CopyrightC 1988,American Societyfor Microbiology
Herpes
Simplex Virus Virion Host Shutoff Function
ANND. KWONG, JOHN A. KRUPER,AND NIZA FRENKELt*
Department of Molecular Genetics and Cell Biology, the University of Chicago, Chicago, Illinois 60637 Received 14August1987/Accepted 2 December 1987
Herpessimplex virus (HSV) virions containone or morefunctions which mediate theshutoffof host protein
synthesisand thedegradation of host mRNA. HSVtype1(HSV-1)mutantsdeficient in the virion shutoff of host protein synthesis (vhs mutants) wereisolated and werefoundto be defective intheir abilitytodegrade host mRNA.Furthermore, itwasfoundthatviral mRNAsincells infectedwiththe vhslmutanthaveasignificantly
longer functional half-life than viral mRNAs in wild-type virus-infected cells. In the presentstudy we have
mapped the vhsl mutation affecting the virion shutoff of host protein synthesistoa265-base-pair NruI-XmaIII
fragment spanning map coordinates 0.604 to 0.606 of the HSV-1 genome. The mutation(s) affecting the
functionalhalf-livesofhost mRNAaswellasthea(immediate-early),
0i
(early),andy(late)viral mRNAswerealso mapped within this 265-base-pair fragment. Thus, the shutoffof host protein synthesis is most likely mediatedbythesamefunctionwhichdecreasesthe half-lifeofviral mRNA.The shorterhalf-life of infected-cell mRNAsmayallowamorerapid modulationofviralgeneexpressioninresponsetochangesinthetranscription of viral genes. Interestingly, the vhsl mutation of HSV-1 maps withina region which overlaps theBglII-N
sequencesof HSV-2 DNAshown previouslytotransformcells in culture. Thepossible relationshipbetween the transformation and hostshutoff functionsare discussed.
Infection of cells with herpes simplex virustypes 1 and 2 (HSV-1 and HSV-2) results in the shutoffof host protein synthesis and the sequential expression of several coordi-nately regulatedgroupsof viralgenes(20). Theinhibition of
host macromolecule synthesis in HSV-infected cells is a
multiphase process (reviewed in reference 7). A primary phase of the shutoffof hostprotein synthesisis mediatedby
a virion component. It occurs in cells infected in the pres-enceofactinomycinD(topreventviralgeneexpression)and in cells infected with UV light-irradiated virus (8, 10, 24, 39-41, 44, 56-58). A late (secondary) shutoff function
re-duces the remaining levels of host protein synthesis and requires the expression ofviralgenes(9, 40,44).
HSV-1and HSV-2 reduce the abundance of host mRNAs inavariety of celltypes (3, 10,21, 24, 34, 36, 38, 40-42, 48, 54-56). However, the mechanism bywhich this occurs has yettobeelucidated. InVero cells, both the shutoffof host protein synthesis and the degradation of host mRNA are
observed in theabsence of viralgeneexpression (3, 10, 21, 24, 41, 44, 48, 56).HostmRNAdegradationmaybe respon-sible for the dissociation of host polyribosomes (56). In contrast,in HSV-infected Frienderythroleukemia cells, the synthesisofglobinis arrested byavirionfunction, whereas hostmRNAdegradationappearstorequiretheexpressionof viralgenes(38-40).
Asetof virion host shutoff(vhs)mutantswerepreviously
isolated in our laboratory (44). Unlike the wild-type (wt)
virus, all of these mutants were deficientin theirability to degrade preexisting
P-actin
and a-tubulin mRNA in the absenceof viralgeneexpression (24, 56). However, the vhs mutantsarenotaltered withrespect tothesecondaryshutoff function. When viralgeneexpressionisallowed,the synthe-sis of host proteins is turned off, albeit in a delayed and incompletemanner (24, 44).Viral protein synthesis begins concomitantly with the shutoff of hostproteinsynthesis.Theprogramofexpression
* Correspondingauthor.
tPresent address: Laboratoryof Viral Diseases, National Insti-tutesofHealth, TwinbrookFacility, Rockville, MD20852.
involves the sequential turning on of the transcription of several groups of viral genes, including the a
(immediate-early), v (early),and -Yl and _Y2 (late) genes(20, 61). As the
later viralgenes are turnedon, thesynthesis of earlier viral
proteins ceases, implying the existence ofa mechanismto turnoff thetranslationof thepreviously synthesizedmRNAs (20, 24). The mechanismof this shutoff is unknown.
We have recently proposed that HSV encodesafunction
which indiscriminately shortens the half-life of host aswell asvirala., 3,and-ymRNAs(24). We havealsoproposedthat
theshutoff ofhost protein synthesisis aconsequenceofthis function. This hypothesis is based on the findingthat both host and viral mRNAs are significantly more stable incells infected with the vhsl mutant virus than in cells infected with the wt virus. However, because this mutant was
derivedby generalbromodeoxyuridine mutagenesis,itcould contain mutationsinseparategeneswhichmightbe respon-sible for these different phenotypes. In fact, as already
noted,in infected Frienderythroleukemiacells the shutoff of host protein synthesis was mediated by a virion function whereas thedegradationof host mRNArequiredthe expres-sion of viral genes(40).
In thisreportwehavemappedthe vhslmutationaffecting the shutoff of hostprotein synthesis and thedegradationof hostmRNA withina265-base-pair(bp) regionspanningmap coordinates 0.604 to 0.606of the HSVgenome. The muta-tion(s)affectingthe functionaldestabilization of thea,
P,)
and y mRNAswas alsomappedwithin thesame 265-bp region.Thus,allof thesephenotypes aremostlikelymediatedbya
single viralgene.
MATERIALSAND METHODS
Cells and virus. Rabbit skin cells and mouse Ltk- cells
were obtained from B. Roizman (University of Chicago,
Chicago, Ill.). Vero monkey cells were obtained from S.
Bachenheimer (University of North Carolina, Chapel Hill), and humanepidermoid-2(HEp-2)cellswerefrom the
Amer-icanTypeCulture Collection. The vhslmutantwasderived by bromodeoxyuridine mutagenesis of HSV-1 (KOS) as
previously described (44). The KOS strain was originally
912
on November 10, 2019 by guest
http://jvi.asm.org/
HSV VIRION HOST SHUTOFF FUNCTION 913
obtained from P. A. Schaffer (Harvard Medical School,
Boston,
Mass.).Preparation of viral andplasmidDNAs. Viral DNAs were
prepared from infectedcells by CsCl density centrifugation
as described
previously
(29). The EcoRI clones of HSV-1strain KOS (16) were a
gift
from M. Levine (University ofMichigan,
AnnArbor).
Transfections weredoneusing CsCldensity-purified plasmid
DNA preparedby
the method ofClewell and Helinski (5). Restriction enzymes and other
enzymes used inthe
cloning
workwere obtained fromNewEngland
BioLabs,
Inc.(Beverly,
Mass.), or Bethesda Re-searchLaboratories(Gaithersburg, Md.).
The enzymeswere used asrecommendedby
thesuppliers.
Marker transfertests. Mixtures of vhslmutantviralDNA andcloned KOS testDNA
fragments
wereusedtotransfect rabbit skin cells in 25-cm2 dishes bythe calcium phosphateprecipitation
method(17,
47).Approximately
5 ,ug of intact viral DNA, 5 ,ug of salmon sperm carrier DNA (Sigma ChemicalCo.,
St.Louis,
Mo.), and 0.1 to 2.0 ,g of testfragment
DNA(excised
from theplasmid
vector) weregently
mixed in 0.5 ml of HeBs buffer(21
mM HEPES[N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid],
0.702 mMNa2HPO4,
0.137 M NaCl, 5 mM KCl, 5.6 mMD-glucose, pH 7.05)
andprecipitated
for 20 min at room temperatureby
the addition of30 p,l of2 MCaCl2.
Mean-while,
the cells were washed twice with Versene (137 mMNaCl,
2.68 mMKCl,
8 mMNa2HPO4,
1.47 mM KH2PO4,0.537 mM
EDTA)
and thentwice with HeBs solution. Theprecipitated
DNAwasaddedtothe cells,and after 30 minat roomtemperature, 4 mlof Dulbecco modifiedEagle
mediumsupplemented
with2% heat-inactivated fetal calfserum wasaddedand thetransfected cellswereincubatedat
37°C.
After4 to 5
h,
the cells were rinsed three times with Dulbeccomodified
Eagle
mediumsupplemented
with6%heat-inacti-vatedfetal calfserumandwerethen incubatedat
37°C
until the viral infection hadspread through
theentire
culture(4
to 5days).
Virus from the transfection(passage
0)
was har-vestedby
threecycles
offreeze-thawing
and wasserially
propagated (at
1:3dilution)
in 25-cm2 cultures of HEp-2 cells.Working
stocks wereprepared
in 150-cm2 cultures ofHEp-2
cellswhichwereinoculated with4/5theyield
ofP2or P3 virus.Trichloroacetic acid assay for the host shutoff function. Vero cells
(24-well cultures)
werepreincubated
for 30 minwith medium199
plus
actinomycin
D. This mediumconsistsof medium199
(KC Biologicals, Lenexa, Kansas) containing
1% heat-inactivated calfserum and 5 ,ugof
actinomycin
D(Calbiochem-Behring,
LaJolla, Calif.)
per ml. The cellswere infected withthetestvirus in medium199plus
actino-mycin
D.After2h of virusadsorption
at37°C,
the cellswererinsed three times and were incubated in medium 199 plus
actinomycin
D.Eight
hours afterinfection,
the cells were washed threetimes with medium199lacking
methionineandcontaining
1%dialyzed
calfserumand 5pug
ofactinomycin
Dper ml. The cells were then labeled for 2 h in
labeling
medium
containing
5 ,ugofactinomycin
Dper ml.Labeling
medium consisted of medium 199 with 1/20 the normal
concentration of unlabeled methionine, 1% dialyzed calf serum, and 50
,uCi
of[35S]methinonine
(New England Nu-clearCorp., Boston, Mass.)
per ml. Theprotein samples
were
prepared
aspreviously
described(44).AnalysesofthefunctionalstabilityofviralmRNAs. For the
ot mRNA
stability
assays(20,
24), Ltk- or Vero cells in24-well cultureswere
preincubated
for 30 min in medium 199plus cycloheximide.
This medium consists of medium 199containing
1% inactivated calfserumand 50 ,ug ofcyclohex-imide (Sigma) per ml. The cells were then infected in medium 199plus cycloheximideasdescribed above. Thirty
minutes before thecycloheximide reversal, actinomycin D was added to the medium at a final concentration of 5 ,ug/ml. After a 30-min incubation, the cycloheximide block was reversed by three washes with mediumcontaining
actinomy-cin D, and the cells either were labeled with [35S]methionine
at thatpoint or were incubated in the presence of actinomy-cin D for 4 h further before the addition of the label. The proteins were labeled as described above.
For the analyses of the functional stability of a and -y mRNAs (24), Vero cells were infected in medium 199 containing 1% heat-inactivated calf serum. At the indicated time, the cells were washed three times with medium con-taining 5 ,ug of actinomycin D per ml. The cells were labeled with [35S]methionine either at that point or after further incubation in the presence of actinomycin D. The prepara-tion of the protein samples and gel electrophoresis were done as previously described (44).
Analyses of total infected-cell RNA. The procedures used in
the RNA preparation and RNA blot hybridization are
de-tailed elsewhere (24, 56).
RESULTS
Design of the marker transfer assay. The vhsl mutation is not lethal to virus growth, although the yield of infectious vhsl virus per cell is two- to fivefoldlowerthanthe yield of wt virus (44). We reasoned that the slight growth advantage of the wt virusmightbeemployed in the mapping of the vhs
lesion. Bythismethod,cells wouldbetransfected with vhsl mutant virus DNA and cloned DNA fragments of the wt virus. Putative recombinants are expected to constitute a small proportion of the initial virus population. However,
recombinants in which the vhs mutation has been repaired may have agrowth advantageand take overthepopulation
during sequential virus propagation. Wecouldthus testfor the transfer of the vhsphenotype by analyzing the
popula-tionenblocrather thanby laboriously screening hundreds of plaque isolates in theoriginaltransfection.
The study summarized in Fig. 1 was designed to test whether the wt virus exhibited a growth advantage during the propagation of mixed virus populations. Specifically,
replicatecultures of rabbit skin cells were infected with wt
virus alone, with the vhsl mutant virus alone, or with
different mixtures of the wtand mutant viruses. A total of
100PFU were inoculated per culture so as to approximate thenumber ofplaques produced during routineDNA
trans-fections. Plaqueswereallowedtoprogressfor severaldays, and the harvested virus stocks (passage 0) were serially propagated in HEp-2 cells. Passages 0through3 were then
assayed for the vhs phenotype to determine whefher this
protocolresulted inselection ofthe wt virus.
Inthe assay, Verocellswereinfected withtestvirusesin the presence of 5 ,ug ofactinomycin D per ml, preventing viralgene expression. Parallel cultures were
mock
infected in the presence of the drug. The cells were labeled with [35S]methioninefrom8to 10hpostinfectioninthepresence ofactinomycin D. The amount of trichloroaceticacid-pre-cipitable
countsin theinfected cellswasthencomparedwith that in the mock-infected cultures which were similarlytreated with actinomycin D. The findings (Fig. 1) can be summarizedasfollows.
(i)Asexpected,infections with the wt KOS virusyielded
virus stocks whichexhibited the virion-associated host shut-offphenotype. In cells infected with the KOS passage 0 VOL.62, 1988
on November 10, 2019 by guest
http://jvi.asm.org/
Passage
FIG. 1. Advantageousgrowth of thewtvirus.Duplicatecultures ofrabbitskincells (25-cm2 dishes)wereinfectedwithatotal of 100 PFUof HSV-1 (KOS)wtvirus, the vhslmutantvirus, andmixtures containing the specified ratios ofwt to vhsl mutant viruses. The infectedcellswere harvested,yielding thepassage 0 stockswhich
were further propagated in HEp-2 cells, as described in thetext. Twoindependent serieswerederivedfrom each virus mixture and wereassayed separatelyfor thevirion-associated host shutoff
phe-notypeasdescribed in thetext.Eachpointonthegraphsrepresents
an average of data obtained from the two separate series. The variations between the duplicate series were less than 5% of the valuesin themock-infected cells.
virus, host protein synthesis was reduced to 25% of the synthesis in the mock-infected cells. The incomplete (75%) shutoffreflected the fact thatpassage0viruswaslow intiter
(not shown). The level of shutoffincreased to greater than 95% with successive passaging duetoincreasing virus titers. (ii) The passaged vhsl virus stocks exhibited a stable
mutantphenotype. Infact, in cells infected with passages 1
through 3 of the duplicate vhsl series, the level of host protein synthesis washigher (130to140%) than theamount synthesized in mock-infected cells. We have repeatedly noted that host protein synthesisincreases in cells infected with highmultiplicities ofinfectionof the vhsl mutantvirus in thepresenceofactinomycin D. Tentatively, this increase maybeattributed totheability of the mutated vhslfunction to protect host mRNA from normal turnover (56; A. D. Kwong and N. Frenkel, manuscript in preparation).
(iii) The mixed (KOS plus vhsl) virus series exhibited increasing levels ofwt host shutoff with successive
passag-ing, revealing the selection of viruspossessingthewtshutoff function. In the series derived from coinfections with equal amounts ofvhsl and KOS viruses (1:1; Fig. 1), significant shutoff (54%) was attained upon infection of cells with passage 0 virus. Furthermore, the shutoff of the host was
almost complete (92%) in cells infected withpassage1 virus. No shutoffwas observedin cellsinfected with passage0of
theseries,derived by infection of the initial cultures with 100 PFU of vhsl and 1 PFU aloneof KOS virus (1:100; Fig. 1). However, by passage 2, the ability to shut off the host
approachedthat of thewtvirus. The advantageous growth of the wt KOS virus was used in the marker transfer
experi-mentsdescribed below.
Mapping of the vhs mutation(s) using EcoRI DNA frag-ments. The EcoRI enzyme cleaves the 150-kilobase (kb)
HSV-1 genome into several large fragments. The wt KOS EcoRI fragments were cloned by Goldin et al. (16) into pBR325. The vhsl mutation was first mapped within these
large fragments. Specifically, duplicate rabbit skin cell
cul-tures were cotransfected with vhsl mutant virus DNA and each ofseveral cloned EcoRI fragments. Control
transfec-tions received vhsl DNA alone or vhsl DNA along with
plasmid DNA (pBR322). Virus stocks derived from these transfections were passaged to enrich for putative
recombi-nant viruses in which the vhs mutation has been repaired. Thevarious transfections were done in duplicate (designated a and b in Fig. 2), and the resultant stocks were passaged separately. The passage 2stockswerethenassayedfor their
ability to shut offhostprotein synthesis in the presence of actinomycin D.Specifically, 24-well cultures of mouse Ltk-cells were infected withsamples of thepassage2 testviruses
in the presence or absence of actinomycin D. Additional
cultures were mock infected or were infected withstandard HSV-1 wt KOS and vhsl viruses (not derived from the transfection experiments). The proteins were labeled from 8 to 10 h postinfection in the presence or absence of actino-mycin D, respectively, and were analyzed by electrophore-sis in sodium dodecyl sulfate-polyacrylamide gels.
Lanes 1 through 14of Fig. 2 show the pattern of protein synthesisin the cultures which were infected in the presence ofactinomycin D. Comparison of the level of protein syn-thesis in the wtvirus-infected cells (lane 2) with that in the mock-infected culture(lane 1) shows thepronounced shutoff ofhost protein synthesis in thepresence of the drug. Lane 3 (Fig. 2) shows the corresponding lack of shutoff by the vhsl mutantvirus. Lanes 5 and6 represent theduplicate indepen-dent transfections (a and b) with the EcoRI-A clone and show thatthis clone wascapable oftransferring the wt host shutoffphenotype to the mutantvirus. Lanes 7 through 14 reveal that the other testedEcoRI clones werenot capable of this transfer.
Lanes 15through 28 of Fig. 2 contain protein samplesfrom
cultures infected in the absence of actinomycin D. They
show that high amounts of viral proteins were produced in cells infected with viruses derived from all of the EcoRI
cotransfections. This result ascertains that sufficiently high
multiplicities of infection were used in the actinomycin D tests shown in lanes 5 through 14.
These results show that the vhsl mutation resideswithin
the 22-kb KOS EcoRI A fragment, spanning map coordi-nates 0.493 to 0.633 of the HSV-1 genome. It is noteworthy
that Read andco-workershave alsomapped the vhsl muta-tion within theEcoRI Afragment(C. R. Krikorian and G. S. Read, unpublished results, cited in reference 41).
Location inEcoRI-A of the mutationaffecting the functional stability ofaand
,Iy
mRNAs. Wenext determined whether the EcoRI-A recombinant viruses resembled the wt or the vhsl mutant viruses with respect to thefunctionalstability ofviral mRNAs (24, 41, 44).
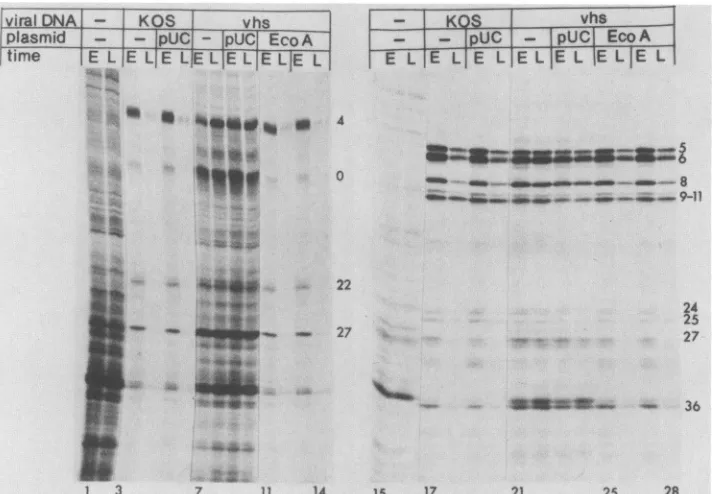
Lanes 1 through 14 of Fig. 3 show theexperimentdesigned
totest the functional stability of a mRNA.Specifically,
Ltk-mouse cells were infected with the second passages oftwo independently derived EcoRI-A recombinant stocks. Con-trol cultures were mock infected or were infected with passage 2 virus derived from transfections which received KOS wt virus DNA alone, KOS plus pUC19plasmid, vhsl DNA alone, or vhsl plus pUC19 plasmid. Infections were done in the presence of cycloheximide, allowing the tran-scription of a genes. At 8.5 h postinfection, actinomycin D was added to the medium. Thirty minutes later the cyclo-heximide was removed, and incubation was continued in the presence of actinomycin D, blocking further transcription,
butallowing the translationof proteins fromthepreformeda mRNA (20). Proteins were labeled with [35S]methionine in thecontinued presence ofactinomycin Deitherimmediately
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.91.282.65.226.2]HSV VIRION HOST SHUTOFF FUNCTION 915
+ Actinornycintl1)
\_K1vK DNA KOSEo I fragments-.-I A I ,F01 0 IpBR afabala blala aib ajb
I
A 5.4..
k
P.9
^.l,S.t ^ ,,, s :4 * L
EcoRi
i .I --...
~ G ,F
*0fr
II.;,,,
A.-¶
,, ,
I . I
--I'
'T-'''-0.493 0.W33
.K
I
S
m
_FIG. 2. ThewtEcoRI Afragment transfers the host shutoff phenotype. Ltk-mouse cellsweremockinfected(lanes 1 and15)or were infected with(percell) 10 PFU ofwtHSV-1 KOS virus (lanes 2 and 16), the vhslmutantvirus(lanes3and17),orvirus stockspropagated from transfected culturesreceiving the vhsl virusDNAalong with thetestEcoRI clones (16),asindicated. Lanes1through 14,Infectionwas doneinthe presence ofactinomycin D, andproteinswerelabeledfrom8 to 10 hpostinfectioninthe presence ofthedrug.Lanes 15through 28, Infectionwasdone inthe absence ofactinomycin D, and proteins were labeled from 8to10 hpostinfection. The map shows the location of theEcoRIwtKOSfragments (51) tested in this experiment. Numberstotheright of the gel designate ICPs.
(0 to 2 h) or after further incubation (2 to 4 h) after the
cycloheximidereversal.
The functional stability of the a mRNAs can be
deter-mined bycomparing the level ofa protein synthesis during the early (0 to 2 h) and late (2 to 4 h) pulses. In the mock-infected cells (Fig. 3, lanes 1 and 2) equivalent
amountsofproteinsweremadeduringtheearly (E)andlate (L)pulses,indicatingthat the bulkofthehost mRNAswere
stableforatleast2to4hrs. Incells infectedwith thewtKOS
virus (lanes 3 and 4) orwith the KOS plus pUC19 control virus(lanes 5 and 6), thesynthesis ofthe aproteins 4, 0, 22, and 27decayedsignificantly 2to4 haftertheamRNAwas
made. In these cells the a proteins were expressed from functionally labile mRNAs. In contrast, a-protein synthesis was stable in the cells which were infected with the vhsl
virus (lanes 7 and 8) or with the vhsl plus pUC19 control
virus (lanes 9 and 10). The EcoRI A recombinant virus
stocksclearly exhibited thewta-synthesis phenotype (lanes
11 through 14). Thus, the EcoRI A fragmentwascapable of
transferring boththe virion-associatedhost shutoff function and theadestabilization function.
Theassayforthetransferofthe,/IymRNAdestabilization function (24) is shown in lanes 15 through 28 of Fig. 3.
Specifically,
Vero cells were infected in the absence of added drugs with virus stocks which were generated as describedabove. ActinomycinDwasaddedtothe cellsat8 hpostinfection. Proteinswerelabeled eitherimmediately (0to 1 hafteractinomycinDaddition)orafterfurther incuba-tion (6 to 7 h) in the presence of the drug. This design allowedus todeterminethe stability of theP and y mRNAs which were made prior toactinomycinD addition.
Thesynthesisof, infected cellproteins(ICPs) (e.g., 6, 8, 36) and y ICPs(e.g., 5, 9, 11, 25) decayed in the cellswhich were infected with the KOS and KOS plus pUC19 viruses
(Fig. 3, lanes 17 through 20). In contrast, the synthesis of these proteins continued at undiminished rates during the late pulse in the vhsl and the vhsl plus pUC19 infections (lanes21 through 24). Thesynthesis ofthe pand yproteins decayed rapidly in the cells thatwereinfected withthe two virus stocks derived from the EcoRI-A cotransfections (lanes25 through 28). These resultsshowthatthemutation which affects the functional destabilization of the P and y mRNAs also resides within the EcoRI A fragment.
Finemapping ofthe hostshutoffmutation.To fine mapthe vhsl mutation, the wt KOS EcoRI A fragment wasfurther subcloned. The subclonescontainingthefragmentsshown in Fig.4 wereeach testedfor theabilitytotransfer thewthost shutoff traittothe vhsl mutant. Thefragments whichfailed to transfer the wt vhs phenotype are shown as solid black
bars;those withwhich transfer wasobtained are shownas hatched bars (Fig. 4). Representative protein profiles are shown in Fig. 5.
On the basis of these results, the vhsl mutation resides within a 265-bp NruI-XmaIIIfragment spanning map
coor-i
VOL.62, 1988
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.169.452.67.383.2]viralDNA KOS vhs plasmid - - pUC _- pUC EcoA
time E L E L E LEL EL
EELIE
L*X:&i,:
_
..m ^
t;. *^
.. w
'|wt
wS
2..
e* >*ji t 300
- ..KOS vhs
-
-Ipu
-IPUC|
EcoA
E L E L E L E L E L E
LIE
L4
- - 8s
_MMM . - _ __ _8
___46qw ___" ____ 9-11
22
24
25 27 27
Agiw..
.War-'sw -pail 36
1 3 7 11 14 15 17 21 25 28
FIG. 3. EcoRI-Acontains thefunction which destabilizes the a, ,, andymRNAs. Ltk-mousecellsweremockinfected (lanes 1, 2, 15, and 16)or wereinfected with stocks propagated from transfectionsreceivingwtKOS virusDNAalone(lanes 3, 4, 17, and 18);wtpluspUC19 (lanes 5, 6, 19, and 20); vhsl virusDNAalone(lanes 7, 8, 21, and 22); vhsl pluspUC19 (lanes9, 10, 23 and24);orvhslplus thewtEcoRI-A plasmid pSG124 (lanes 11 through14and 25through 28).Lanes 1through 14, Infections involvedacycloheximidereversal intoactinomycin Dasdescribed in thetext.Labeling with[35S]methioninewasfrom 0to2 h(early[E])and2 to 4h(late [L]) after thecycloheximidereversal. The aproteins are indicated. Lanes 15 through 28, Infectionsweredone in the absence of drugs upto8 h postinfection, atwhich point actinomycinDwasadded.Labelingwasfrom0 to 1h(E) and6 to 7 h(L)after the addition ofactinomycinD.Exemplary 3 andyICPsare indicated.
dinates 0.604to0.606. Thisregion containssequences com-monto all thecloneswhich successfullytransferred thewt phenotype. Most notably, positive transferwasattained with theHpaI-to-NruI clone (pNF630) (Fig. 5,lanes 17 through 20) and the BamHI-to-XmaIII clone(pNF633) (lanes21 and 22).The resultsshowingtransferorlack of transfer of thewt vhs traitbytheremaining fragmentswereall consistentwith thismapassignment.
Mappingofthe functionresponsiblefor thephysical degra-dation of host mRNA. Ourpreviousstudies have shown that host mRNA was rapidly degraded in cells which were infected withwt virus in thepresenceofactinomycin D. In contrast, host mRNA was stable in the cells which were infected with the vhsl mutant in the absence of viral gene
expression (24, 56).
The passagedvirus stocksdescribed above were usedto determinewhether the mutationaffectingthestabilityof host mRNAalso mapped within the NruI-XmaIIIfragment. Du-plicateseries(aandb)weretested for each of theplasmids.
Cellsweremock infectedorinfected with thetestviruses in thepresenceofactinomycinDfor6.5 h. The cellswerethen
harvested, and the nucleic acids were prepared and trans-ferredtoNytran (Northern[RNA])blots. Before transfer of the RNA to the blot the gel was stained with ethidium
bromideto ascertain that theamountsof RNA loadedwere
similar in all thelanes. The blots wereprobed with 1-actin (18)anda-tubulin (6) probes. The results obtained with the twoprobesweresimilar,andonly the 13-actinhybridizations
areshown(Fig. 6).The data revealed host mRNAinstability
in the cellswhichwereinfectedwith thewtKOSpropagated virus (Fig. 6, lanes 2 and 3). In contrast, host mRNA was
stable in cells infected with the stocks derived from the
transfections receiving vhsl DNA or vhsl plus the pUC8 plasmid vector (Fig. 6, lanes 4 through 7). Some of the
recombinantstockstransferred thewthostmRNA degrada-tion trait. Most notably, the BamHI-to-XmaIII clone (pNF633; Fig. 6, lanes 14 and 15) and the HpaI-to-NruI clone (pNF630; lanes 12 and 13) rescued the vhsl defect. These results demonstrate that the mutation affecting the
ability of vhsltodegrade the
P-actin
and a-tubulin mRNAsalso resides within the265-bpNruI-to-XmaIII fragment.
Tests fortransfer ofthe viral mRNA destabilization func-tion. The virus stocks derived from the cotransfections described above were next assayed with regard to the functional stability of the a and
B/,y
viral mRNAs. Asdescribed above, to test the functional stability of the a mRNAs, cells were infected with the test viruses in the presenceofcycloheximide, allowingamRNAtranscription.
At 7 h
postinfection
the cycloheximide was removed, andincubationwascontinued in the presence ofactinomycinD. Theamountofaproteinsmadeimmediatelyafter addition of
actinomycin D (Fig. 7, lanes E) was compared with that made 4 h after addition of the drug (lanes L). The same clones which transferred the trait of destabilizing host mRNA also transferred the functional instability of the a mRNA (Fig. 7, lanes 1 through 18). Therefore, the vhsl
mutation which affects the stability of the a mRNA also mapswithin the 265-bpNruI-to-XmaIIIfragment.
Finally, the functional stability of 13 and y mRNA was
assayed by allowing infected-cell mRNA to accumulate in the absence ofdrugs. At 11 hpostinfection, actinomycinD wasadded to thecultures andPand ryproteinsynthesiswas
At..;.
AN
.4w.
1
MW
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.131.487.65.312.2]HSV VIRION HOST SHUTOFF FUNCTION 917
1.493 22 kb
5133
pSG124 EcoRI A Eco HI
-Ik
pNF533
EcoRI A/Hindill
A 1kb595
HindIll
K H H532 EcoRIA/Hindlil L
627 BamHI-Hpal B Hp
Bam Nru Xma X PsaPV Hpa
629 BamHI-PstI ...
636 Hpal- Nrul __-_p
134 Hpal- Pstl p Hp
632 Hpal-XmailI X Hp
B X
633 BamHI-Xmalil
631 BamHI-NrulI
265bp
[image:6.612.121.495.64.334.2].614
g.6igFIG. 4. Mapsof the various clones tested in the vhs marker transfertests.The EcoRI-A clonepSG124wasderivedbyGoldinetal.(16) and wasfurther subcloned as follows: pNF533, -595, and -532 were subcloned in pBR325; pNF627 and pNF629 through -633 were subcloned intoamodified form pUC9inwhich anHpaI linker had been inserted into the PstI site in thepolylinker region. Each ofthe subcloneswastested for theabilitytotransfer thewthost shutoff traittothe vhslmutant.Thefragmentswhich failedtotransfer thewtvhs phenotypeareshownassolid black bars. Thesubclones with which thewttransferwasobtainedareshownbyhatched bars. The HSV-1map
coordinates for the 22-kb EcoRI A DNAfragment (0.493-0.633)and the265-bp NruI-XmaIII DNAfragment (0.604-0.606)are indicated. Restrictionenzymesites: E, EcoRI; H, HindIII;B, BamHI; N, NruI;X,XmaIII;P,Pstl; andHp, HpaI.
\1 K% _ htlsviralD' AKODA+ SDN)A¾ nxr Kg'
lt( Eco BamHpa Barn Banl HIpa B.1111 i
--fin -Itpa -Pst -PSI -Nru -rAT11a
aa
~~~~~a
1,S
3, Ihat,IId' T''tTt t 1l Td1|!|hi
#1
a
ts
'
_~
j~~~~~
-4
1 4 9 13 17 21 26 29
FIG. 5. Finemapping of the virion host shutoff mutationto a265-bpNruI-XmaIIIDNAfragment. Vero cells weremockinfected (lanes 1 and27)orinfected with passage4of virus stocks derived from transfections receiving KOS viral DNA alone (lanes2 and28), vhslviral DNAalone(lanes3 and29),vhsl DNAplus pUC8 (lane 4),or vhsl DNAplusthe indicatedKOSDNAfragments (lanes5through 26).Lanes athroughddenoteseparately propagated series from replicate culturesreceivingthe indicated DNAs. The Eco-Hin clone showninlanes 5 and6isthepNF532subclone ofHindlIl fragmentL.Theinfections wereperformedin thepresenceofactinomycin D, and the cells were labeled with[35S]methioninefrom9 to 12 hpostinfection.Theproteinsampleswereelectrophoretically separatedin a9.25%sodiumdodecyl sulfate-polyacrylamide gel.
VOL.62, 1988
Ir
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.157.447.424.653.2]'1 KOS hsDNA+ KOSDNA fragmennis hi
pUC Bam Bam Hpa Bam |ipa
1
| .-Hpa -Pst, -Nru -Xma -Xmaa b-a bb ababab ab
*
_MNM
'.'.x..
l 4 8 12 16 18
FIG. 6. The mutation affecting the stability of host mRNA maps in the 265-bpNruII-XmaIII DNA fragment. Vero cells were mock infected (lanes 1 and 18) or infected with passage4 virus derived from transfections receiving wt KOS virus DNA alone (lanes 2 and 3), vhsl mutant virus DNA alone (lanes 4 and 5), vhsl plus pUC8 vector DNA (lanes 6 and 7), or vhslvirusDNAplus the indicated test KOS DNA fragments (lanes 8 through 17). The cells were infected in the presence of actinomycin D, and the total cellRNA washarvestedat6.5hpostinfection. The RNAwas electrophoreti-cally separated in a 1.2% agarose-2.2 M formaldehyde gel and electrotransferredtoNytran(Schleicher and Schuell). The blotwas probed with
0-actin
(18). Thesmearin lane10of the blot represents thehybridization of bacterial plasmid andnotactinDNAsequences, inasmuchas asimilarpatternofhybridizationwas seenwithapUC9 plasmidDNAprobe (datanotshown). Further analyses of this virus stock(datanotshown) revealed the presence of defective genomes containing plasmid DNA sequences. These genomes most likely arosebyrecombination betweenthepNF629constructand thevhsl genomeduring the cotransfection. Thetranscriptionof the resultant defective genomes apparentlygives risetoRNAsof variablelengths containing plasmid DNA sequences. Studies in progress are de-signedtocharacterize these genomes further.compared early (0 to4 h; Fig. 7, lanesE) orlate (4to 8 h; lanes L) after the addition of the
drug.
The/Iy
mRNAstability phenotypeswere not asclear-cutasthehost shutoff
andamRNAstability phenotypes. We attributethis
behav-iorto thefactthat thesecondary shutoff function (9, 40, 44)
was expressed in the cells which were infected in the absence of drugs to allow the
synthesis
ofB/Iy
mRNA.Nevertheless, all of the constructs which transferred the
abilityto shut offhostprotein synthesiswere also foundto
transfer the functional instability ofthe
P
and y mRNAs. These results suggest thatthe vhsl mutant virus containsasingle mutation which affectsa
single
gene productrespon-sibleforthedestabilization ofhost, ax,and
P/y
mRNAs.This mutation maps within the265-bpNruI-XmaIII fragment.DISCUSSION
Finemapping of the vhsl mutation. Two
approaches
have been used to map the virion-associated hostshutoff function of HSV. The first was based on thefinding
that HSV-2inhibited host protein synthesis more rapidly than HSV-1.
Using a battery of HSV-1 x HSV-2 recombinant viruses,
Morse et al. (35) and Fenwick et al. (8) mapped the"rapid
host shutoff function" withinaregion spanningmap
coordi-nates 0.52 and 0.59 of the HSV-2 genome. A second
ap-proach, used in the present study, has mapped the vhsl
mutationwithin the
265-bp
NruI-XmaIII fragmentspanning
mapcoordinates0.604 to 0.606 of the HSV-1 genome. The
rapid shutoff by HSV-2 and the vhs function most
likely
represent the same gene, since the 265-bp fragment lies at the
right-hand
border ofthe 11-kbregion
identifiedbyMorseet al. (35). The small discrepancyin map coordinates most
likely reflects theways in whichthe mapcoordinates have
beencalculated.Itis alsonoteworthythatOroskar and Read haverecentlymappedthe vhslmutationwithin the EcoRI A
fragmentof HSV-1 DNA(41).
Role of theshutoff function in virusreplication. We have
found that the same DNA
fragment
which transferred theabilitytoshut off hostprotein synthesiswasalsocapable of
transferring
the functionaffecting
thephysical integrity
ofhost mRNA. This
finding
suggests that in Verocells,
the shutoff of hostprotein synthesisismechanisticallylinkedtothe physical degradation of host mRNA. As elaborated elsewhere (56), there are three alternative mechanisms to
explain
thislinkage. First,
the vhs locus could encode anuclease,
which isbrought
intothe cellsas avirion compo-nentandwhichreduces thehalf-life ofmRNAin theinfected cell.Second,
the vhs function could activate apreexisting
host nuclease. Finally, the vhs function could
modify
the translationalmachineryso as torender mRNAmorevulner-abletonucleases.Atpresentwecannotexcludeanyof these hypotheses. However, the virionRNase
hypothesis
appearsunlikely
inlight
ofrecentstudies which have revealedthat the mutated vhsl function canirreversibly
protect hostmRNAfrom
degradation by
thewtvhsfunction(Kwong and Frenkel,manuscript
inpreparation).
We havepreviously proposedthatthevirionhost shutoff functionplaysarole in thetransienceof translation of viral
mRNA. This
hypothesis
issupported by
severalobserva-tions, asfollows. (i) Prolonged synthesis ofa proteins was
observed in cells infected with several
independently
de-rived vhs mutants (44).
(ii)
The destabilization ofa-protein
synthesis
coincides withshorter half-lifeofa mRNA(24, 41,
56). Furthermore,
the decreasedstability
oftheamRNAis mediatedby
a virion component(24).
(iii)
The functionalstability
aswellasabundance ofP andy mRNAsarelowerin cells infected with the wt virus
compared
with theircounterpartsinfected withthevhslmutantvirus
(24,
56).
(iv)
Most
conclusively,
as shown in the presentwork,
the wtvirus function which decreases the functional
stability
of viral mRNAs comaps within the265-bp
NruI-XmaIIIfrag-ment which contains the host shutoff mutation.
However,
we have not shown
directly
that a virion component is involved in thePl-y
mRNAdegradation,
and atpresent we cannot rule outthehypothesis
thatthisphenotype
ismedi-ated
by
aseparatemechanism.Forexample, changes
in viral mRNAstability
coulddepend
on theprior
modification of host cellmachinery by
the virion host shutoff function.Furtherstudies will be needed to resolve this issue
unam-biguously.
Regardless
of whether the hostandviral shutoff functionsareidenticalor
merely interdependent, they
bothoperatetolimitthe
expression
ofgenesintheinfected cells. Thevirion function whichmediates this effect isclearly
notessentialfor virus growth in cultured cells. However, it does conferadvantageous growth
tothewtvirus,
asshownby
therapid
selection ofwt virus
during
serialviruspropagation.
More-over, a minimal level of host and viral shutoff may beessentialforvirus
growth,
since the vhslmutanthasretainedthe
secondary
host shutofffunction,
and thesynthesis
ofhost,
a, andp
proteins
iseventually
turnedoff(24,
44, 56).
Lastly,
theeconomyimplied
in thetimely
transitions in thesynthesis
of viralproteins
mayplay
a more critical role in viralreplication
invivo, during
which thesynthetic
machin-ery may be more
limiting. Thus,
the initial shutoff ofhostprotein
synthesis
may allow the maximal translationalca-pacity
ofthecelltobe used in theexpression
of viral genes.on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.109.276.64.199.2]HSV VIRION HOST SHUTOFF FUNCTION 919
HSV(Y niRNA Fuictional StabilitN
\l KOS vhs DNA+KOS DNA fra-mne%s: pUC Bam Bain Hpa Barn] Hpa
-Hpa -Pst -Nru -Xma -
Xmna
L[ILI,EIL ELE E|L
m
@3~~~0
;;
- * ~°27* ,d.
.. *
+ + ..a t#:~~~~
w tj-*_2
1 3 5 9
HiSV Ph imiRNA Funictional Stability
.
...
M KOS vhs DNA+KOS DNA fragments: . PUC B;an Bamn Hpa
Baam
Hpa-HETL
I I -Pst -Nru -Xma -XmaI
EI
LI
EI
L1E{L1 EIL{7LF
fEI
jLI*m
b. qmva.:
_m AM m 911
wqp*4w+ _ *..- :.4,* ,Rm15
. "" - 20,
_ .A - 3_
_
_0
_-0 _ I 0 3625--j In N,27
i. -- _ t _1 _ 33
__
do_R A- 4 36l8 19 21 23 27 36
FIG. 7. Themutation affecting the functional stability of a,13, and -y viral mRNAs maps in the 265-bp NruI-XmaIII DNA fragment. Vero cellsweremock infected(lanes 1, 2, 19, and 20)or wereinfected with passage 4of virus stocks derived from transfections which received KOS viralDNAalone(lanes 3, 4, 21, and 22), vhsl viralDNAalone(lanes 5, 6, 23, and 24), vhsl pluspUC8 (lanes 7, 8, 25, and 26), or vhsl viralDNAplus the indicated test KOS DNA fragments (lanes 9 through 18 and 27 through 36). Lanes 1 through 18, Test for HSV a mRNA functional stability. Vero cellswereinfected in the presence ofcycloheximide.At 7 hpostinfection, actinomycinDwasadded, and the cells werelabeled with[35S]methioninefrom0to4(early[El)or from4to8 (late[L]) after the addition of the drug.TheaICPs are indicated. Lanes 19through 36, Testfor HSV
Ply
mRNAfunctional stability. Vero cells were infected in the absence of drugs.At 11h,actinomycin D was added, and the cellswerelabeled with[35S]methioninefrom0 to 4h(E)orfrom4to 8h(L) after addition of thedrug.Representative ,B andyHSV ICPsareindicated.
Inaddition, limiting theexpression of a and 1 genes (when -y transcription is turned on) may allow the maximum synthesis of the structuralproteins when virion maturation begins.
Tentative identification ofthe vhs gene product and rela-tionship between host shutoff and malignantcell transforma-tion. Frink etal. (11)have mappeda single mRNA species overlapping the BamHI-to-XmaIII fragment spanning map
coordinates 0.602to0.606.ThismRNAis1.9kb in sizeand
encodes a 58-kilodalton protein. Nucleotide sequence data
(D. J. McGeoch, personal communication) are consistent with the transcriptional data. Taken together, these data suggest that the vhs function is mediated bythe 58-kilodal-ton-protein. Studies designed to identify the vhs protein moredirectly arecurrently inprogress.
The assignment ofthe vhs function tothis region ofthe
HSV-1 genome is especially intriguing inasmuch as this
region overlaps the
BglII
N fragment of HSV-2 DNA,representing
oneofseveral HSV DNAregions exhibitingtheability totransform cells in culture (12, 13, 45). The
trans-formingsequences within the
BglII
Nfragment were shown to reside in an open reading frame which encodes a61-kilodalton protein (15). This reading frame of HSV-2 is
homologousto thatfor the58-kilodalton HSV-1polypeptide
which we havetentatively identified as the vhsprotein. Theparadoxical comapping ofthetransforming and host
shutoff functionscanbeinterpreted in at least twoways: (i)
the twofunctionscould involve differentgene products, e.g., if the observed cell transformation involved a promoter
insertion mechanism; and (ii) transformation could be an
indirect consequence of the transient inhibition of host
protein synthesis. By this model, the vhs activity could
result intransientcell stress alterationsleadingto amplifica-tion, translocaamplifica-tion,orotherrearrangementsofoncogenes or cellcycle regulatory genes. Suchgene amplificationmay in turnleadtocelltransformation (reviews in references1, 27,
and 53). Indeed, amplification of simian virus 40 (SV40)
DNA sequences was shown to occur in SV40-transformed cells after infection with HSV (32, 33, 50). Furthermore, HSV inducestheamplification of cotransfectingSV40 DNA sequences in cells whichare
nonpermissive
for SV40DNAreplication (R. R. Danovich and N. Frenkel, submitted for publication), althoughit is as yet unknownwhetherthe vhs
functionplaysanyrole in this amplification.
Withregardtothis model,itis noteworthy that treatment
of cells with inhibitors ofprotein synthesis such as
cyclo-heximideandpuromycinwaspreviouslyshown to be
accom-panied
bytheformation ofsmallpolydisperse circularDNA structures. Gene amplification and gene rearrangements were shown to be induced by transient treatment of cells withinhibitors ofDNAreplicationsuch asmethotrexate and hydroxyurea (4, 22, 31, 49, 60) and upon stress conditionssuch astransient hypoxia(46).Furthermore,amplificationof
specificDNAsequences was inducedbyphysicaland
chem-icalcarcinogens (2, 23, 25, 26, 37) which interfere with the normal progression of the S phase, as shown by Lavi and co-workers (23). It has been suggested thattransient treat-VOL.62, 1988
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.113.502.67.342.2]mentof cells with such agents may lead to DNA amplifica-tion by inducing replicon misfiring andthe repeated replica-tion of specific DNAsequences (23, 49, 52, 59, 62). Inline
with this hypothesis istheobservation that the vhs function ofHSV-2 ismoreefficient than that of HSV-1 (8, 19, 35, 43,
56). This may account for the fact that transformation has been observed only with the HSV-2BgII Nfragment, and notwith the corresponding sequences of HSV-1 DNA (45).
Because the vhs function is expected tobe incompatible with long-range cell survival, itcan be predicted that DNA sequences located in the proximity of the HSV-2 BglII N transformingfunction willnotberetained in the transformed cells. This reasoning may explain previous findings that
DNAsequencesfrom this regionof the HSVgenome arenot
retained in transformed cells and are preferentially lost
during cell propagation (12, 14, 28, 30). Itremains tobeseen
whether further studies of the vhs function can add to the unresolved issue oftransformation by HSV.
ACKNOWLEDGMENTS
These studies weresupported byPublicHealth Serviceresearch
grantsCA19264, A115488,andA123015 from theNationalInstitutes ofHealth. A.D.K. isaLeukemia Society of America Fellow.J.A.K. was apredoctoraltrainee supported byUniversity of Chicago viral oncology traininggrantCA-09241.
We thank Jennifer Olerand George C. Katsafanas for excellent technicalhelp.WethankM.Levine (University of Michigan)for the giftofthe EcoRI clones, and E. Fuchs(University ofChicago)for the ,-actin clone and a-tubulin clone. We thank D. J. McGeoch (Institute forVirology, Glasgow, Scotland) for communicatingtous DNAsequencedatabeforepublication.
LITERATURE CITED
1. Alitalo, K., and M. Schwab. 1986. Oncogene amplification in tumorcells. Adv. CancerRes. 47:235-281.
2. Baran, N., A. Lapidot, and H. Manor. 1987. Unsual sequence
element found at the end ofanamplicon. Mol. Cell. Biol. 7: 2636-2640.
3. Bastow, K. F., J. Bouchard, X.-J. Ren, and Y.-C. Cheng. 1986. Synthesisofdihydrofolate reductaseand metabolism of related RNA inamethotrexate resistant human cell line infected with herpes simplexvirus type2. Virology 149:199-207.
4. Brown, P.C., R. S. Tlsty, and R. T. Schimke. 1983. Enhance-ment of methotrexate resistance and dihydrofolate reductase gene amplification by treatment ofmouse 3T6 cells with hy-droxyurea. Mol. Cell. Biol. 3:1097-1107.
5. Clewell, D. B., and D. R. Helinski. 1970. Properties of a
supercoiled deoxyribonucleic acid-protein relaxation complex and strandspecificity of therelaxation event. Biochemistry9: 4428-4440.
6. Cowan, N. J., P. R.Cobner,E. V. Fuchs,and D. W. Cleveland. 1983. Expression of human alpha-tubulin genes: interspecies conservation of 3' untranslatedregions.Mol. Cell.Biol. 3:1738-1745.
7. Fenwick, M. 1984. The effects of herpesviruses on cellular macromolecular synthesis, p. 359-390. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology, vol. 19. PlenumPublishing Corp.,NewYork.
8. Fenwick, M., L. Morse, and B. Roizman. 1979. Anatomy of herpes simplex virus DNA: apparent clustering of functions effecting rapid inhibitionof host DNA andproteinsynthesis. J. Virol. 29:825-827.
9. Fenwick, M. L., and J. Clark. 1982. Early and delayed shut-off ofhostprotein synthesis in cellsinfected with herpes simplex virus. J.Gen. Virol.61:121-125.
10. Fenwick, M. L., and M. J. Walker. 1978. Suppression of the synthesisofcellularmacromoleculesbyherpes simplexvirus. J. Gen.Virol. 41:37-51.
11. Frink, R. J., K.P.Anderson, and E.K. Wagner. 1981. Herpes simplex virus type 1 HindIll fragmentLencodes splicedand
complementary mRNA species. J. Virol. 39:559-572.
12. Galloway, D. A., C. D. Copple, and J. K. McDougall. 1980. Analysis of viral DNA sequences in hamster cells transformed by herpessimplexvirus type II. Proc. Natl. Acad. Sci. USA 77: 880-884.
13. Galloway, D. A., and J. K. McDougall. 1981. Transformation of rodent cells by a cloned DNA fragment of herpes simplex virus type2. J. Virol.38:749-760.
14. Galloway, D. A., and J. K. McDougall. 1983. The oncogenic potential of herpes simplex viruses: evidence for a "hit and run" mechanism.Nature(London) 302:21-24.
15. Galloway, D. A., J.A.Nelson, and J. K. McDougall. 1984. Small fragments ofherpesvirus DNA with transforming activity con-tain insertion sequence-like structures. Proc. Natl. Acad. Sci. USA 81:4736-4740.
16. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C. Glorioso. 1981. Cloning of herpes simplex virus type 1 se-quencesrepresenting the whole genome. J. Virol. 38:50-58. 17. Graham,F.L.,and A.J. Van der Eb.1973. A new technique for
the assayof infectivity of human adenovirus DNA. Virology 52: 456-467.
18. Hlanokoglu,I., N. Tanese, and E.Fuchs. 1983. Complementary DNAsequences of ahuman cytoplasmic actin. J. Mol. Biol. 163:673-678.
19. Hill,T.M., J.R.Sadler,andJ.L.Betz. 1985. Virion component of herpessimplex virus type1KOS interferes with early shutoff of hostprotein synthesis induced by herpes simplex virus type 2186. J. Virol. 56:312-316.
20. Honess, R. W., and B.Roizman. 1974. Regulation of herpesvirus macromolecular synthesis.I. Cascade regulation ofthe synthe-sis ofthreegroupsof viral proteins. J. Virol. 14:8-19. 21. Inglis, S. C. 1982. Inhibition of host protein synthesis and
degradation of cellular mRNAs during infection by influenza andherpessimplex virus. Mol. Cell. Biol. 2:1644-1648. 22. Johnston,R.N., J. Feder, A. B. Hill, S. W. Sherwood, and R.T.
Schimke. 1986. Transientinhibitionof DNA synthesis results in increased dihydrofolate reductase synthesis and subsequent increasedDNAcontent percell. Mol. Cell. Biol. 6:3373-3381. 23. Kleinberger, T., S. Etkin, and S. Lavi. 1986.
Carcinogen-mediated methotrexate resistanceanddihydrofolate reductase amplification in Chinese hamster cells. Mol.Cell. Biol. 6:1958-1964.
24. Kwong, A. D., and N. Frenkel. 1987. Herpes simplex virus infected cells contain function(s) which destabilize both host andviralmRNAs. Proc.Natl. Acad. Sci. USA 84:1926-1930. 25. Lavi, S.,andS.Etkin. 1981.Carcinogen-mediated induction of
SV40 DNA synthesis in SV40 transformed Chinese hamster embryo cells.Carcinogenesis2:417-423.
26. Lavi, S.,N.Kohn,T.Kleinberger,Y.Berko,andS. Etkin.1983. Amplification of SV40 and cellular genes in SV40-treated Chi-nese hamster cells treated with chemical carcinogens, p. 659-670. In E. C. Friedberg andB. A. Bridges (ed.), Cellular responsesto DNAdamage. AlanR. Liss, Inc.,NewYork. 27. LeBeau, M. M., andJ. D. Rowley. 1984. Recurring
chromno-somalabnormalitiesinleukaemiaandlymphoma. Cancer Surv. 3:371-394.
28. Leiden,J.M.,N.Frenkel,and F. Rapp.1980. Identification of theherpessimplex virusDNAsequences present insixherpes simplexvirusthymidinekinase-transformedmousecell lines. J. Virol. 33:272-285.
29. Locker, H.,N.Frenkel,andI.Halliburton. 1982.Structure and expressionof class II defectiveherpes simplexvirusgenomes encoding infectedcellpolypeptide number 8. J. Virol. 43:574-593.
30. Manservigi, R.,E.Cassai,L.P.Deiss,D. DiLuca, V.Segala,and N.Frenkel.1986.Sequences homologousto twoseparate trans-forming regionsofherpes simplex virusDNAarelinked in two humangenital tumors.Virology 155:192-201.
31. Mariani,B.D.,and R. T.Schimke. 1984.Geneamplificationin asinglecellcycle inchinese hamster ovary cells. J. Biol. Chem. 259:1901-1910.
32. Matz, B. 1987. Herpes simplex virusinfectiongenerateslarge tandemly reiteratedsimian virus 40DNAmolecules ina
on November 10, 2019 by guest
http://jvi.asm.org/
HSV VIRION HOST SHUTOFF FUNCTION 921 formed hamster cell line. J. Virol. 61:1427-1434.
33. Matz, B., J. R.Schlehofer,andH. zur Hausen. 1984. Identifica-tion of a gene funcIdentifica-tion of herpes simplex virus type 1 essential for amplification of simian virus 40 DNA sequences in trans-formed hamster cells. Virology 134:328-337.
34. Mayman, B. A., and Y. Nishioka. 1985.Differential stability of host mRNAs in Friend erythroleukemia cells infected with herpes simplex virus type1.J.Virol. 53:1-6.
35. Morse,L.S., L. Pereira,B.Roizman, andP.A.Schaffer. 1978. Anatomyof herpes simplex virus (HSV) DNA. X. Mapping of viral genes byanalysis of polypeptides and functions specified byHSV-1 x HSV-2 recombinants. J. Virol. 26:389-410. 36. Nakai, H., I. H. Maxwell, andL. I. Pizer. 1982. Herpesvirus
infection alters the steady-state levels of cellular polyadenylated RNA in polyoma virus-transformed BHK cells. J. Virol. 42: 1131-1134.
37. Neer,A.,N.Baran, and H.Manor. 1977. In situ hybridization analysis ofpolyoma DNA replication in an inducible line of polyoma-transformed cells. Cell 11:65-71.
38. Nishioka, Y., and S.Silverstein. 1977. Degradation of cellular mRNA duringinfection by herpes simplex virus. Proc. Natl. Acad. Sci. USA74:2370-2374.
39. Nishioka, Y., and S. Silverstein. 1978.Alteration in theprotein synthetic apparatus of Friend erythroleukemia cells infected with vesicular stomatitis or herpes simplex virus. J. Virol. 25: 422-425.
40. Nishioka, Y., and S. Silverstein. 1978. Requirementof protein synthesis for the degradation of hostmRNAin Friend erythro-leukemia cells infected with herpes simplex virus type 1. J. Virol.27:619-627.
41. Oroskar, A. A., and G. S. Read. 1987. A mutant of herpes simplex virus type1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 61:604-606.
42. Pizer, L. I., and P. Beard. 1976. The effect of herpes virus infectiononmRNA inpolyoma virus-transformed cells. Virol-ogy75:477-480.
43. Powell, K.L., and R.J. Courtney. 1975. Polypeptides synthe-sized in herpes simplex virus type 2-infected HEp-2 cells. Virology 66:217-228.
44. Read, G. S., and N. Frenkel. 1983. Herpes simplex virus mutantsdefective in the virion-associatedshutoff of host poly-peptide synthesis and exhibiting abnormal synthesis ofa (im-mediateearly) viralpolypeptides. J. Virol. 46:498-512. 45. Reyes,G.R.,R. LaFemina, S.D.Hayward,andG.S.Hayward.
1979. Morphological transformation by DNA fragments of hu-manherpesviruses: evidence fortwodistinct transforming re-gions in HSV-1 and HSV-2 and lack of correlation with bio-chemical transfer of the thymidine kinase gene. Cold Spring HarborSymp. Quant. Biol.44:629-641.
46. Rice, G. C., C. Hoy,and R. T.Schimke.1986.Transienthypoxia
enhances thefrequency ofdihydroflolate reductase gene ampli-fication in chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 83:5978-5982.
47. Ruyechan,W.T., L. S.Morse,D. M. Knipe, andB.Roizman. 1979.Molecular genticsof herpes simplex virus. II.Mapping of themajor viralglycoproteins andof the genetic locispecifying thesocialbehavior of infected cells. J. Virol. 29:677-697. 48. Schek, N., and S. L. Bachenheimer. 1985. Degradation of
cellularmRNAs induced bya virion-associated factor during herpes simplex virusinfection of Vero cells. J. Virol. 55:601-610.
49. Schimke, R. T. 1984. Gene amplification in cultured animal cells. Cell37:705-713.
50. Schlehofer, J. R., L. Gissmann, B. Matz, and H. zur Hausen. 1983. Herpessimplex virus-induced amplification of SV40 se-quences intransformed Chinese hamster embryo cells. Int. J. Cancer 32:99-103.
51. Smiley, J. R.,B. S.Fong,andW.-C. Leung. 1981.Construction of a double-jointed herpes simplex viral DNA molecule: in-verted repeats are requiredfor segment inversion, and direct repeats promotedeletions. Virology 113:345-362.
52. Smith, C. A.,andJ.Vinograd. 1972.Smallpolydisperse circular DNAof HeLa cells. J. Mol. Biol. 69:163-178.
53. Stark,G.R.,andG.M.Wahl.1984.Geneamplification.Annu. Rev. Biochem. 53:447-491.
54. Stenberg, R. M., andL. I. Pizer. 1982. Herpes simplex virus-induced changes in cellular and adenovirusRNAmetabolism in anadenovirus type 5-transformedhumancellline.J. Virol.42: 474-487.
55. Stringer, J. R.,L. E. Holland, R. I. Swanstrom, K. Pivo,and E.K. Wagner.1977. Quantitation of herpes simplex virus type 1RNAininfectedHeLacells. J.Virol. 21:889-901.
56. Strom, T.,andN. Frenkel.1987.Effects ofherpessimplex virus onmRNAstability.J. Virol. 61:2198-2207.
57. Sydiskis,R.J., and B.Roizman. 1966. Polysomes andprotein synthesis in cells infected withaDNAvirus.Science 153:76-78. 58. Sydiskis, R.J., and B. Roizman. 1967. The disaggregationof hostpolyribosomes in productive andabortive infection with HSV.Virology 32:678-686.
59. Varshavsky,A.1981. On thepossibility of metabolic control of replicon "misfiring": relationship to emergence ofmalignant phenotypes in mammalian cell lineages. Proc. Natl. Acad. Sci. USA 78:3673-3677.
60. Varshavsky,A.1981. Phorbolesterdramatically increases inci-denceof methotrexate resistant mouse cells; possible mecha-nism and relevance to tumorpromotion. Cell 25:561-572. 61. Wagner,E. K. 1985. Individual HSVtranscripts:
characteriza-tionofspecificgenes, p. 45-104.InB. Roizman(ed.), Herpes-viruses, vol.3.PlenumPublishingCorp., New York.
62. Yamagishi, H.,T.Kunisada,andT. Tsuda.1982.Small circular DNAcomplexes ineucaryotic cells. Plasmid 8:299-306. VOL.62, 1988