Vol. 63, No. 4 JOURNALOFVIROLOGY, Apr. 1989, p. 1743-1755
0022-538Xl89/041743-13$02.00/0
CopyrightC) 1989, American Society for Microbiology
Bovine Papillomavirus
Type
1Encodes
Two
Forms
of
aTranscriptional
Repressor: Structural and Functional Analysis
of
New
Viral
cDNAs
JOONHO CHOE,t PETERVAILLANCOURT, ARNE
STENLUND,t
AND MICHAEL BOTCHAN* Department ofMolecular Biology, University of California, Berkeley, California 94720Received23 November1988/Accepted6January 1989
Genetic and biochemical evidence has established that the E2 open reading frame (ORF) of bovine
papillomavirustype1 encodesatleasttwodifferentsite-specificDNA-binding proteins,onewhichactivates and
the other whichrepressesexpression fromaviralpromoter(P. F. Lambert, B. A. Spalholz,and P. M. Howley, Cell50:69-78, 1987). Wehave obtaineddata which show thatasecondform of therepressor geneisexpressed intransformedcells harboring stable viral plasmids. Thestructuraldetailsofthisgenehave been discerned by cDNAcloning, by RNase protection, andby primer extension analysis of in vivo RNA. Moreover,datafrom invitro transcriptionexperiments supportthe notion that this form of the E2repressoris expressedfrom a
novelviral promoter and thatasmallexonfrom anotherORF is linkedtoanactiverepressordomain in E2.
Thus,twodifferentforms of therepressorareexpressedfrom differentpromotersand might beindependently regulated either in the cell cycleor indifferent tissue types. We show byfunctional in vivoassaysutilizinga cDNAvectorencoding thisgenethat thetrans-actingfactorhas invivo activitiessimilartothose of the known
repressor. OurscreenofacDNA library for cDNA clones representing bovine papillomavirus transcripts has
also revealed a number ofother novel structures defining new donor and acceptor RNA-processing sites.
Notably, cloneswhichconceptuallycanbe translatedtoyieldanE7 protein, theviral Mgene, and the entire E2 ORFhavebeen characterized. Finally, truncatedversionsofputativeE8cDNAs werealsoobtained.
The life cycles of the DNA viruses which infect animal
cellscanbebroadlyplaced intotwocategories. Some DNA viruses infect cells and rapidly enter into productive
repli-cation with ensuing cell death, while others establish their genomesasepisomesand havealong latentperiod.(We use the wordepisome to mean anadded genetic element which
can reproduce itself in the cell indifferent waysdepending
onthenatureof the element and the cell type. Thus the word
episomecanrefer toanelementacquired by infection which
iscarried by the cell in an integrated state or as aplasmid. For further details, see reference 20.) This latency even persists throughmany celldivisions if theinfected cells are dividingor areinducedtoproliferate by virallyencoded gene
products. The viruses which cause warts are interesting
members of this latter class because the viral episomes establish asstable nuclearplasmidsandmustreplicatetheir genomesincoordinationwith the cell through a large
num-ber (54) of cell divisions. Bovine papillomavirus type 1
(BPV-1) in particular causes massive fibropapillomas soon after infection (25), and in these cells the viral genome is carried as a high-copy-number plasmid. Vegetative and
exponential replication ofthisvirus, however,occursonlyin
terminallydifferentiated keratinocytes. Thus, ahighly regu-lated process coordinating viral gene expression and cell
state must playamajorrole in the maintenance oflatency. Along theselines, it is thought thata majorviral promoter
(PL)
is active only indifferentiating keratinocytes (2).We have been interested in the study of BPV-1 plasmid replication with the view that anunderstanding of how the
*
Corresponding
author.tPresent address: Department of Biology, Korea Institute of Technology, Taejon 302-338, Korea.
tPresentaddress: Cold SpringHarbor Laboratory, Cold Spring Harbor,NY 11724.
replication system is regulated might give some clues into how transcription and replication processes interplay in eucaryotes. Therodent C127 cellsprovidewhatwe
optimis-tically believe to be a good model for this latent plasmid replication in vivo. Viral infection of the cells leads to oncogenic transformation with single-hit kinetics (11), and the viral genome is carried as a stable multicopy nuclear
plasmid (26, 27). The facts that the virus transforms with
single-hitkinetics and that the copy number of 100to 200is detectable after only a limited number of cell doublings implythataninitialamplificationof viralDNA occursbefore
astableregulated copy numberis achieved. This concept is
reinforced by transient replication studies (28) which
indi-catethat viral DNAreplicatesfaster than cellular DNA upon initial entryintothecell. Afterestablishment,the viral copy number staysconstant.
Thegenetic analysis of the BPV-1 genome reveals thata
large network of viral factors and promotersmustinteractto
achieve the switch from this initiation mode ofreplicationto
the homeostasis apparent in the transformed cells. The
somewhat surprising feature of this system seems to be its
complexity, as at least six different gene products are
included in this process. How these factors interact in a
kinetic sense and in maintenance has not been elucidated. The El open readingframe
(El
ORF) ofBPV-1 is theonly
ORF within which mutation absolutely abolishes transientreplication in C127cells. Within this ORF,
genetic
analysis
indicates thatatleasttwogeneproductsarerequiredintransfor establishment of the stable
plasmid
state. The R gene encoded in part by the 3'portion
of the ORF(4,
29)
isabsolutely required for transient
replication
and has been postulatedtotherefore play adirect role in viralreplication
(4). Incontrast,frameshift mutations in the 5'
portion
of theEl ORF donot seem to affecttransient
replication.
These mutations define agene in anothercomplementation
group,1743
on November 10, 2019 by guest
http://jvi.asm.org/
M. This M gene is
required
for the maintenance of stableplasmids
and has thegenetic
properties
ofa repressor ofreplication. Thus,
cellsharboring
low levelsofBPV-1 plas-mids are immune toamplification
of wild type uponsuper-infection. Yet M- mutants
replicate
asefficiently
in these cells asthey
do in uninfected cells(4, 29). Furthermore,
Roberts andWeintraub
(39)
haveshown thataproduct(s)
of BPV-1likely
to include M canrepress intrans simianvirus 40 runawayamplification,
if the BPV-1origins
are linked incistosimian virus 40. These results
together imply
thatMisarepressorof
amplification
or apositive
factor forregulated
low-level
replication
whichisantagonistic
towardamplifica-tionorboth.
Recently,
Thorneretal.(49)
haveidentified theM
product
as asmallphosphoprotein
whosesize iscompat-ible with its gene
being
encodedmainly by
the 5'portion
of the El ORF.To define with more
precision
the structures of these genes encodedby
the ElORF,
we havesought
to obtain cDNAcopies
of theirtranscripts.
Inthis report,wedescribe thestructures ofseveralnewBPV-1transcripts
asdeducedfromthe sequences of cDNAs andfromthedirect
analysis
ofin vivo viral RNAs. One of these cDNAs is indeed a
potential
candidate foranMmessenger.Surprisingly,
how-ever,themostabundant RNAs which contain El sequences
detectedinour
analysis
initiatefroma newBPV-1promoterwithin the ORF. The
major
product
ofthis promoter is aspliced
RNA which encodes another form of the BPV-1transcriptional
repressor(23).
MATERIALS ANDMETHODS
Cell culture. C127 cells
(11),
ID13 cells(26),
and HeLa cells were maintained in Dulbecco modifiedEagle
mediumsupplemented
with 10% fetal calf serum andpenicillin-streptomycin.
cDNA
library.
ID13cellsweretreated withcycloheximide
(35 .ig/ml) prior to being harvested. A cytoplasmic extract
wasobtained
by lysis
of cells in thedetergent
Nonidet P-40 andasubsequent
pelleting
of nuclei via standardprotocols.
Thisextractwas thenprocessed
via the standardguanidium
isothiocyanate-cesium
chloride method(30). Poly(A)
RNAwas
prepared by
passage ofcytoplasmic
RNA overoli-go(dT)-cellulose
twice. A cDNAlibrary
wasconstructedby
using
poly(A)
RNA and theX590
vector(9).
Ten ,ug ofpoly(A)+
RNA was used tosynthesize
the first strand ofcDNAin a
50-pl
mixture of56 mMTrishydrochloride (pH
7.5)-85
mM KCl-3 mMMgCl2-0.5
mMdeoxynucleoside
triphosphate-2
mMMeHgOH-30
mM,B-mercaptoethanol-5
p.g
ofoligo(dT) (Collaborative
Research, Inc.)-30
U ofRNasin
(Promega
Biotec)-1,000
U of murine reversetran-scriptase (Bethesda
ResearchLaboratories, Inc.).
Thesolu-tionwasincubatedfor 60 minat
37°C.
Themixturewasthen usedtomakethe second strandofcDNAina150-pl
volume of 85mMTrishydrochloride
(pH 7.5)-15
mM(NH4)2SO4-30 mM KCl-0.17mMdeoxynucleoside triphosphate-100
,uCiof[cx-32P]dATP
(Amersham Corp.,
400Ci/mmol)-l
Uof RNaseH
(P-L Biochemicals,
Inc.)-4.5
U of DNA polymerase I(New
England
BioLabs, Inc.).
Second-strandsynthesiscon-tinued for16hat
16°C.
Afterelongation
of the second strandof
cDNA,
1 ,ulof100 mM ATP and 1 U ofT4 DNAligasewereadded and incubated for 15 minatroom temperature. The mixture was extracted with phenol:chloroform and
double-stranded cDNA was precipitated with ethanol. cDNAwastreated withT4DNA
polymerase
(New EnglandBioLabs)
tomake bluntends.Subsequently,
HindIIIlinkerswere
ligated
to double-strandedcDNA,
using
T4 DNAligase. The cDNAs were then completely digested with HindlIl to remove excess linkers from the ends at the cDNAs, and the cDNAswere separated on Bio-Gel A-50M (Bio-Rad Laboratories). cDNAs larger than 500 base pairs
werepooled and ligated with
X590
arms. AfterligatedXDNA was packaged by using Gigapack plus (Stratagene), the cDNA librarywas amplified, using C600Hflhost bacteria. The cDNAlibrarywasscreenedby theplaquehybridizationmethod (3), using nick-translated BPV-1-specificprobes(see
Fig. 1). DNA fragments were sequenced by the dideoxy
sequencing method(40) afterbeing subcloned into
M13mpl8
ormpl9 vectors.
Plasmids.Theplasmid CMV E8/E2 contains a 1.2-kilobase (kb) insert of clone N7-3 subcloned into theHindIll site of pON260 (43) in the same transcriptional orientation as the cytomegalovirus (CMV) promoter. pON260 contains a 760-base-pair fragment of the CMV promoter and enhancer and
aninternal ,-galactosidasecassette.pON260 is a derivative ofpON240. Oligonucleotide-directed (in vitro) mutagenesis
was carried out as follows to construct the plasmid CMV E8*/E2. The Hindlll fragment (1.2 kb) of clone N7-3 was
subcloned intoM13mpl9. The oligonucleotide(5'-CCGTGT
TCTAACGCCCCTC-3')wassynthesized by a DNA synthe-sizer(Applied Biosystems, Calif.). By using this
oligonucle-otide and the method ofNakayame and Eckstein (34), codon 7 of the E8/E2ORF has beenchanged from leucine (TTA)to atermination codon (TAA). The mutatedHindlll fragment
inM13mpl9wasisolated and insertedinto pON260,aswas the wild-type cDNA. Plasmid pUC C59 is a derivative of
pC59 (51). TocreatepUC C59, the DNA fragment contain-ing the early promoter and the polyadenylation sequences of simian virus 40 and BPV-1 cDNA was subcloned into the HindIII and SmaI sites of pUC18. Plasmid pMLBPV100 contains the entire BPV genome linearized at the BamHI site cloned into vectorpML and has beendescribed elsewhere (27). Plasmids used as probes for the RNase protection
experiments and for in vitro transcription are described in thefigure legends.
S1 nucleaseandRNase protection analysis of RNA. For S1
nuclease, RNase protection, andprimer extension analysis of in vivo RNA, poly(A)-selected cytoplasmic RNA was prepared as described previously (5). Unless otherwise
indicated,cellsweretreated with 30
p,g
ofcycloheximide per mlfor 4 hpriortobeingharvested. S1 nucleaseanalysis was carried out as described elsewhere (13). Preparation ofuniformlylabeled SP6probes andRNaseprotection analysis
were performed as described previously (33), with the
fol-lowing modifications. Hybridizations were carried out in 40
mMPIPES (piperazine-N,N'-bis(2-ethanesulfonicacid) (pH
6.7)-0.4 M NaCl-1 mM EDTA in a final volume of30 ,ul.
Following denaturation for 5 min at 85°C, hybridizations
were carried out at 68°C for 3 h. RNase digestions were
performedat25°Cfor 30 min unless otherwise indicated. All
digestion productswerefractionatedon5% denaturing
poly-acrylamide gels. Cordycepin A sequencing ladders were made by preparing uniformly labeled SP6 probes as de-scribedpreviously (33) in the presenceof 125 p.MdATP.
Invitrotranscriptionandprimer extension. Primer exten-sionwasperformedasdescribed elsewhere (46). Whole-cell
extracts of HeLa cells were prepared as described
previ-ously (31), and invitrotranscription wascarried out by the
procedure ofDynanandTjian (12). All reactions contained 25 p.l ofan optimal amount of HeLa cell extract in TM.1 buffer (50 mM Tris [pH
7.9]-0.1
mM EDTA-12.5 mMMgCl2-100
mM KCl-2mMdithiothreitol-17% glycerol),2%polyvinyl alcohol, 250 p.M each of the four
on November 10, 2019 by guest
http://jvi.asm.org/
NEW BPV cDNAs 1745
A.
OPEN READING 2 FRAMES
3
49 501 1099 1479 2581
449 659
1 ---EEl
813 2663
i a CY
ZL I fL8 2 £
x 0 ux u mco
a I I I a I
tz
01 I
ml
S.PROBES
PP1
PE9
PP2 PE7 PE1J
PE6 PE8 PEIA
C. cDNA
J9-3
N7-3
1235 3225 4203
N15-2
2556 , N12-2
18"1940
b1m31613
J9-2
---1940
N7-1
138 304 528
12-4
7840 7905 528
N19-1
73477385 547 8641032
P19-1
7224 7385 558 864 1032
27-3 7430 7905 556 6641024
26-1
7438 7905 528
N16-1
72917385
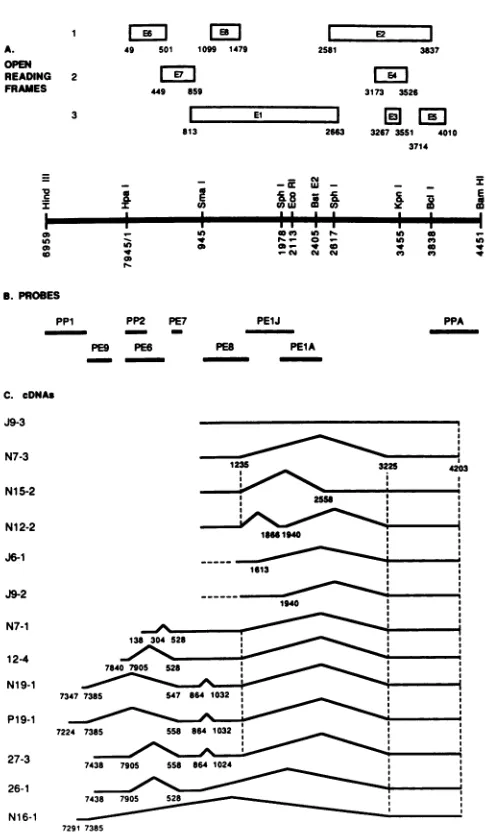
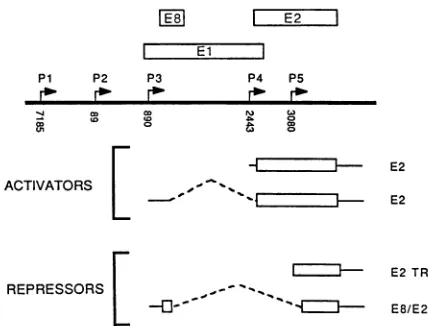
FIG. 1. Structure of BPV-1early ORFs and cDNAs. (A) Map of the BPV-1 69%(HindIII-BamHI) fragment. Early ORFsare repre-sentedbyopenboxesdesignated EltoE8 andarealigned according
toreading frame. (B) Probes used for screening the cDNA library. The DNAfragments shownweresubcloned into pBR322 orpML2, werenick translated, andwereusedtoprobe the cDNA library. The nucleotidepositions of each of the probesare asfollows:PP1, 6958 (HindIII)to7476(ClaI); PE9, 7476to7760(RsaI); PP2, 1 (HpaI)to
240 (RsaI); PE6, 1 to 470(RsaI); PE7, 697 (TaqI) to 767 (TaqI); PE-8, 1010 (Hindll) to 1515 (BglII); PE1J, 1515 to 2113 (EcoRI); PE1A, 1978 (SphI) to 2450 (RsaI); PPA, 3838 (BcIl) to 4451 (BamHI). (C) Structures of the cDNAs. cDNAs are arranged accordingtothemajorgroupsdefinedby the hybridization profiles asdescribed in the text. The structures of all cDNAs were com-pletely determined by nucleotide sequencing. Exons ( ) and intervening sequences (A) are shown. Numbers beneath the lines indicate the ends and splice junctions of the cDNAs. Thetopfour cDNAs(J9-3, N7-3, N15-2,andN12-2)have 5' endsatnucleotides 1070, 1012, 930, and 948, respectively, and are likely to be
tran-scribed from the P3promoter.cDNAsJ6-1 and J9-2 have5' endsat
nucleotides 1379 and 1410, respectively. As no5'ends have been mappedin thisregionthusfar,we assumethat thesearetruncated cDNAsrepresenting transcripts which initiate atthe P3 promoter.
Thisassumption is depictedin thediagram bythe dashedhorizontal lines whichextendtothe P3capsite. The 5' ends of cDNAs N7-1 through N16-1 are labeled, and their probable promoters are de-scribedinthetext.
E2 J
otides,
1.0Vtg
ofsupercoiled plasmidDNA, and H20 to a 3837 final volume of 50,ul.
Where indicated, 4 pg ofa-amanitin
LE4
| per ml was added to the reaction mixtureprior
to the 3173 3526 addition of DNA. Reactions were incubated for 30min
atla
El
30°C and were terminated by the addition of 90 ,u1 of stop326730S1 4010 solution containing 1% sodium dodecyl sulfate-20 mM
3714 EDTA-200 mM NaCl-250 ,ug of tRNA per ml. Reactions were extracted once withphenol:chloroform, were
precip-E itated with ammonium
acetate,
and weresuspended
in l hybridization buffer for primer extension reactions. Primer extension products were fractionated on an 8% denaturingc 0 V polyacrylamide gel.
CAT (chloramphenicol acetyltransferase) assay. HeLa cells were transfected by the calcium phosphatemethod (15) with modifications. Each 60-mm plate of HeLa cells received 10
P
,g
with the total amount of DNA adjusted by the addition ofcarrier DNA. Unless otherwise indicated, cotransfection included 2 ,ug of upstream regulatory region (URR) CAT expression plasmid to provide the CAT activity. The DNA-calcium precipitate was applied to the cells for 12 h followed
I---a
by 1)V7o glycerol snock
Cr)f1 -.... 1 -C__, r)__ o : __11- '51ILfior
3
min.tells
werenarvestea3t
naftertransfection. CATassay wascarried out as previously described (14). The amount of all extracts used for the assays was50 ,ug of protein, and the assays were run for 1 h at37°C.
RESULTS
Identification ofBPV-1 cDNA clones. AcDNAlibrary was constructed fromcytoplasmic poly(A) RNA extracted from virus-infected ID13 cells. Prior to RNA extraction, the cells weretreatedwith cycloheximide to increase our chances of
netting viral RNAs with short half-lives (22). cDNAs were synthesized via modifications of the standard protocols (35), using oligo(dT) as a primer for first-strand synthesis, and the library size, which was measured to be 1.5 x 106, was subsequently amplifiedonce (see Materials and Methods).
To screen this library for cDNAs which might represent El transcripts, the series of hybridization probes shown in
Fig. 1 below a physical map ofthe early region of BPV-1 wereconstructed. Thelocation oftheprobes with respect to theORFsof theearly regionareshown and in thelegend to Fig. 1B, the precise BPV-1 nucleotide positions are given.
Previous RNA mapping experiments indicated that a BPV mRNA exists which contains an exon encoded by a 5'
portion ofthe ElORF(47). As outlined in the introduction, we wereinterested inobtaining cDNAs which might encode for the M gene and thus RNAs with codingsequencesfrom
this 5' Elregionweresought.Towardthisend,we used the PE8probe inaprimaryscreenof thelibrary. Approximately
0.01to0.05%of the lambdaplaqueshybridizedtothisprobe.
The abundance of such inserts was approximately two- to
threefold lower than those frequencies estimated with the PE6probe.
Forty-five PE8-positive plaques were picked, and the inserts were then analyzed in detail. The first
characteriza-tion was to hybridize the cDNAs with each ofthe
probes
shown inFig. 1B. Theinsertswerethusclassified intomajorgroupsaccording to theirhybridization profiles. The struc-turesof nine representatives of each classof cDNA charac-terizedbythisscreen areshown in
Fig.
1C. Theyarenamed clones J9-3through N19-1;clonesP19-1through
N16-1wereobtainedbyanotherscreendescribed below.To
analyze
thestructures of these clones in
detail,
the inserts were sub-cloned into M13 and pBR322 vectors. Restriction analysesand Southern blots were usedtoadd information about the various families. The DNA sequencesof the
representative
members shown inFig. 1 were completelydetermined.I 9 I I
VOL.63, 1989
I. .I I
I* r-:: 0 ;
co ob 10
CY cm W
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.52.295.70.487.2]Themostabundant suchfamilyofcDNAs obtainedbythis
primary screen is represented by clone N7-3 (Fig. 1C). Twenty-four such clones were obtained. These cDNAs
hybridized only tothe PE8 and PPA probes. Clones of the N7-3 family had 5' ends clustered between BPV-1 nucleo-tides 900to 1000.These clonesterminated, asdid allothers in ourstudy, at a3' positionof 4203 followed byapoly(A)
tail. cDNA clones with the hybridization profiles of N7-1 wereobtainedeighttimes while thoseof the J9-3familywere obtained seven times. All others were obtained as unique
members.
ThecDNAs thatarebelievedtobegeneratedbycommon
promoters are grouped together in Fig. 1. As discussed
below, webelieve it is likelythat abundant clones J9-3and
N7-3,aswellastherelativelyrarerclonesN15-2andN12-2,
represent RNAs produced from a previously unreported BPV-1promoternearthe5' endofthe ElORF.The5'ends of these clonesarenot labeledin thefigure but aregiven in thelegend. The abundant cDNA N7-1 probably represents RNAs initiatingfrom the previously characterized P2 (P89)
promoter,as it hasa5'endonly49 nucleotides downstream from this cap site. Two of the clones obtained via this
primaryscreencontainedBPV-1sequenceswithexonsfrom theURR;12-4 andN19-1. N19-1 is 161 nucleotides short of the previously characterized P7186 (P1) promoter, and 12-4
may represent a severely truncated transcriptfrom this P1 promoteror anas-yet-unidentified promoterin theURR.
To obtain other clones containing sequences from the
URR, two separate primary screens of the library were
done, usingthe PP1 and PE9probes.Withbothprobes,two million bacteriophage were independently screened. P19-1
wasobtained(Fig. 1C), whichhasastructureverysimilarto thatofN19-1, althoughadifferentacceptor links theposition
7385 URR donor to the downstream coding sequences. Three otherclones, 27-3 through N16-1, were also charac-terized. Two clones, 27-3 and 26-1, both have 5' ends at
preciselythe same nucleotides. Thisposition 7438is atthe endofaT8stretchinthecodingstrand andmayrepresenta
hot spot forcleavage with RNase H in the preparation for second-strand cDNAsynthesis, thuscreating artificially the chanceforcommon5'ends in afamily of cDNAs.
We will discuss in another section of this article the
potential coding capacitiesof the variouscDNAsdescribed here.However,atthispointit isinterestingtopointoutthat withthe possible exceptionof cDNAcloneJ9-3, nocDNAs
specifically isolated could encode for the R gene and only
this clone contained a nucleotide sequence between
posi-tions 1940 and2558. This isparticularly intriguingas muta-tionaldatamentioned above shows that mutationsatBPV-1
position 2113 inactivate transient as well as stable plasmid replication. To directly screen for cDNAs which might
contain sequences from this region, we probed the cDNA
librarywiththeprobePE1J(Fig. 1B). Fromascreenofmore than 5 x 106phage, no BPV-1cDNAs werefound that did notfall intothe families definedby J9-3, J6-1, orJ9-2.
To summarize this structural information briefly, a large
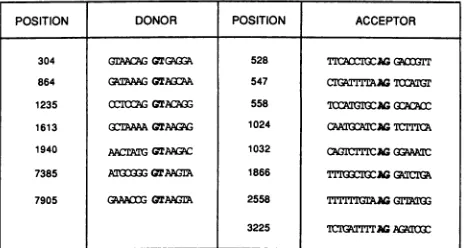
numberofnewdonor andacceptorsiteswithinBPV-1 have beenestablishedbythese cDNAs,and the sequencesfound atthesesitesareshown inFig.2. Initially,wedidnot expect to find such a large number of different types of cDNAs whichspannedthe 5' endof the ElORF. However,recently
published Northern (RNA) blot analysis of BPV-1 RNAs with probes similarto those used here (6, 46) substantiate
thiscomplexityinthatalargenumberoftranscripts ranging from 1.2 to 2 kb in length were detected. Clearly, more detailed genetic analysis will be necessary to correlate the
POSITION DONOR POSITION ACCEPTOR
304 GrAALA GTGUQZ 528 TICAMOWAGG(IT
864 [email protected] 547 CITGAT=AG700
1235 0IO;GGTAMGG 558 lCAGGAGGC8A
1613 G(3AAGTAaG 1024 CA :MCr=Il
1940 AArGGTAC 1032 AGICTlCMAGGAN 7385 AT3G= QrAMLG 1866 Tl'uxG=oGGATIG
7905 GPAACO GTAA 2558 rGAmyGGTrkI1
3225 ¶CIGfTIITG AGA FIG. 2. Positions and surrounding DNA sequences of splice
junctionsdeduced from the viral cDNAs sequencedin thisstudy.
Consensus sequencesarein boldface.
physical RNA map with functional genes. The majority of cDNAs founduse acommon splice donor in the El ORFat
nucleotide 1235. The position of this donorwas first estab-lished by electronmicroscope analysis of BPV-1 RNAs(47). However, the complexity of the genetic organization of the virus in this region isillustrated by the findings reported here thatthis donor can join exons to either the mostcommonly used acceptor in the virus at position 3225 (2, 51) or to
acceptorsatnucleotide 1866or 2558.
Identification of a promoter within the El ORF. The abundant cDNAs described in the previous section all have their 5' ends near the beginning of theEl ORF. Neverthe-less, it is impossible to prove via cDNA cloning where the bona fide 5' ends of the actual RNAs are by this sort of analysis. Even with model systems such as globin 9S mRNA, methods which enrich forfull-length clones gener-ally yield cDNA which are truncated at their 5' ends (36). We had anticipated that the RNAs represented by the cDNA clone N7-3 would be initiated by a promoter close to the beginning of the El ORF.Alternately, these RNAs could be initiated upstream and a small exon generated by splicing might have been lost in cDNA synthesis. This assumption wasbased on the facts that the cDNA inserts in thelibrary werelarge and that sequenceanalysis of cDNAs from known abundant BPV-1 RNAs terminated close to known start sites (data not shown). Thus, to determine where the likely initiation sites are, it was necessary toanalyze in vivo RNA by a variety of nuclease protections and primer extension methods.
A series of probes either 5' end labeled or uniformly labeledwereused, and their structures are displayed in Fig. 3. First, a DNA fragment from nucleotides 576 to 1010was
5' end labeled on thenoncoding strand and was annealedto cytoplasmic poly(A)RNAfromID13orC127 cells and was digested with Si nuclease (Fig. 3). Two protected fragments
areclearly seen in the lanemarked ID13 and aremissing in theC127 controlpanel; theyarelabeled P3a and P3b. From the lengths of these fragments measured relative to the sequence ladderand size markersruninparallel, the 5' ends maptoBPV-1positions 896 and 886. TheBPV-1sequence at these positions does not show splice acceptor sites, so we
tentativelyassumedthatthese positions demarked bona fide 5' ends. To substantiate thisconclusion, synthetic primers extending fromBPV-1nucleotides 925 to 945 or from 1038 to 1058(Fig.4)were synthesized,5'endlabeled, and annealed
to ID13 cytoplasmic poly(A)-selected RNA, and primer extension analysis was performed. The extension products were fractionated by gel electrophoresis and separated
alongside a sequence ladder of BPV DNA obtained by
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.322.556.79.203.2]NEW BPV cDNAs 1747
22>
-22
d:
'>Probes
'.55 - 4
E8
, El
P3
-.
rl.;
ffi ! > A .
__
__ t.. _
:-<t re4$ _
..=.>.
>_ rO
_..:>:s
X .^.s
s I;>s>t .,.
.. +:
^..b. ..
_..:::
s .. __ o4-X 4w ....
XF
.
X...
:
..:
Prolectedfragments
.:5
9
FIG. 3. Si nucleasemappingofP3 capsites. Poly(A) cytoplasmic RNA from 2 x 107ID13cells or C127 cells was hybridized to the 5'-end-labeled noncoding strand of a DNA fragment from positions 576 to 1010 (probe I)anddigested with
Si
nuclease. The hybrids were fractionatedon a5%denaturingpolyacrylamidegel.Lanes: M,5'-end-labeledfragments from aBstNl-EcoRIdigest of pBR322;G,A, T, and C, size markersgenerated bydideoxysequencing of asingle-stranded M13 bacteriophage template. Size markers (in nucleotides) are shown atthe left.dideoxy sequencing, using this same primer annealed to BPV DNA. The lane marked in vivo in the autoradiogram
shown in Fig. 4 maps two discrete extension products to nucleotides 886 and 896, thus corroborating the
Si
analysis. Tofurtherextend the notion that a stable 5' end maps tothese nucleotides, a plasmid containing the BPV-1 PstI
fragment from nucleotides 576 to1299 was assayed in vitro
forpromoteractivityin whole-cell extractsofHeLacells in
thepresence orabsence of4,ugof a-amanitin per ml. This is
alevel ofdrug whichinhibitsRNApolymerase II
transcrip-tioninvitro.Theinvitroproducts were analyzed via primer
extensionanalysisasdescribed forthein vivoRNA.Thein
vitro products produced by thisfragment have the same 5'
endsasdotheinvivoRNA, andtheirsynthesis is sensitive
to the RNA polymerase II inhibitor(Fig. 4). We will here-after refer tothis regionof DNA asthe P3 promoter, using
theconventionfromourlaboratory firstdescribed for the P1 promoter (46). We use this convention rather than naming
the promoter after putative cap sites (2) for two reasons.
First, promoters usually refer to cis-acting regulatory
se-quencesandweassumethat thestarts atnucleotides 886 and 896 share common cis control elements. Second, for very
heterogeneousends (e.g., those aroundnucleotide 2440), it
is extremely cumbersome to label each nucleotide as a
promoter. The disadvantage, of course, isthat new
promot-erswhich may be found would require differentnamessince the orderfromP1 would be shifted.
To determine the structure of the exons of the mRNAs
initiating atthe P3 promoter, a series ofRNase protection
experiments were performed, using the uniformly labeled cRNAprobesdepictedinFig. 3.Cytoplasmic poly(A)RNAs
extractedfromID13orC127cellswerehybridizedto anSP6
polymerase-generated probe which contained BPV-1
se-quencesfrom nucleotides 576 to 1299 (probe II in Fig. 3).
Aftertreatmentwith RNase Aand RNaseT1, the protected fragmentswereseparated byelectrophoresis. The
autoradio-gram of this gel is shown in Fig. 5A; lane C is the C127 control,and lanes A and B are identical experimentsexcept for the RNasedigestion conditions. Bythe mobilities ofthe fragments we could tentatively ascribe each band to a
particular transcript. The model for these assignments is
shown diagrammatically in Fig. 3. The topmost protected
band is attributed to full-length probe protection and
repre-sentsunspliced (withintheproberegion) RNAs;this bandis
unlabeled. The next band down represents RNAs initiating upstreamof the 576positionandsplicingat1235;RNAs such
as cDNA clone N7-1 and 12-4would generate such bands.
Thetwobands labeledP3b and P3acorrespondtofragments produced bytranscripts initiatingatnucleotides 886and 896 and splicing at nucleotide 1235; specifically, the length of
these bands corresponds to 338 and 348 nucleotides, the
length from the P3 promotertothedonor. Themostfrequent
cDNA clone that we obtained in our screen N7-3 would
generate suchfragments,and these resultscorroboratethose findings. The band labeled 576-865 is ascribed to all RNAs initiating upstream of 576 (e.g., E6 and E6-7 messengers) whichusethe 865 donor(47, 51).Totesttheseassignments,
probe IIwashalvedtogenerateSP6probesIII and IV. Each
ofthese probes was annealed to cellular RNAs and was
analyzed. The predicted fragment lengths were detected
with each probe(Fig. SB). Specifically, the protected frag-mentfromprobeIV(nucleotides945to1299)representsthe
cumulative signal of all exons which cross nucleotide 945
anduse the 1235 splice donor(i.e., P3aand P3b as wellas N7-1 and 12-4 transcripts). Probe III detects the RNAs
which usethe865 donor.
Fromthe intensities of these bands inFig.5,it would seem
that the ratios of the P3
transcripts
totranscripts
initiating VOL.63, 1989TI
Z.
S-: !,-; ,
.r
Z,.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.65.553.81.326.2]in vitro
0[E E E
> r,
* 0:
V
_; ::.s
_7~1
_ ~_...~ ~ ~
~~-_~~~~
_O*P3b..
I I
:"ro<:r: 7.Ie:- r~losA.-IASSs-h
FIG. 4. Primer extensionanalysis of in vivo- andinvitro-transcribedRNAfromthe P3 promoter.TheBPVPstIfragmentfrompositions
576to1299, cloned intothePstI site ofpSP65,wastestedinaninvitrotranscriptionassayusingManleyextractsof HeLa cellsinthepresence (+ a-am) orabsence(-a-am)of4,ugof a-amanitinper ml(see Materials andMethods),followedbyprimerextensionusinga5'-end-labeled oligonucleotidecontaininganoncoding-strandsequencefrom nucleotides 1038to1058. (Twoprimerswereused in thissortofexperiment,
onefrom nucleotides 925 to 945 and one fromnucleotides 1038to1058. The resultsfromthelatterareshown.)Primerextensionwasalso performedonpoly(A)cytoplasmicRNAfrom2 x 107ID13 cells. LanesG,A,T, and C containdideoxysequencingladdersusingboththe primerandtemplateused forthe invitrotranscriptionexperiment. Extensionproductswerefractionatedon an8%denaturingpolyacrylamide
gelandarelabeledasshown on the right.
upstream(such as those at P2)is roughly 1:5. (Forexample,
comparetheintensities ofbands labeled P3a and P3btothe bandat576-865 in Fig. 5A.)This number agrees reasonably
with the resultsof the cDNA cloning experimentsdescribed
above. Furthermore, a significant number of transcripts probably initiate at P2 and generate RNAs which use the 1235donor(e.g.,N7-1).Thiscanbe estimatedfromthe band labeled 576-1235 in Fig. 5A.
As the RNAs used astemplates forcDNAsynthesiswere
prepared fromcycloheximide-treated cells,itwasofinterest to address the questionofwhether drug treatment changes
qualitativelythe RNAprofileinthis region.The results(Fig.
5C) show that no qualitative differences can be discerned,
although there is a large increase in signal specifically from the P3 promoter with cycloheximide treatment. (Note that
10-fold more RNA was used in the cycloheximide-minus experiment.)
Clone N7-3 encodes a repressor of BPV gene expression.
Conceptual translation of the sequences encoded by clone N7-3predicts a 24,000-dalton protein. This putative polypep-tidelinks11 amino acids from the E8 ORF to the C-terminal domain of the BPV-1 E2 ORF. Interestingly, it has recently been shown (32) that the carboxy-terminal domain of the E2 protein contains sequences both necessary and sufficient for
site-specific binding to BPV-1 DNA (17). Further
heighten-ingourinterest in the functional significance of clones such
as N7-3 and the P3 promoter in general was the work of Lambert et al. (23, 24). In their original work defining
functionally a BPV-1 transcriptional repressor, Lambert et al. (23) assayed the repressor by placing a surrogate
pro-moter upstream of BPV-1 sequences included within the
clone pCW1-28 which contains BPV sequences from posi-tion 845 continuously to 4203 (Fig. 1). On the basis of the available genetic evidence at that time (deletion and termi-nation mutants withinpCW1-28), theytentativelyconcluded that theenhancer in the surrogate promoterwas stimulating transcription from a distal internal promoter which initiated
starts more than 2 kb away. The promoterwas positioned around BPV nucleotide 3080. Indeed, surrogate promoters placed just upstream of 3080 did produce vectors which functionally producedarepressor. Supportingevidence that
a truncated form of E2 was actually expressed by virus-infected cells was found in the literature. RNA mapping experiments which showed that 5' ends could bemappedto nucleotide 3080(47) and thecloningofacDNAwhich could representsuch RNAs,p1153, produced byBaker and
How-ley (2) from infected bovine
fibropapillomas,
made it seemlikely that such arepressor protein speciesprobablyexists. However, certain deletions upstream of the putative start site at nucleotide 3080 in the promoted pCW1-28 clone
eliminated theabilityof this vectortoexpress the repressor (24). These lattermutants could have deleted an upstream
exon of the repressor gene encoded by pCW1-28 or a
promoter element for the putative p3080 promoter. From thesefindingsandourresults, wesuspected thattwoforms of the repressor gene areexpressed: one initiatedfromthe P3 promoter, and the other initiated from within the E2 ORF. Accordingly, the surrogate enhancers in pCW1-28 wereprobablyinitiating transcriptsfrom P3.
Totestthispossibility directly,weplacedthe N7-3 cDNA clone downstream of theCMV promoterto create aputative repressorexpression vector called CMV E8/E2. As a
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.63.553.78.331.2]NEW BPV cDNAs 1749
A
Probe11
m
IA
B CI999 - _
__ 5* S-1235
516>
506 0t
ai.::
396 - _
- P3b
34 4-
-0
;S 4 P298 - .4-576-865
221>*
225 ,
B
ProbeI11PObe IV
D V a:
--576-865- j 4- 945 1235
-I
_U
As
[image:7.612.116.490.74.258.2]h
FIG. 5. RNaseprotection mapping of P3 transcripts. Poly(A)cytoplasmic RNAfrom2 x 107ID13cellsorC127 cellswashybridizedto uniformly labeledantisense SP6 probes containingsequencesdepictedinFig. 3asprobes II,III,orIV andwasdigested withRNase Aand
RNase T1. Hybridswerefractionated on5%denaturing polyacrylamidegels. (A) RNAsedigestion of hybrids made between probeIISP6 RNA and ID13 RNA (lanes A and B)orC127 RNA (lane C)wascarried out at10°C(lanes B and C)or25°C(lane A). End-labeled DNA
markers (lane M)werepreparedasdescribed in the legendtoFig.3. (B)Protectedfragments derived fromRNasedigestionofhybridsformed between probe IIIorprobe IV SP6 RNA and ID13orC127RNA,asshown(arrows).CordycepinAsequencingladder usingastemplate the
BPVSmaI-PstI fragment cloned into the SmaI andPstIsites in the polylinker of pSP65 (probeIV)is shown (laneAtrack). (C) RNAfrom 2x 107ID13orC127 cells whichweretreated with 30 ,ug of cycloheximidepermlfor4hpriortoharvest(lanes +CH and C127, respectively) orfrom 2 x 108ID13 cells whichwerenotcycloheximide (lane -CH) treatedwere hybridizedtoprobeIISP6RNAandweredigested.
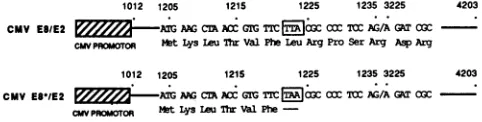
trol,toassessboth the transeffects of the repressorvector dueto itspotential protein production, as opposed to pro-moter competition effects, and to test the notion that the specific protein is an E8/E2 fusion, the clone CMV E8*/E2 wasconstructed (Fig. 6). By oligonucleotide-directed
muta-genesis, the leucine codon (TTA) at position 1223 was
changed to a termination codon (TAA). This single point mutation eliminates only the coding potential of all E8
proteinsin the vectorand leaves intactpossibleexpression
from other frames. (The transversion introduces a silent mutation in the El ORF).
We firstasked ifthe CMV E8/E2vectorcouldrepressthe
transactivation of the E2 product upon the inducible
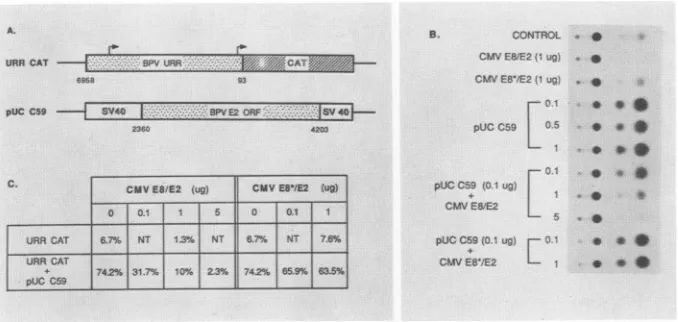
en-hancer in the BPV-1 control region (23, 24, 49). Figure 7A showsthereporterplasmid,URRCAT, which providesthe
cis-binding sites for the E2products, andtheplasmid pUC C59,whichprovidesthetransactivator forthereporter.pUC
C59 isaderivativeof the C59 E2vectorkindly provided by
P. M. Howley (51). The URR CAT construct can produce
the protein CAT, and its activity can be quantitatively assayedasdescribed elsewhere (14). Alltransfections were
carriedoutbyusingHeLa cellsasrecipients,and the results of the CAT assays are shown as raw data in Fig. 7B and
1012 1205 1215 1225 1235 3225 4203
CMV E8/E2 GAAGCI AC GIG TrC7 rC(TOI AG/AGATCGC
CPAOFOt MetLysLeuTtwVal PheLeuArgPro Ser Arg AspArg
1012 1205 1215 1225 1235 3225 4203
CMVES1/E2 E AIA AAG CM NMGIG CC COCTOCAG/AGATa3c
CwPROMTOR MtLysIouThrValPhe
FIG. 6. Structure ofplasmids CMV E8/E2 and CMV E8*/E2.
The CMV promoter-enhancer ( ) and sequences representing
the cDNA sequences cloned into the vectorwith HindIll linkers ( )areshown. CriticalBPVnucleotide and amino acidsequences
are given, and the codon which has been altered by site-directed mutagenesis (r)isshown.
quantitativelyinFig.7C. The table shows the percentageof
acetylated chloramphenicol foreach particular experiment. For each datum point shown, 2 ,ug of CAT reporter DNA
was either transfected alone or with combinations of the
various trans effector constructs. Interestingly, we found
that the CMV E8/E2 vector represses the basal activity of
the reporter as shown at the top ofFig. 7B. This effect is
measuredtobeatleastfivefold(Fig. 7C,6.7%conversionin contrast to 1.3% conversion) and clearly is not due to
artifactual plasmid competition since the pointmutant
con-structCMVE8*/E2 has no effecton the reporter.Thenext
three experiments (Fig. 7B)repeatthe classic transactivation
experimentswhich show thatevenatverylowdoses the E2 vectorcanevokealarge stimulationof theability of theBPV
URR to promote gene expression (16, 17, 45, 48). In our
hands in this particular system, the stimulation is about 10-fold. The results shown illustrate that the E2 stimulation saturates at levels below or at 0.1 ,ug of transactivator vector. Wenext mixedtogetherreporterCAT vector,
satu-rating levels of transactivator E2 (0.1 p.g), and increasing
levels of thetestplasmidCMV E8/E2. Asshown,the CMV E8/E2 construct counteractsthetrans-positiveeffects ofE2 and lowers the basal activity of the reporter. Again, the CMVE8*/E2vectorhasnoeffectinthistype oftriple mixing experiment (e.g., BPV URR CAT, pUC C59, and CMV
E8*/E2), illustratingthat thenegativeeffectsof therepressor vector must be duetothe transaction of its product.
Overexpressionofaproteinwhichcancounterthe effects of the E2proteinwouldbeexpectedtointerferewithBPV-1
focus-forming capacity (23). We tested this hypothesis by mixingtogether increasingconcentrationsof either the CMV E8/E2 or CMV E8*/E2 vector with a constant amount of
wild-type BPV-1 DNA. In our assays, 1 ,ug of BPV DNA induced about 175 foci ina60-mm culture dish. The repres-sorvectordid indeedinhibit BPV-1 foci inadose-dependent
C
Proe
,.r
393
398-
I
344
-298
-* P3a
22C
VOL.63, 1989
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.53.294.609.670.2]URRCAT - B URR..i:K.. XCAT' :
6908 93
pUCC59 S640- - Y40
2360 4203
CMVES/E2 (us) CIAVE5/E2 (ug)
0 0.1 0 0.1 1
URR CAT 6.7'S6 NT 1N3%NT 67% NT 76%
URRCAT
+c 774.2% 31.7% 10% 2.3% 742% 66.6% 63.6%
PUGCSO
B. CONTROL *
CMV EW1E2(Iug ,
CMV EB'IE2 t
ug)-pUCC59 . 0
0O.0 * ., PUCCC9(0.1 ug)
+ 1
L5
pUL C59 0.1 ug r 0.1 9 CMVES'/E2 - * * *
FIG. 7. Inhibition ofE2 transactivity bytheE8/E2repressor.(A) Structure ofreporterplasmidand E2 trans-activatorexpressionplasmid.
ThereporterplasmidURR CATcontainsafragmentfromnucleotides6958to93, whichincludes theentireupstreamregulatoryregionaswell astheP2 promoter atnucleotide 89, linkedtotheCATgene(14).ThepositionsoftheP1and P2 promotersareindicatedbyarrows.pUC C59 isaderivative ofC59 (51;seeMaterials andMethods). (B)Autoradiogramof CATassayspreparedfromextractsofHeLa cells which weretransfected with2 ,ugofURRCATalone(control) ortogether withtheindicatedamountsofexpression plasmid. (C) Quantitationof resultsof CATassayshown inpanelB.Values areexpressedas apercentconversion of[14C]chloramphenicoltotheacetylatedforms.NT,
Nottested.
way, while the mutant vectorhad no measurable effectsin
trans (Fig. 8).
DISCUSSION
BPV-1has a relatively small genome, yetalargeportion of
the sequence hasnotbeenfoundexpressedasmRNA. Most
conspicuously missing fromthe maps of RNAsproducedin
either productively infected fibropapillomas or latently
in-fected cultured cells are mRNAs from parts of the highly conservedEl ORF and fromthe URR. Togetherthese two
regions constitute morethan one-third of the 8-kb genome. Astrans-actinggeneproducts have been definedgenetically fortheEl ORF, it isgenerally assumed that thereason for this lack of detection is the low abundance ofthese tran-scripts. Using specific probesfrom both the El ORF and the
URR,wehave been abletoisolate and characterizeavariety
ofnew cDNAs and define a new transcription unit in the virus.
The mainpoints of this article concern the functional and structural analysis of the transcripts from the P3 promoter. The major transcript from this promoter encodes a repres-sor,andfromthe resultspresented aboveweestimate that it
is as abundant as the individual transcripts from the
up-stream promoter P2. This estimate is based on both the
frequency ofclones such as N7-3 relative to E6 clones in the
libraryand onthedirectRNAmappingdata shown inFig. 5. Before we discuss specific results relevant to the rarer cDNAs, two caveats should be mentioned. First, we used
cyclohexamideprior to harvesting cellular cytoplasmic RNA
toincrease our chances of obtaining mRNAs which might be
degraded rapidly orexpressed rarely. The block to protein synthesis which slows down mRNA degradation increases BPV-1 transcript abundance but potentially augments very weak promoters (e.g., theP1promoter) by lowering repres-sor levels. Thus, we may have obtained cDNAs in some
caseswhich areextremely rare in stably transformed cells. We do not, however, knowofa situation wherein the drug
creates newpromoters or causesaberrant splicing. A second limitation concerns the promoter assignments for the rare cDNAs. As the cDNAs were in general truncated by 50 to
300 base pairs, itwasrelatively straightforwardforcDNAs
suchasN7-3tobeassignedtopromoterP3, forweobtained
manycDNAs ofthis type, all ofwhich had 5' endscloseto
themapped P3 end. Furthermore, wecould mapan exonof
expected size with RNase protection experiments by using RNAs harvested fromnon-drug-treated cells. However,for
cDNAs obtained only once (e.g., 26-1 and 27-3), the pro-moterassignment is clearlyspeculativeandis simplybased
on our currentknowledge ofBPV-1 promoters. Thus in all
cases weascribedthenearest5' promoterto agivencDNA. TheP1 promoterisweakly expressed fromstableplasmids
in ID13 cells(2, 46), and severalofthe cDNAscharacterized
here are likely to be expressed from this promoter (Fig. 1;
see Fig. 9). Inparticular, P19-1 can be conceptually
trans-lated to yield an E7 protein. This 102-amino-acid peptide
wouldutilize an ATG codonjust two codons upstream of the nucleotide 7385 donor sequence. Given the central role that E7 plays in human papillomaviruses (38, 42; Banks and
Crawford, personal communication), itispossiblethat such
aprotein mightbe involved insome stageof the BPV-1 life
cycle. Recentlywehavefound that certain BPV-1 mutations in the P1 promoter reducetransformation frequencies of the DNAs (G. L. Breamand M. Botchan,unpublished results)
and that thesemutantscanbecomplemented bythe
expres-sion oftheP19-1 cDNA. Whether this effect is dueto only
thedirect effect of E7 or tothe effectof the genesoncopy numberstability isnot known. ThecDNA 27-3, whichuses
the same E7 acceptor site as P19-1, cannot encode such a protein since in-frameterminators exist 5' totheE7 ORF in this cDNA and no in-frame ATGs are present in E7. The cDNA clone 27-3, however, has the capacity for a small 100-amino-acid 5' Elprotein, since this cDNA contains the conserved El ATG at position 838. This putative polypep-tide isnotlikelytobe theMprotein,asmutations within the intron (864 to 1024) affect the M complementation group.
NeitherN19-1 norP19-1 can code forthis putativeElgene since the splice acceptorat nucleotide
1032
(as opposed tothe acceptoratnucleotide 1024 in27-3) leadstocodon usage
outof the El ORF.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.145.484.73.234.2]NEW BPV cDNAs 1751
A
a
c
0
U-ugof DNA
Ourcharacterization of severalnewdonors andacceptors within the El region raises the possibility that othergenes
might overlap El. In particular, recentgenetic studies
sug-gest that the E8 ORF encodes a trans-acting gene product (M. Lusky and J. Choe et al., manuscript in preparation).
Point mutations in the E8 ORF either 5' or3' to the splice
donor at1235 lead to BPV DNAs which integrate infocus assays. Ifone assumesthatbothJ6-1andJ9-2 aretruncated
cDNAs whose RNAs initiated from P3, either of these
products could code for an E8 protein. Indeed, these E8
mutationscanbecomplemented byavectorwhich carriesa surrogate promoter linked to clone J6-1 (Fig. 1) if the E8 ATG startregion is reconstructed.
One of themajor objectives ofourcDNA screen was to findclones which couldrepresentputativemRNAsfor theM
gene product. We had anticipated that eithera small exon
would be spliced in frame into the El ORF or that a promoterwould be foundjust5'totheORF.This is because the limited genetic studies on the M gene define coding sequences uptothe 5' end of the ORF(29). Chowetal. (8)
have characterized an abundant 5' end at an analogous
position in human papillomavirus types 6 and 11 RNAs extracted from infected tissues. One ofthe RNA structures defined by Chow et al. (8) which has this 5' end position (their species b)hasan exon overthe El ORFregionwhich
overlaps withtheM-codingsequencesin BPV-1and
resem-FIG. 8. The toppanel showstheresults of a focus assay in which 60-mm platescontaining C127 cells were transfected with 1 ,ugof pMLBPV100 alone (plate A) ortogether with 1 or 5 ,ug of CMV E8/E2(plates B andC, respectively). Plates were fixed and stained with methylene blue 2 weeks after transfection. The bottom panel shows the results of an experiment in which C127 cells were transfected with 1 ,ugofpMLBPV100 alone or cotransfected with 0.1, 0.5, 1.0, or5.0 ,ugof either CMV E8/E2orCMV E8*/E2. For each of the transfections, the efficiency of focus formation is expressedas apercentage of the valueobtainedwith pMLBPV100 in theabsence ofexpression plasmid.
blesthe BPV-1 species (2b) characterized by Stenlundetal. (47). Whileourdatacannotexclude the existence of such an analogous promoteratthe end of the El ORF, ourdata do show that suchamessagemustbeatleast 10-to20-fold less abundant than other RNA species which can be detected from thisregionandcancodeforM.(See, forexample, Fig.
SA; suchputativestructureswith endsjustatthe endof the El ORF would produce bandsjust above the P3a and P3b exon bands.) The abundant cDNA family represented by clone N7-1cancodefor the M gene ifoneallows forasingle
RNAspecies to have the coding potential fortwo separate
proteins. Indeed,there isample precedentfor suchpotential polycistronic RNA in the papillomavirus literature. For example, the RNA likely to encode the E2 protein in the
cottontailrabbitpapillomavirussystemhas the E6ORF 5'to
thetransactivator sequences (50; M. S. Barbosa and F. 0.
Wettstein, personal communication). Recently, Thorner et
al. (49) have identified the BPV-1 M protein, using
serolog-ical techniques. Various animal cell expression vectors
which have the N7-1 cDNA insertedcanindeedtransiently produce a nuclear phosphoprotein which
comigrates
withprotein detected in virus-infected cells (L. Thorner and M. Botchan, unpublished results). Thus, we conclude that at
leastonewaytomakeMwouldbetoinitiate from P2 andto
generate an RNA with the structure of N7-1. Other less
abundantmRNAssuchasthoserepresented bycDNAclone VOL. 63, 1989
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.62.543.71.465.2]12-4
could,
ofcourse, alsoprovide
alternativepathways
forM
production.
The
genetic analysis
of the R gene is at thispoint
too uncertain to make clearpredictions
about arole inreplica-tion for a P3 mRNA which
might
be colinear with the ElORF,
such as one thatmight
berepresented
by
clone J9-3. On theonehand,
wepoint
outthat allpublished
mutations in the El ORF between the donor siteat nucleotide 1235 and theBglII
site at nucleotide 1515 affectnotonly
El but also E8.Thus,
thepossibility
existsthataspecies
such as J9-3 couldencode foran Rfactor,
ifonespeculates
that it is E8 ORF functions thatareaffectedby
thesemutants.However,ontheother
hand,
recentfindings
suggest that stopcodonsin thisregion
ofEl thatdonotaffect E8 codons leadtomutantsthat are carried
only
asintegrated
genomes in transformed cells(M.
Lentzand M.Botchan,
unpublished observations).
This would suggest that the R gene must utilize a startmethionine in the El ORF 5' to any carried
by
clone J9-3.These results donot
preclude
thepossibility
thattwoformsof the R message
exist,
onerequired
for stableplasmid
maintenance and
represented
by
cloneJ9-3,
and anotherexpressed transiently
uponinitial infection.Thus,
while thegenomic
organization
ofthe M gene seems to be clarifiedsubstantially by
the presentdata,
the hunt for thestructuresofthe R
message(s)
continues.DirectRNA
mapping experiments
and invitrotranscrip-tion data
presented
above establish the existence ofthe P3 promoter. Themajor
mRNAproduct
ofthis promoterex-pressed
in culturedmousecells islikely
toberepresented by
cDNA clone N7-3. About one-half of the cDNA clonesisolated from our
primary
screen ofthelibrary
with 5' Elprobes yielded
structuresessentially
identicalto N7-3. Theabundance of this
particular
5' exon in total BPV RNA issubstantiated
by
the SP6protection experiment
shown inFig.
5. As indicated in a summarydiagram
in Fig. 9, theproduct
of mRNAsrepresented by
N7-3 isan E8/E2fusionprotein
and has been shown here to be a repressor of thetransactivatorfunctionsof the E2
proteins.
RecentlyChinetal.
(7)
haveprovided
conclusiveevidence forasimilargeneencoding
repressoractivity
in humanpapillomavirus
type11.
The basic assay that we relied on was to show that the
E8/E2gene could counteract the
positive
effects ofthe E2 geneproduct
inexpression experiments.
Lambertetal. (23) have discussed various models which one can envision to accountfor suchanantagonism
offunctions, given
that bothforms ofthe repressor share
along
with thetransactivatora commoncarboxy-terminal DNA-binding
domain. Thus, ifone
postulates
that the amino terminus of E2 contains an essential domain fortranscriptional activation,
the expres-sion ofthecarboxy-terminal
domain alonemight
directlycreate a
competitive
situation forbinding
to DNA sites ormight
createpoisoned
protein aggregates with the positive effector which would inhibit itsactivity.
Interestingly, weshow here that the E8/E2 gene product can act directly to
negatively
regulate
thebasal geneexpressionfrom the URRCAT construct.
Thus,
the repressor proteins are likely to interferedirectly
with the transcription apparatus, perhapsby
making
abortive interactions with other site-specificDNA-binding
proteins
or by simply blocking contactsbe-tween the
polymerizing
apparatus and the DNA (see, forexample,
Keleheretal.[21]).
This laterfindingregarding the effects of the E8/E2geneonbasal levelexpressiondoes notpreclude
the models suggested by Lambertet al. (24) vis avis the
antagonism
between E2 and the repressor butsug-gests, as onewould
certainly predict,
that interactions withE-8
IE27
El
I
P1 P2 P3
ri re rw
cotD oa
_ tD to
ACTIVATORS
REPRESSORS [
P4 PS
rol rill
E2 E2
E2 TR E8/E2
FIG. 9. Summary of the transcription units expressing the E2 transactivatorandrepressor genesin BPV-1. Thetopof the figure showstheearlyregion of BPV-1 and demarks the known promoter regions; the relevant viral ORFs discussed in the text are also shown.Theactivatorproteincanbe expressedfrom either P4orP3 and the "generic" cDNAs which have been shown to have the
abilitytoproduce such activities areC59 (51) and N15-2 (Vaillan-court and Botchan, unpublished observations), respectively. The repressor genes canbeexpressed from P3orP5.The cDNAclones whichhave been showntoproduce such activitiesarecalledN7-3 (this study) and p1153 (23). Notice that the proteins encoding sequences(indicated bytheopenboxes above thetranscript) predict differentstructuresfor therepressorforms and identicalstructures for the activator.
cellular proteins must be critical. More importantly, this
observation suggests that the family of repressors have transcriptional roles other than to simply antagonize the
action oflarge E2proteins.
A general strategy of autoregulation used by viruses in both procaryotes andeucaryotes involves expression early
in theinfection ofproteinswhich can stimulatetranscription, followed by expression of proteins which modulate down this activation. This strategy is even used in acutely lytic
systemssuch as arefound inadenoviruses and simianvirus
40polyomaviruses, and it makesperfect sense that viruses with long latencies, such as BPV-1, should have such systems. If one assumes that a single bovine wart with a massof 1.5 kgis the result of oneviralinfection, this leads
tothe calculation thatthe virusmustmaintain its latency in
a transformed cell through at least 43 cell doublings. A complicated circuitry of factors and cis-regulatory sites
mightbepresupposedtoexist to ensurecontinued growth of
such a massivestructure. Thus, theproblem ofmaintaining homeostasis with regard to cell division is a much more
important probleminvivo than isusually addressed in focus
formation and expansion to a cell line in culture in which
routinely only 20to 25 celldivisions occur before analysis. We can formally suggest two discrete but not exclusive
reasonsforwhythe twodifferentforms of the repressor gene
arearranged the waythey are in BPV-1. On the one hand, theproteins predicted by their gene structures are different and thus they may have different functions. Both proteins have a common 204-amino-acid carboxy terminus and di-vergeat anamino-terminal domain defined by the acceptor siteatBPV-1 nucleotide 3225 (Fig. 9). The amino terminus of theE8/E2proteinhas11amino acids from an upstream exon while E2TR has an additional 35 amino acids which are encodedbyacontinuation of the E2 ORF5'to the acceptor. Anotherdifference between the proteins may involve their
..
1% ".,.,.-=..
on November 10, 2019 by guest
http://jvi.asm.org/
[image:10.612.331.547.70.233.2]NEW BPV cDNAs 1753
putative interactions with cellular factors mediated by their amino-terminal domains. We might point out that overex-pression of either of the gene products for repressor function through surrogate promoters may obscure differences in function between the two proteins. For example, one protein may have higher affinities for a particular binding site than does another, and overexpression might cancel such differ-ences. These points focus on structural differences in the proteins. Alternatively, as the repressor genes are linked to separate promoters, they are perhaps independently regu-lated. Thus, one or the other gene may function as the important negative regulator in a particular cell type (or time in the cell cycle). Thus, much more work must be focused on how these promoters for the repressors are expressed and where these proteins are expressed in situ, and more specific assays (e.g., with different promoters) may be called for to distinguish the putative differences between the genes. In-terestingly, the recent work of Hubbert et al. (19) shows via serological methods that both forms of the repressor protein coexist in unsynchronized cultures of virus-transformed cells. These workers have shown that the E2TR (or long form of the repressor protein) is more abundant than is the
E8/E2form in the C127 cell system. Finally, it is possible that the putative different functions of the genes alluded to above cannot be addressed in short-term cultures of heter-ologous mouse cells. In this system, either repressor may suffice to maintain appropriate levels of gene expression.
We were surprised to find cDNA clone N15-2 because it encodes for a function which is antagonistic to the function of theE8/E2 repressor gene regulated by the action of the P3 promoter. As indicated in Fig. 9, the potential promoter indicated for clone N15-2 is the P3 promoter; this clone can code for the entire E2 ORF since the splice acceptor at nucleotide 2558 is just upstream of the likely ATG initiator codon in E2. Moreover, recent unpublished work withN15-2 shows that it can functionally make an E2 transactivator when linked to surrogate promoters (P. Vaillancourt, J. Choe, and M. Botchan, manuscript in preparation). How-ever, by far the bulk of the RNAs initiated from P3 must splice from the common donor at nucleotide 1235 to the acceptor at 3225 rather than to the acceptor at nucleotide 2558. Thus, splicing as well as promoter utilization may contribute to the balances between positive and negative factors. Similarly, the vast majority ofstarts from P4 splice to yield a putative E5 message (1, 18, 47, 51), and it is questionable whether the bulk of the E2 protein is indeed expressed from this promoter. Stenlund et al. (47) charac-terized a BPV RNA (their species 3) which links theinternal E6 donor to an acceptor just upstream of the E2 ORF, and we may speculate, on the basis of the relative abundance of this transcript, that the P2 promoter may be the strongest contributor to the production of the transactivator E2. Again, however, we would like to emphasize the extremely low abundance of RNAs such as N15-2 and point out that genetic studies assessing the role of theacceptor at nucleo-tide 2558 will be needed to begin to test critically the complexities implied by Fig. 9. In summary, then, the structurally differenttranscriptional repressor proteins seem to be expressed from two different promoters while the putatively unique activator may be expressed from three different promoters. While on the surface it may seem bewilderingly complex to have so many different ways to express similar activities, given the complexity of the life cycle of the virus mentioned above and the range of cell types it can infect, this isperhaps expected. Recentfindings on the expression patterns of a single cellular locus which
codes for a family of transcription factors shows a similar level ofcomplexity (41).
Ifthe plethora of promoters and transfactors discussed above does not as yet describe atranscriptional regulatory
system for the BPV-1 genome, some of the complexities
concerning BPV-1 E2 genetics may be clarified by the discovery of the E8/E2 gene. Lusky and Botchan (28)
showed that an E2 mutant,d1211,wasseverelycrippledina
transient replication assay. Moreover, in side-by-side
as-says, the mutantBAL15replicatedtransiently as
efficiently
as didwild-type BPV-1 eventhough it is deletedforallofthe E2ORF (Lusky andBotchan[29]).
Itseemslikelyto usthat d1211, although destroyingthepositivetransactivation func-tionof the large E2 transactivator anddeletingthe promoter for the negative repressorE2TR, leftintact theE8/E2 gene. Thus, expression of only the negative regulator in the absence of a positive activator may shut thereplication
genes off too severely. This hypothesis would thereforeinclude the speculation that in any short-term assay, the completeabsenceof bothnegative andpositivegenes
might
be roughly equivalent to that ofthe wild type if the initial expression of the gene(s) being assayed is E2independent
and onlybecomes E2 regulated at alater stageofinfection. Similar ideas may be relevanttothegeneticdatafromother groups. For example, DiMaio and Settleman (10) have defined a ts mutant in the E2 ORF wherein the lesion wasengineered 5' to thenucleotide 3225 acceptor in the E2 ORF. This temperature-sensitive mutation
potentially
inactivates both the E2 transactivator and the E2TR repressor, yet it leavesintact the E8/E2 gene. This situation may accentuatesome ofthe phenotypestheymeasured;
specifically,
itwould increase the apparent requirementfor thepositive
E2 prod-uct itself. Finally, Hermonat andHowley
(18)
showed that certainE2 mutantssustained in the ORF 3'totheacceptoratnucleotide 3225 could not be complemented
by
the C59 E2expression vector (Fig. 9). C59 cannot
provide
an E8/E2product and thus might notbe
expected
tocomplement
any E2 mutants which affect this member of the E2family.
Furthermore, it is conceivable that the C59 cDNA may not
provide E2 itself in precisely the
right
way.Perhaps
E2 provided by P2 or P3 is needed to achieve most-efficientvirustransformation and plasmid establishment.
ACKNOWLEDGMENTS
Thisresearch wassupported byPublic HealthService grants CA 42414andCA30490fromtheNationalInstitutesof Health. J.C. was supported by apostdoctoral fellowshipfrom the Cancer Research Coordinating Committee of the University ofCalifornia.
We thank Virginia Staubach-Summa for typing the manuscript,
P. M.Howley forprovidingthe E2expressionclone C59,and E. S. Mocarskifor thepON260vector.
LITERATURE CITED
1. Ahola, H., A. Stenlund, J. Moreno-L6pez, and U. Pettersson. 1987. Promoters and processing sites within the transforming regionof bovinepapillomavirus type 1. J. Virol. 61:2240-2244.
2. Baker, C. C., and P. M. Howley. 1987. Differential promoter utilizationby the bovinepapillomavirusintransformed cells and in productively infected warttissues. EMBO J. 6:1027-1035. 3. Benton, W.D., and R. W. Davis. 1977. Screening
Xgt
recombi-nant clones by hybridization to single plaques in situ. Science 196:180-182.
4. Berg, L., M. Lusky, A. Stenlund, and M. R. Botchan. 1986. Repressionof BPVreplicationismediated byavirally-encoded
trans-actingfactor. Cell 46:753-762.
5. Brawerman, G., J.Mendecki, andS. Y. Lee. 1972. A
procedure
for the isolation of mammalian messenger ribonucleic acid. VOL.63,1989