents shown are in addition to that contributed by the buffer in which the 3AB is dissolved (0.05% Triton X-100; final concentration of ;0.004% in the 25-ml assay).
on November 9, 2019 by guest
http://jvi.asm.org/
PAGE. No incorporation of label into protein with a mobility similar to that of 3AB was detected (data not shown). Similar results were obtained when viral RNA, either with or without added oligo(U), was utilized as the template. Thus, the stim-ulation by 3AB of transcription by the polymerase in vitro occurs by a mechanism that does not include incorporation of 3AB into the RNA product.
DISCUSSION
We have shown that poliovirus proteins 3Dpoland 3AB form a detergent-sensitive complex in vitro that results in the stim-ulation of the enzymatic activity of the polymerase by a mech-anism that may involve enhanced binding to a template-primer. While the polymerase-3AB complex does not appear by itself to discriminate between viral and nonviral RNAs in vitro, other viral and host proteins may contribute in vivo to the observed specificity. 3AB, which is found tightly associated with newly synthesized membranous vesicles in infected cells (24, 81), may act to direct the polymerase to sites on these vesicles where viral RNA synthesis occurs. This trafficking event may occur after 3AB is already bound to membranes or
may be subsequent to initial complex formation in the cytosol. Alternatively, 3AB may act in vivo by binding to the 3D moiety of 3CD, which in turn is bound in a proposed initiation com-plex to the 59end of plus-strand RNA (4, 30). This would bring the initiation complex and 3AB into close proximity in the membranous vesicles, with the consequent exclusion of nonvi-ral RNAs from the replication machinery.
[image:8.612.69.552.68.419.2]In addition to the lack of specificity observed in vitro, no incorporation of 3AB into product RNA was detected. With plus-strand RNA as the template, hairpin priming was appar-ently observed. Because the major product of transcription in the infected cell is plus-strand RNA linked at its 59end to VPg, some mechanism for suppressing hairpin priming must exist. One way this might be accomplished is if the 39 end of the minus strand was closely juxtaposed to the 3CD-bound struc-ture at the 59end of the plus strand. This structure might have the dual role of blocking hybridization between the 59end of the plus strand and the 39end of the minus strand and at the same time preventing, in trans, hairpin formation in the minus strand. The resulting constraints may then favor 3AB (or VPg)-initiated, rather than hairpin-primed plus-strand synthe-sis. VPg could be generated from the membrane-bound 3AB
FIG. 8. Glycerol-gradient sedimentation of 3Dpoland 3AB. 3Dpoland 3AB were centrifuged either alone (A, B, D, and E) or together (C, F, and G) through 15
to 30% glycerol gradients. (A to C and G) Without detergent; (D, E, and F) with 0.1% Triton X-100. Gradients in panels A and D contained 6mg of 3Dpol; panels
B and E contained 2.4mg of 3AB; panels C and F contained 6mg of 3Dpolplus 2.4mg of 3AB; panel G contained 6mg of 3Dpolplus 6mg of 3AB. After 23 h at 40,000
rpm, gradients were fractionated and aliquots were removed and analyzed by SDS-PAGE and then by Western blotting with polyclonal antibodies specific for either 3Dpol(gradients in panels A and D) or VPg (gradients in panels B and E) or both (gradients in panels C, F, and G). Fraction 1 was the top of the gradient. In gradients
in panels F and G, fraction 1 was lost. In a separate gradient (not shown), protein standards cytochrome c (14 kDa) sedimented to fractions 1 to 2, bovine serum albumin (68 kDa) sedimented to fractions 4 to 5, and aldolase (160 kDa) sedimented to fractions 7 to 9. In lane 15 of panels A, D, and G and in lane 14 of panels C and F, protein 3Dpol
was added as a mobility marker. In lane 14 in panel B and lane 15 in panels C, E, and F, protein 3AB was added as a marker.
on November 9, 2019 by guest
http://jvi.asm.org/
by the proteolytic activity of the proximal RNA-bound 3CD. Recent work has shown that 3CD can cleave 3AB, releasing 3A and VPg, only if 3AB is presented in a membrane-bound form; unbound 3AB is not a substrate for 3CD (or 3C) (40). The autocleavage of 3CD into 3C and 3Dpolwould presumably also be necessary for the initiation of transcription, because 3CD lacks transcriptase activity (29, 89). The capability to carry out this autocleavage may reside in the components of the initia-tion complex. 3AB has been shown to stimulate the autocleav-age of 3CD (50). Either the membrane or RNA-bound 3AB could promote the release of active 3Dpol. It is possible, how-ever, that when bound in the initiation complex, 3CD may have the ability to catalyze the uridylylation of 3AB or VPg, thereby generating the primer needed for further transcription by 3Dpol, either produced by the autocleavage of 3CD or present in trans.
Although we and others (30) have not detected the incor-poration of 3AB or VPg into newly synthesized RNA in vitro, evidence for such incorporation has been presented. With plus-strand RNA as the template, a transesterification mech-anism has been proposed linking VPg to the 59 end of newly synthesized minus-strand sequences by cleavage at the junction of a poly(A)-poly(U) hairpin generated by the action of a host factor with TUTase activity (84). We did not examine the effects of host factor preparations in this study, and it would be of great interest to determine, using snap-back double-length RNA synthesized in the presence of TUTase, whether and with what efficiency 3AB also exhibited the cleavage and transes-terification activities ascribed to VPg. One reasonable predic-tion drawn from these studies is that 3AB would further en-hance the stimulatory effect of TUTase on the transcription of viral RNA in the absence of an oligo(U) primer.
A recently proposed model attempts to unify mechanisms for both plus- and minus-strand synthesis by suggesting that transesterification by VPg (specifically VPgUpU) may also oc-cur during the initiation of plus-strand synthesis (55). Poliovi-rus polymerase was shown to possess a terminal adenylyl trans-ferase activity which could add A residues to a synthetic oligoribonucleotide having the same sequence as the 39end of minus-strand RNA (we previously detected such an activity in 3Dpol using synthetic plus-strand RNA containing plasmid-derived sequences at its 39end [65]). If an oligo(A) sequence was added to the 39end of full-length minus-strand RNA in vivo, it might form a hairpin for template-primed synthesis of plus-strand RNA. The cleavage of the hairpin by VPgUgU by a transesterification mechanism at the border between the A residues in the loop and those base paired to the U residues in the template would generate the correct 59-terminal sequence in the plus-strand RNA. The binding of 3AB to the polymerase could facilitate some or all of the steps in this reaction mech-anism.
The precise mechanism whereby 3AB stimulates the poly-merase has not been determined. The apparent increase in productive transcription complexes suggests that 3AB acts to enhance the affinity of the polymerase for the template-primer. The fact that the relative degree of stimulation by 3AB is greater at low template RNA concentrations is consistent with enhanced binding. That 3AB can enhance the binding of 3Dpol to RNA is supported by experiments showing that the binding of 3CD and 3Dpolto the pseudoknot structure at the 39end of poliovirus genomic RNA requires the presence of 3AB (30). In addition, the ability of 3AB to bind both poly(A)zoligo(dT) and 3Dpolsuggested a mechanism by which enhanced binding and the consequent stimulation of the activity of 3Dpolon these template-primers could occur (60).
In our experiments, 3AB apparently binds to the polymerase
as a multimer. The precise composition of the multimer has not been determined, but from its sedimentation rate in glyc-erol gradients, it appears to be at least a tetramer. We do not know whether the formation of this multimer is a specific stoichiometric association or is merely a nonspecific aggrega-tion that occurs as the protein sediments through the gradient, leaving behind the detergent in which it was prepared. How-ever, the stimulation of the polymerase by 3AB occurs only at concentrations of detergents that are below their critical mi-celle concentrations, suggesting that the disruption of the mul-timer by high levels of detergent is directly correlated with preventing stimulation. This does not exclude the possibility that the 3AB monomer may bind to the polymerase if it could be generated in the absence of detergent. Immobilized GST-3AB binds the polymerase; its physical state is not known, but it is unlikely to adopt the same conformation as that of 3AB in solution. In vivo, 3AB is membrane bound and the nature of its association with the polymerase is likely to be strongly influ-enced by its surrounding hydrophobic environment.
3AB is not the only protein capable of specifically binding to the polymerase. In addition to the 36-kDa host protein that binds to the 59end of genomic RNA in association with 3CD (4, 30), a 68-kDa host protein, identified by its ability to stim-ulate transcription by the polymerase of viral RNA in the absence of an oligo(U) primer, was previously shown to bind to polymerase immobilized on CNBr-activated Sepharose (11). A protein of similar size was shown to coimmunoprecipitate with 3Dpol(19). It would be of interest to determine whether this protein(s) modulates 3AB binding to the polymerase.
The binding of 3AB to the polymerase appears to be medi-ated by both the VPg and 3A moieties of the protein. Site-directed mutagenesis of both 3AB and the polymerase should facilitate the identification of the amino acid residues involved in this interaction. The transfection of cells with genomic RNA containing the appropriate mutations will allow the evaluation of the role this interaction plays in the replication of poliovirus in vivo.
ACKNOWLEDGMENTS
We thank Ellen Baum and Ian Mohr for their suggestions and careful reading of the manuscript.
REFERENCES
1. Adler, C. J., M. Elzinga, and E. Wimmer. 1983. The genome-linked protein of picornaviruses. VIII. Complete amino acid sequence of poliovirus VPg and carboxy-terminal analysis of its precursor P3-9. J. Gen. Virol. 64:349– 355.
2. Aldabe, R., and L. Carrasco. 1995. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 206:64– 76.
3. Ambros, V., and D. Baltimore. 1978. Protein is linked to the 59-end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem.
253:5263–5266.
4. Andino, R., G. E. Rieckhof, P. L. Achacoso, and D. Baltimore. 1993. Polio-virus RNA synthesis utilizes an RNP complex formed around the 59-end of viral RNA. EMBO J. 12:3587–3598.
5. Andino, R., G. E. Rieckhof, and D. Baltimore. 1990. A functional ribonucle-oprotein complex forms around the 59end of poliovirus RNA. Cell 63:369– 380.
6. Andrews, N. C., and D. Baltimore. 1986. Purification of a terminal uridylyl-transferase that acts as host factor in the in vitro poliovirus replicase reac-tion. Proc. Natl. Acad. Sci. USA 83:221–225.
7. Andrews, N. C., D. Levin, and D. Baltimore. 1985. Poliovirus replicase stimulation by terminal uridylyltransferase. J. Biol. Chem. 260:7628–7635. 8. Baltera, R. F., Jr., and D. R. Tershak. 1989. Guanidine-resistant mutants in
poliovirus have distinct mutations in peptide 2C. J. Virol. 63:4441–4444. 9. Baron, M. H., and D. Baltimore. 1982. Anti-VPg antibody inhibition of the
poliovirus replicase reaction and production of covalent complexes of VPg-related proteins and RNA. Cell 30:745–752.
10. Baron, M. H., and D. Baltimore. 1982. In vitro copying of viral positive strand RNA by poliovirus replicase. J. Biol. Chem. 257:12359–12366.
on November 9, 2019 by guest
http://jvi.asm.org/
11. Baron, M. H., and D. Baltimore. 1982. Purification of a host cell protein required for poliovirus replication in vitro. J. Biol. Chem. 257:12351–12358. 12. Baum, E. Z., G. A. Bebernitz, O. Palant, T. Mueller, and S. J. Plotch. 1991. Purification, properties, and mutagenesis of poliovirus 3C protease. Virology
185:140–150.
13. Bernstein, H. D., P. Sarnow, and D. Baltimore. 1986. Genetic complemen-tation among poliovirus mutants derived from an infectious cDNA clone. J. Virol. 60:1040–1049.
14. Bienz, K., D. Egger, and L. Pasamontes. 1987. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic im-munocytochemistry and autoradiography. Virology 160:220–226. 15. Bienz, K., D. Egger, M. Troxler, and L. Pasamontes. 1990. Structural
orga-nization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J. Virol. 64:1156–1163.
16. Burns, C. C., M. A. Lawson, B. L. Semler, and E. Ehrenfeld. 1989. Effects of mutations in poliovirus 3Dpol
on RNA polymerase activity and on polypro-tein cleavage. J. Virol. 63:4866–4874.
17. Caliguri, L. A., and I. Tamm. 1970. The role of cytoplasmic membranes in poliovirus biosynthesis. Virology 42:100–111.
18. Cho, M. W., O. C. Richards, T. M. Dmitrieva, V. Agol, and E. Ehrenfeld. 1993. RNA duplex unwinding activity of poliovirus RNA-dependent RNA polymerase 3Dpol
. J. Virol. 67:3010–3018.
19. Dasgupta, A. 1983. Antibody to host factor precipitates poliovirus RNA polymerase from poliovirus-infected HeLa cells. Virology 128:252–259. 20. Datta, U., and A. Dasgupta. 1994. Expression and subcellular localization of
poliovirus VPg-precursor protein 3AB in eukaryotic cells: evidence for gly-cosylation in vitro. J. Virol. 68:4468–4477.
21. Flanegan, J. B., and D. Baltimore. 1977. Poliovirus-specific primer-depen-dent RNA polymerase able to copy poly (A). Proc. Natl. Acad. Sci. USA
74:3677–3680.
22. Flanegan, J. B., R. F. Pettersson, V. Ambros, M. J. Hewlett, and D.
Balti-more.1977. Covalent linkage of a protein to a defined nucleotide sequence at the 59-terminus of virion and replicative intermediate RNAs of poliovirus. Proc. Natl. Acad. Sci. USA 74:961–965.
23. Flanegan, J. B., and T. A. Van Dyke. 1979. Isolation of a soluble and template-dependent poliovirus RNA polymerase that copies virion RNA in vitro. J. Virol. 32:155–161.
24. Giachetti, C., and B. L. Semler. 1991. Role of a viral membrane polypeptide in strand-specific initiation of poliovirus RNA synthesis. J. Virol. 65:2647– 2654.
25. Gorbalenya, A. E., V. M. Blinov, A. P. Donchenko, and E. Koonin. 1989. An NTP-binding motif is the most conserved sequence in a highly diverged monophyletic group of proteins involved in positive strand RNA replication. J. Mol. Evol. 28:256–268.
26. Gorbalenya, A. E., and E. V. Koonin. 1989. Viral proteins containing the NTP-binding sequence pattern. Nucleic Acids Res. 17:8413–8440. 27. Guinea, R., and L. Carrasco. 1990. Phospholipid biosynthesis and poliovirus
genome replication, two coupled phenomena. EMBO J. 9:2011–2016. 28. Hanecak, R., B. L. Semler, C. W. Anderson, and E. Wimmer. 1982.
Proteo-lytic processing of poliovirus polypeptides: antibodies to polypeptide P3-7c inhibit cleavage at glutamine-glycine pairs. Proc. Natl. Acad. Sci. USA 79: 3973–3977.
29. Harris, K. S., S. R. Reddigari, M. J. Nicklin, T. Hammerle, and E. Wimmer. 1992. Purification and characterization of poliovirus polypeptide 3CD, a proteinase and a precursor for RNA polymerase. J. Virol. 66:7481–7489. 30. Harris, K. S., W. Xiang, L. Alexander, W. S. Lane, A. V. Paul, and E.
Wimmer.1994. Interaction of poliovirus polypeptide 3CDpolwith the 59and
39termini of the poliovirus genome. J. Biol. Chem. 269:27004–27014. 31. Hey, T. D., O. C. Richards, and E. Ehrenfeld. 1986. Synthesis of plus- and
minus-strand RNA from poliovirion RNA template in vitro. J. Virol. 58: 790–796.
32. Jablonski, S. A., and C. D. Morrow. 1993. Enzymatic activity of poliovirus RNA polymerases with mutations at the tyrosine residue of the conserved YGDD motif: isolation and characterization of polioviruses containing RNA polymerases with FGDD and MGDD sequences. J. Virol. 67:373–381. 33. Johnson, K. L., and P. Sarnow. 1991. Three poliovirus 2B mutants exhibit
noncomplementable defects in viral RNA amplification and display dosage-dependent dominance over wild-type poliovirus. J. Virol. 65:4341–4349. 34. Jore, J., B. de Geus, R. J. Jackson, P. H. Pouwels, and B. E. Enger-Valk.
1988. Poliovirus protein 3CD is the active protease for processing of the precursor protein P1 in vitro. J. Gen. Virol. 69:1627–1636.
35. Kirkegaard, K. 1992. Genetic analysis of picornaviruses. Curr. Opin. Genet. Dev. 2:64–70.
36. Kitamura, N., B. L. Semler, P. G. Rothberg, G. R. Larsen, C. J. Adler, A. J.
Dorner, E. A. Emini, R. Hanecak, J. J. Lee, S. van der Werf, C. W. Andersen, and E. Wimmer.1981. Primary structure, gene organization and polypeptide expression of poliovirus RNA. Nature (London) 291:547–553.
37. Kuhn, R. J., H. Tada, M. F. Ypma-Wong, B. L. Semler, and E. Wimmer. 1988. Mutational analysis of the genome-linked protein VPg of poliovirus. J. Virol. 62:4207–4215.
38. Kuhn, R. J., and E. Wimmer. 1987. The replication of picornaviruses, p.
17–51. In J. Rowlands, M. A. Mayo, and B. W. J. Mahy (ed.), The molecular biology of the positive strand RNA viruses. Academic Press, Inc., London. 39. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of
the head of bacteriophage T4. Nature (London) 227:680–685.
40. Lama, J., A. V. Paul, K. S. Harris, and E. Wimmer. 1994. Properties of purified recombinant poliovirus protein 3AB as substrate for viral protein-ases and as co-factor for RNA polymerase 3Dpol. J. Biol. Chem. 269:66–70. 41. Larsen, G. R., A. J. Dorner, J. R. Harris, and E. Wimmer. 1980. The structure of poliovirus replicative form. Nucleic Acids Res. 8:1217–1229. 42. Lee, Y. F., A. Nomoto, B. M. Detjen, and E. Wimmer. 1977. A protein
covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. USA
74:59–63.
43. Li, J. P., and D. Baltimore. 1988. Isolation of poliovirus 2C mutants defective in viral RNA synthesis. J. Virol. 62:4016–4021.
44. Lubinski, J. M., G. Kaplan, V. R. Racaniello, and A. Dasgupta. 1986. Mech-anism of in vitro synthesis of covalently linked dimeric RNA molecules by the poliovirus replicase. J. Virol. 58:459–467.
45. Lubinski, J. M., L. J. Ransone, and A. Dasgupta. 1987. Primer-dependent synthesis of covalently linked dimeric RNA molecules by poliovirus repli-case. J. Virol. 61:2997–3003.
46. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual, p. 200–201. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
47. Maynell, L. A., K. Kirkegaard, and M. W. Klymkowsky. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 66:1985–1994. 48. Mirzayan, C., and E. Wimmer. 1992. Genetic analysis of an NTP-binding
motif in poliovirus polypeptide 2C. Virology 189:547–555.
49. Mirzayan, C., and E. Wimmer. 1994. Biochemical studies on poliovirus polypeptide 2C: evidence for ATPase activity. Virology 199:176–187. 50. Molla, A., K. S. Harris, A. V. Paul, S. H. Shin, J. Mugavero, and E. Wimmer.
1994. Stimulation of poliovirus proteinase 3Cpro-related proteolysis by the
genome-linked protein VPg and its precursor 3AB. J. Biol. Chem. 269: 27015–27020.
51. Molla, A., A. V. Paul, M. Schmid, S. K. Jang, and E. Wimmer. 1993. Studies on dicistronic polioviruses implicate viral proteinase 2Apro in RNA repli-cation. Virology 196:739–747.
52. Molla, A., A. V. Paul, and E. Wimmer. 1993. Effects of temperature and lipophilic agents on poliovirus formation and RNA synthesis in a cell-free system. J. Virol. 67:5932–5938.
53. Morrow, C. D., and A. Dasgupta. 1983. Antibody to a synthetic nonapeptide corresponding to the NH2terminus of poliovirus genome-linked protein VPg
reacts with native VPg and inhibits in vitro replication of poliovirus RNA. J. Virol. 48:429–439.
54. Morrow, C. D., G. F. Gibbons, and A. Dasgupta. 1985. The host protein required for in vitro replication of poliovirus is a protein kinase that phos-phorylates eukaryotic initiation factor-2. Cell 40:913–921.
55. Neufeld, K. L., J. M. Galarza, O. C. Richards, D. F. Summers, and E.
Ehrenfeld.1994. Identification of terminal adenylyl transferase activity of the poliovirus polymerase 3Dpol. J. Virol. 68:5811–5818.
56. Neufeld, K. L., O. C. Richards, and E. Ehrenfeld. 1991. Purification, char-acterization, and comparison of poliovirus RNA polymerase from native and recombinant sources. J. Biol. Chem. 266:24212–24219.
57. Nicklin, M. J. H., H. Toyoda, M. G. Murray, and E. Wimmer. 1986. Proteo-lytic processing in the replication of polio and related viruses. Bio/Technol-ogy 4:33–42.
58. Nomoto, A., B. Detjen, R. Pozzati, and E. Wimmer. 1977. The location of the polio genome protein in viral RNAs and its implication for RNA synthesis. Nature (London) 268:208–213.
59. Pallansch, M. A., O. M. Kew, B. L. Semler, D. R. Omilianowski, C. W.
Anderson, E. Wimmer, and R. R. Rueckert.1984. The protein processing map of poliovirus. J. Virol. 49:873–880.
60. Paul, A. V., X. Cao, K. Harris, J. Lama, and E. Wimmer. 1994. Studies with poliovirus polymerase 3Dpol
: stimulation of poly(U) synthesis in vitro by purified poliovirus protein 3AB. J. Biol. Chem. 269:29173–29181. 61. Pettersson, R. F., V. Ambros, and D. Baltimore. 1978. Identification of a
protein linked to nascent poliovirus RNA and to the polyuridylic acid of negative-strand RNA. J. Virol. 27:357–365.
62. Pilipenko, E. V., S. V. Maslova, A. N. Sinyakov, and V. I. Agol. 1992. Towards identification of cis-acting elements involved in the replication of enterovirus and rhinovirus RNAs: a proposal for the existence of tRNA-like terminal structures. Nucleic Acids Res. 20:1739–1745.
63. Pincus, S. E., D. C. Diamond, E. A. Emini, and E. Wimmer. 1986. Guanidine-selected mutants of poliovirus: mapping of point mutations of polypeptide 2C. J. Virol. 57:638–646.
64. Pincus, S. E., and E. Wimmer. 1986. Production of guanidine-resistant and -dependent poliovirus mutants from cloned cDNA: mutations in polypeptide 2C are directly responsible for altered guanidine sensitivity. J. Virol. 60:793– 796.
65. Plotch, S. J., O. Palant, and Y. Gluzman. 1989. Purification and properties of poliovirus RNA polymerase expressed in Escherichia coli. J. Virol. 63: 216–225.
66. Porter, A. G. 1993. Picornavirus nonstructural proteins: emerging roles in
on November 9, 2019 by guest
http://jvi.asm.org/
virus replication and inhibition of host cell function. J. Virol. 67:6917–6921. 67. Racaniello, V. R., and D. Baltimore. 1981. Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc. Natl. Acad. Sci. USA 78:4887–4891.
68. Reuer, Q., R. J. Kuhn, and E. Wimmer. 1990. Characterization of poliovirus clones containing lethal and nonlethal mutations in the genome-linked pro-tein VPg. J. Virol. 64:2967–2975.
69. Richards, O. C., and E. Ehrenfeld. 1990. Poliovirus RNA replication. Curr. Top. Microbiol. Immunol. 161:81–119.
70. Richards, O. C., T. D. Hey, and E. Ehrenfeld. 1987. Poliovirus snapback double-stranded RNA isolated from infected HeLa cells is deficient in poly(A). J. Virol. 61:2307–2310.
71. Richards, O. C., P. Yu, K. L. Neufeld, and E. Ehrenfeld. 1992. Nucleotide binding by the poliovirus RNA polymerase. J. Biol. Chem. 267:17141–17146. 72. Rodriguez, P. L., and L. Carrasco. 1993. Poliovirus protein 2C has ATPase
and GTPase activities. J. Biol. Chem. 268:8105–8110.
73. Rothberg, P. G., T. J. R. Harris, A. Nomoto, and E. Wimmer. 1978. The genome-linked protein of picornaviruses. V. O4
-(59-uridylyl)-tyrosine is the bond between the genome-linked protein and the RNA of poliovirus. Proc. Natl. Acad. Sci. USA 75:4868–4872.
74. Semler, B. L., C. W. Anderson, R. Hanecak, L. F. Dorner, and E. Wimmer. 1982. A membrane-associated precursor to poliovirus VPg identified by immunoprecipitation with antibodies directed against a synthetic heptapep-tide. Cell 28:405–412.
75. Semler, B. L., C. W. Anderson, N. Kitamura, P. G. Rothberg, W. L. Wishart,
and E. Wimmer.1981. Poliovirus replication proteins: RNA sequence en-coding P3-1b and the sites of proteolytic processing. Proc. Natl. Acad. Sci. USA 78:3464–3468.
76. Spector, D. H., and D. Baltimore. 1974. Requirement of 39terminal poly-adenylic acid for infectivity of poliovirus RNA. Proc. Natl. Acad. Sci. USA
71:2983–2987.
77. Studier, F. W., and B. A. Moffatt. 1986. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189:113–130.
78. Takeda, N., R. J. Kuhn, C.-F. Yang, T. Takegami, and E. Wimmer. 1986. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J. Virol. 60:43–53.
79. Takeda, N., C.-F. Yang, R. J. Kuhn, and E. Wimmer. 1987. Uridylylation of the genome-linked protein of poliovirus in vitro is dependent upon an en-dogenous RNA template. Virus Res. 8:193–204.
80. Takegami, T., R. J. Kuhn, C. W. Anderson, and E. Wimmer. 1983.
Mem-brane-dependent uridylylation of the genome-linked protein VPg of polio-virus. Proc. Natl. Acad. Sci. USA 80:7447–7451.
81. Takegami, T., B. L. Semler, C. W. Anderson, and E. Wimmer. 1983. Mem-brane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virology 128:33–47.
82. Tershak, D. R. 1982. Inhibition of poliovirus polymerase by guanidine in vitro. J. Virol. 41:313–318.
83. Teterina, N. L., K. M. Kean, E. Gorbalenya, V. I. Agol, and M. Girard. 1992. Analysis of the functional significance of amino acid residues in the putative NTP-binding pattern of the poliovirus 2C protein. J. Gen. Virol. 73:1977– 1986.
84. Tobin, G. J., D. C. Young, and J. B. Flanegan. 1989. Self-catalyzed linkage of poliovirus terminal protein VPg to poliovirus RNA. Cell 59:511–519. 85. Toyoda, H., M. J. H. Nicklin, M. G. Murray, C. W. Anderson, J. J. Dunn,
F. W. Studier, and E. Wimmer.1986. A second virus-encoded proteinase involved in proteolytic processing of poliovirus polyprotein. Cell 45:761–770. 86. Toyoda, M., C.-F. Yang, N. Takeda, A. Nomoto, and E. Wimmer. 1987. Analysis of RNA synthesis of type 1 poliovirus by using an in vitro molecular genetic approach. J. Virol. 61:2816–2822.
87. Tuschall, D. M., E. Hiebert, and J. B. Flanegan. 1982. Poliovirus RNA-dependent RNA polymerase synthesizes full-length copies of poliovirion RNA, cellular mRNA, and several plant virus RNAs in vitro. J. Virol.
44:209–216.
88. Van der Werf, S., J. Bradley, E. Wimmer, F. W. Studier, and J. J. Dunn. 1986. Synthesis of infectious poliovirus RNA by purified T7 RNA poly-merase. Proc. Natl. Acad. Sci. USA 83:2330–2334.
89. Van Dyke, T. A., and J. B. Flanegan. 1980. Identification of poliovirus polypeptide p63 as a soluble RNA-dependent RNA polymerase. J. Virol.
35:732–740.
90. Van Dyke, T. A., R. J. Rickles, and J. B. Flanegan. 1982. Genome-length copies of poliovirion RNA are synthesized in vitro by the poliovirus RNA-dependent RNA polymerase. J. Biol. Chem. 257:4610–4617.
91. Yogo, Y., and E. Wimmer. 1972. Polyadenylic acid at the 39terminus of poliovirus RNA. Proc. Natl. Acad. Sci. USA 69:1877–1882.
92. Young, D. C., D. M. Tuschall, and J. B. Flanegan. 1985. Poliovirus RNA-dependent RNA polymerase and host cell protein synthesize product RNA twice the size of poliovirion RNA in vitro. J. Virol. 54:256–264.
93. Ypma-Wong, M. F., P. G. Dewalt, V. H. Johnson, J. G. Lamb, and B. L.
Semler.1988. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166:265–270.
![FIG. 2. Binding of 3Dpol(A) [ to immobilized GST-3AB and affinity purification.35S]methionine-labeled bacterial extract containing 3Dpol (lane 13) was](https://thumb-us.123doks.com/thumbv2/123dok_us/1282991.80965/4.612.83.274.74.371/binding-immobilized-afnity-purication-methionine-labeled-bacterial-containing.webp)