Copyright() 1972 American Society for Microbiology Printed in U.S.A.
In
Vivo and In Vitro Synthesis of Human
Rhinovirus Type 2
Ribonucleic
Acid'
F. H. YIN AND E. KNIGHT, JR.
Central ResearchDepartment,Experimental Station, E. LduPont de Nemours and Company,
Wilmington,Delaware19898
Received for publication 3April 1972
HeLa cells
infected
with human rhinovirus type 2 synthesize a mixture of single-and double-strsingle-anded ribonucleic acid (RNA). The RNA synthesized by themem-brane-bound
RNA polymerase complex in vitro is also a mixture of single- anddouble-stranded RNA, whereas the deoxycholate-treated RNA polymerase complex
synthesized
onlydouble-stranded
RNA. Although twice as much cell-associatedviral RNAis
synthesized
invivo
at 34 C than at 37 C, there is nodifference in therate
of
RNAsynthesized in vitro
at 34 C and 37 C by the polymerase complex.TheRNA
polymerase complex,
after treatment with deoxycholate, sediments as abroad
peak
with anaveragesedimentation
value of120S.Theinfection of ahost cell withapicornavirus results in
the
appearance of a ribonucleicacid
(RNA)-dependent RNA polymerase complex inthe
cytoplasm
of the host cell. Poliovirushas been
the prototype
of
this group of viruses.The
viralpolymerase
is attached to theparticulate
mem-branestructuresandcanbe
released
by
treatmentofthesestructureswithananionic-nonionic deter-gent mixture (3). The polio RNA polymerase complex solubilized bydetergenthas a
sedimenta-tion coefficient of
70S,
and thecomplex
synthe-sizespoliovirus-specificRNAin vitro.
Human rhinoviruses contain single-stranded RNA
of
30 to 32S(8). The
virionsare acid-sensi-tive, and their buoyant densities in CsCl arehigher than the pH3stable
poliovirus (4,
12). Inthis
reportwedescribe
the in vivo RNAsyn-thesis of the
human rhinovirus type 2(HRV-2)
and the isolation ofa RNA
polymerase
complexfrom
HeLa cellsinfected
withthesamevirus. Thiscomplex
synthesizes
viral RNA in vitro.MATERIALS AND METHODS
Cells and virus. A continuous line of HeLa cells,
"HeLa-Ocells," wasobtained from FlowLaboratory, Rockville, Md. They were propagatedinmonolayer
cultureat37 C inMcCoy's5Amedium(6)containing 10%calf serum, 100
jAg
ofneomycin perml,100,ug ofpenicillin perml, 100,ug ofstreptomycinperml,and 25 ,ugoffungizoneperml.
Thevirus used wasHRV-2, strain HGP, obtained
fromR. R. Grunertof StineLaboratory,E.I. du Pont
deNemoursandCo.,Newark, Del.
1Contribution no. 1906, Central Research Department,
Ex-perimentalStation,E.I. duPont de Nemours andCo.,
Wilming-ton,Del.19898.
Virus infection. Monolayers of HeLa cells were grown toabout80%", confluency. They were washed once with calcium- and magnesium-free phosphate-buffered saline, then infected with virusat a multiplic-ity of infection of 40 plaque-forming units (PFU) /cell. Virus was adsorbed for 30 min at 34 C. McCoy's 5A medium containing 5% heat-inactivated fetal calf serum and antibiotics at concentrations identical to that used for cells was then added to the monolayers. Virusmultiplicationwascarried out at 34 C. Actino-mycinD wasadded to a concentration of5
j.ug/ml
inexperiments where it was desirable to inhibit host RNAsynthesis.
Polyacrylamide gel electrophoresis of viral RNA. Cylindrical gels (0.8 by 10 cm) were preparedwith
re-crystallized acrylamide by using the base-soluble
ethylene diacrylatein thebuffer system described by Loening (6). Electrophoresis was carried out in a
buffer containing 0.04
tris(hydroxymethyl)-amino-methane(Tris), 0.2 M sodium acetate, 0.002 M ethyl-enediaminetetraacetic acid (EDTA), and 0.5% sodiumdodecyl sulfate (SDS), pH7.7.Thegels were
fractionated into 2-mm fractions with a mechanical
cutter (5) in a hexane-dry ice bath maintained at
-20C. Thefractionswerefirstsolubilized with0.5ml of concentrated NH4OH, then counted in Bray's
countingsolution inaliquidscintillationcounter.
Phenol extraction of RNA. Infected cells were
scrapedfrom culture dishes and collectedby centrifu-gation. The cellsweresuspendedat 4 Cinabufferof
0.01 M Tris, (pH 7.2), 0.01 M NaCl, and 2.001 M
MgCl2,andwereruptured with14strokes ina stainless-steel Dounce homogenizer. Thenuclei wereremoved by low-speed centrifugation (800 X g, 5 min), the supernatant fraction(crudecytoplasm)wasmade 0.05 Min asodiumacetate(pH5.0),0.01MinEDTA,0.5'%
in SDS, and the mixture was then extracted with
phenol at 45 C as previously described (10). RNA produced by the polymerase complex in vitro was
extractedbythe sameprocedurebut at 25 C. 93
on November 10, 2019 by guest
http://jvi.asm.org/
Preparation of crudepolymerase. HeLacell
mono-layers were infected withHRV-2 virus in the presence ofactinomycin D. At the endof7hr, thecellswere
removed byscraping,collected bylow-speed centrifu-gation, and the cell pellet was frozen at -70 C. At this stage,the invitro RNApolymerasecanretain its
ac-tivity for at least a month. Approximately 4 X 108 frozen cells were suspended in 10 ml ofcold buffer containing 0.05 M Tris (pH 7.2), 0.002 M MgCl2, and 0.1MNaCl.Thecells wereallowed toswellat4Cfor 10min; they were ruptured with 14 strokes ina stain-less-steel Dounce homogenizer. Unbroken cells and nuclei were removed bycentrifugation at 800 X gfor 5 min. The supernatant fraction (the cytoplasmic ex-tract) wascentrifugedat30,000 X gfor 20 min, and
thepellet was suspendedin2mnl of buffercontaining 0.05 M Tris (pH 8.0) and 0.01 M NaCa.This fraction
containedcytoplasmic membrane and wasdesignated
the crude polymerase complex, and the supernatant fraction was designated the cytoplasmic soluble
frac-tion. The nuclei were suspendedin the same buffer. A1 :1 mixture of10%Nonidet P-40 and 5%sodium deoxycholate (DOC) wasadded in the ratio of 0.2ml of detergentmixtureper 1 mlof suspended nuclei. A
detergent wash of the nuclei was prepared by the method of Ehrenfeld et al. (3).
Invitro RNA polymerase assay. A 0.1-mlsample of enzyme wasusedfor assaying polymerase activity. It was incubated at 34 Cfor 30 min with 0.1 ml ofa
mixture containing the following constituents: 0.25
JAmolesof the5'-triphosphates of adenine, cytidine, and uridine (ATP, CTP, and UTP, respectively); 10 ,umoles of Tris buffer (pH 8); 1 ,umoleof MgCl2; 0.25 ,umoles ofphosphoenolpyruvate; 5,ugof phosphoenol pyruvate kinase; 1jAg of actinomycin D; 1.3,Amoles of dithiothreitol; and 2.5 ,ACi of 3H-guanosine-triphos-phate (GTP, specific activity 1.3 Ci/mmole). The reaction was terminated by adding 0.5 ml of ice-cold
0.1MNa4P207and 0.5mlof 50% trichloroacetic acid. Theprecipitate was collected on a filter, and radio-activitywascountedinascintillationcounter.
Reagents. Actinomycin D was purchased from
Mann Research Laboratory, Inc., New York, N.Y.;
3H-uridine (36.8 Ci/mmole) and 3H-GTP (1.3
Ci/
mmole) werepurchasedfrom New England Nuclear, Boston, Mass. Unlabeled ATP, CTP, and UTP werepurchased from SchwarzBioResearch, Inc., Orange-burg, N.Y. Bovine pancreatic ribonuclease, pyruvate kinase (rabbit muscle), and 2-phosphoenol-pyruvate
werepurchasedfrom Calbiochem, Los Angeles, Calif.
3H-labeled virion RNA was kindly supplied by K.
K. Lonberg-Holm of the Central Research Depart-ment, E.I.du Pont deNemours and Co., Wilmington,
Del. (8).
RESULTS
Rate and temperature dependence of the in vivo
synthesis of HRV-2 viral RNA. When HeLa
cells
are infected with HRV-2 at a multiplicity of 40PFU/cell,
one cycle of virus growth is completedat 12 hrat 34 C (F. H. Yin, unpublished data). The rate of viral RNA synthesis during one-cycle
growth
was monitoredby
the incorporation of3H-uridine into viral RNA. The synthesis of HRV-2 RNA can be detected 3 hr after
infection,
and therateofsynthesisreaches apeak 7 hr after infection and then declines. Viral RNA synthe-sizedat7 hrafter infection isusually 15to20 times morethan the actinomycin D-treated HeLa cell background. Since rhinoviruses growto ahigher titerat 34C thanat 37 C(11), the effect of tem-perature on the synthesis of HRV-2 RNA was
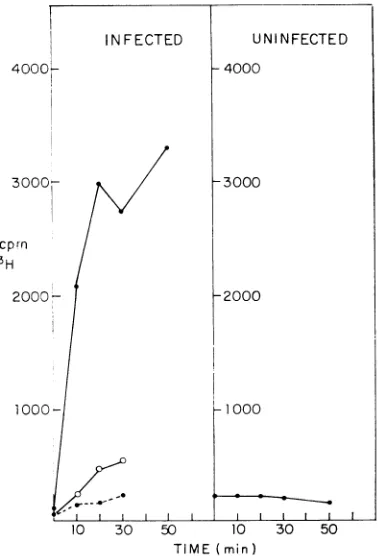
examined.Figure 1 showsthattheamountof viral
RNA synthesized at 34 Cat8 hrafterinfection is twice theamountsynthesizedat37 C.HeLa host
cell RNA issynthesizedat afasterrate at37 C. Products in in vivo RNA synthesis. To examine
the
products
of invivo
HRV-2 RNAsynthesis,
the
RNAsynthesized
ininfected cells
waslabeled
with 3H-uridine for
2hr,
starting
5hr
after
infec-tion. The products
wereanalyzed by
centrifuga-tion
through
a sucrosegradient
andby
electro-HRV-2 INFECTED CELLS HOST HELA CELLS
34°C
cpm
3H
37C/
34I
//e
/
_=
I,
0 2 4 6 8 0 2 4 6 8
[image:2.493.264.454.274.472.2]HOURS AFTER INFECTION
FIG. 1. Effect oftemperature onHR V-2 RNA
syn-thesis in vivo. Duplicate sets ofHeLa cells were in-fected in thepresence ofactinomycinD with HRV-2
atamultiplicityof40PFU/cell.3H-uridinewasadded
to each monolayer to a concentration of5 ACi/ml at the time of infection. One series of plates was
in-cubatedat34Cand theotherat37C. At various times
after infection, the total label incorporated into viral
RNA was determined asfollows. Thecellpellet was
suspendedin I mlof0.01 M sodium acetate (pH 5.0)
containing1% SDS, tolyze the cells. Aftercelllysis,
I ml of cold 50% trichloroacetic acid was added to
precipitatethe RNA. Theprecipitatewascollectedona
filterandwashed three times withcold5%
trichloro-acetic acid, and the radioactivity was counted in a
liquid scintillation counterwith Bray's liquid
scintilla-tor solution. Uninfectedcells represent cellularRNA
synthesized in the absence of actinomycin D. (0) RNA synthesizedat 34 C;
(O----)
RNA synthe-sizedat37C.on November 10, 2019 by guest
http://jvi.asm.org/
SYNTHESIS OF HUMAN RHINOVIRUS TYPE 2 RNA
phoresis on polyacrylamide gels. Figure 2 shows the classes of HRV-2-specific RNA when ana-lyzed by electrophoresis on polyacrylamide gel. Peak III is the single-stranded virion RNA since it
comigrates
with the virion RNA and is completelydigested
by pancreatic ribonuclease at 40Ag/ml
in2.25 SSC, (0.34 M NaCl plus 0.34 M sodium
cit-rate).
Peak II is the double-stranded replicativeform
since it
isthe
only peak thatremains
afterribonuclease digestion (Fig. 2). Peak I is
tenta-tively designated
the
double-stranded replicativeintermediate since (i)
itbarely enters the gel,anal-ogous topolio replicative
intermediate
(9); (ii) it is precipitable by 1 M NaCl; and (iii) it disappearsafter ribonuclease digestion.
When
the3H-uridine-labeled
in vivo RNA isanalyzed
by
sedimentation
through a sucrose10,000
_
9,000I
8,000 7,000
'o28S
6,0001
1 RIBOSOMALcpm ,~RNA
[image:3.493.244.446.182.417.2]FRACTION NUMBER
FIG. 2. Composite profile of gel electrophoresis of
HRV-2 RNA. HeLa cells were infected with HRV-2
and viralRNA waslabeledby theaddition of
3H-uri-dine (20
pCi/ml)
for2hrat5 hrafter infection. Theinfectedcellswere collected, lyzed,andthelysatewas
extractedwithphenolat45CasdescribedinMaterials
and Methods. The RNA was analyzed by
electro-phoresis on2.3%polyacrylamide gels (0.8 by 10cm).
Electrophoresis wasfor 16 hrat 4ma/gel. Migration
ofRNA isfrom left toright. (-0-) HRV-2 RNA
frominfected cells; (-o-)HRV-2RNAfrom infected cells treatedfor30 minat37Cwith pancreatic
ribonu-clease (40 MAg/ml) in 0.34m sodium chloride-0.034m
sodium citrate(2.25 X SSC).(--0--)HRV-2virion
RNA.
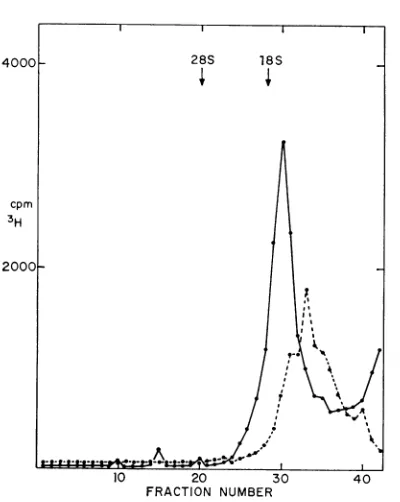
gradient,
two peaks of RNA with relativesedi-mentation values of
18and
28S are obtained(Fig. 3). The 18S is
completely
ribonuclease-resistantwhen the ribonuclease
digestion
is doneafter centrifugation.
If thedigestion
isperformed
prior
tocentrifugation,
however,
theribonuclease-resistant
peak sediments
with a sedimentationvalue
of about
14S.The
28Speak
and asmall
shoulder
at 32Sconsist
mainly of
single-stranded
10 20 30 40
FRACTION NUMBER
FIG. 3. Sucrosegradient analysis ofHR V-2 RNA extractedfrom infected cells. The RNA of infected
cellswaslabeled with 3H-uridine (20 uCi/ml) starling
5 hr after infection. At 7 hr after infection, a
cyto-plasmic extract wasprepared as described in Mate-rials and Methods and was extracted withphenol at room temperature.
Thle
RNA was divided into threeequalportions. Two oftheportions werelayeredonto
two,37-ml, 15 to30% (w/w) linearsucrosegradients (0.01 M Tris [pH 7.2], 0.01 M EDTA, 0.1 M NaCI,
0.2%SDS) andcentrifugedat 20,000 rev/minfor 16 hr at25 Cin aSpinco SW27 rotor. Anotherportion
was treated with pancreatic ribonuclease (40
Mg/imn)
in 2.25 X SSCfor30 min at 37 C. SDS wasadded
to 0.5%, and the RNA was
layered
on a sucrosegradient identical to that for the nontreated RNA. Fractions (1 ml) were collectedfrom the bottom of
the gradient. To eachfraction ofone ofthe untreated gradients 4 mlof2.25 XSSC containing40,gof
pan-creaticribonucleasewasadded. Thefractionswere
incu-batedfor 30 min at 37 C. RNA in allfractions was
precipitated with 20% trichloroacetic acid and col-lected and countedas in Fig. 1.
(-0-)
RNAfrom
infected cell, no ribonuclease treatment;(-O-)
ribonucleasetreatmentaftercentrifugation;
(--0--)
ribonuclease treatment before centrifugation.
VOL. 10, 1972
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.493.49.240.260.514.2]RNA and double-stranded replicative
interme-diateRNA.
Propertiesof the RNA-polymerasecomplex.The isolation of the crude polymerase complex was
described in Materials and Methods. Figure 4
shows the incorporation invitro of3H-GTP into
trichloroacetic acid-insoluble material by various cell fractions. The fraction which contains the cytoplasmic membranes of infected cells is the
only fractionthathassignificantactivity (Fig. 4). The incorporation of 3H-GTP by the polymerase complexislinear for 30minat34C,andtherate ofincorporation of3H-GTPis thesameatpH 7
and 8. The amount of theincorporation of
3H-GTP is a function of the concentration of the
crudepolymerase complex.
Therequirements for 3H-GTP incorporation by the membrane-associatedRNA polymerase
com-plex are shown in Table 1. All four nucleoside
triphosphates and magnesium are required. The
magnesium ion concentration for optimal
reac-tion is 5mm.Manganeseionat4mminthe
pres-10 30 50 10 30 50
TIME(min)
FIG. 4. Time course of RNA synthesis by
polym-erase complex in vitro. Each point represents dupli-cateassays asdescribed in Materials andMethods.
In-jected cells: (-*-) cytoplasmic membrane
frac-lion (crude enzyme); (-O-) detergent wash of
tinu-.clei; (---)cytoplasmic soluble fraction. Uninfected
cells: (-*-) cytoplasmic membrane fraction.
ence
of
5 mm magnesiuminhibits 3H-GTP incor-poration (Table 1).Sedimentation characteristics of detergent-treated RNA polymerasecomplex.
The
polymerase
complexretains
greaterthan90%
of theoriginal
activity
aftersolubilization
ofthemembranes with0.5% DOC. All
the activity
isretained
in the supernatantfraction
after
centrifugation
at30,000
X gfor 20 min. Thisactivity sediments inasucrose
gradient
as abroadpeak
witha sedimen-tation valueof approximately
120S(Fig. 5).RNA products of the in
vitro
polymerase reac-tion. An in vitro reaction wasperformed
with3H-GTP,
and theproducts
were firstextracted
with phenol as described abovethen treated with 0.3 M KOH for 16 hr at 37 C. This results in a 75% loss of trichloroacetic
acid-insoluble
3Hradioactivity indicating
that mostof 3H-GTPwasincorporated into RNA.
Figure 6 shows the in vitro products
of
the crudemembrane-bound
polymerasecomplex after
sedimentation
ofthe
RNAproducts
through
asucrose
gradient. There
is a heterogeneouspeak
from 16 to28S,
with a broadshoulder
greater than 28S. Treatment of the RNA with ribonu-clease beforesedimentation produces
alargepeak
which sediments at about 14S (Fig.6).
Incontrast to the RNA products of the crude polymerase complex, the RNA products of the complex aftersolubilization
with 0.5c% DOCsedimented
asshown in
Fig.
7.There isonlyonepeak
sediment-ing
atabout16S
and no peakat28S. Treatmentof the RNA products with ribonuclease beforecen-trifugation
produces a peak whichsediments
atabout 14S
(Fig.
7),indicating
thatthesolubilized
polymerase complex
is unabletosynthesize
[image:4.493.60.249.307.585.2]repli-cativeintermediate and
single-stranded
RNA.TABLE 1.
Requiremenits
for 3H-GTPinicorporation
by theRNApolymerase complexa
3H1-GTP
Reaction mixture incorporated (counts/min)
Completeb 2,100
-ATP,-CTP, and -UTP 150
-Mg2+ 150
+0.002M Mg2+ 1,600
+0.005 MMg2+ 2,050
+0.010MMg2+ 1,950
+0.020M Mg2+ 1,500
+0.005 MMg2+, 0.004 M Mn2+ 150
aAbbreviations: GTP, guanosinetriphosphate;
ATP, adenosine triphosphate; CTP, cytidine
tri-phosphate; UTP, uridine triphosphate.
b Thecompletesystem isdescribedinMaterials
and Methods.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.493.264.456.488.603.2]SYNTHESIS OF HUMAN RHINOVIRUS TYPE 2 RNA
[image:5.493.41.248.60.302.2]FRACTION NUMBER
FIG. 5. Sedimentationiof RNApolymerase complex.
A 2-ml amount of the crudepolymerase complex was made to 0.5% in sodium deoxycholate and layered onto thefollowing gradient: a 37-ml linear, 7-47% (w/w) sucrose gradient in 0.01 M Tris (pH 7.2)-0.01 M
NaCl. Sedimentationt was performed in a Spinco SW27rotorfor3hr at 37,000rev/min at4 C.
Frac-tions (I ml) were collected from the bottom. A0.5-ml
sample of each fraction was assayed for enzyme
activ-ity. The concentration of each component in the en-zyme assay was the same as thatdescribed in Materials andMethods, but the total assay volume was 1.0 ml. Sedimentation isfrom right to left.
DISCUSSION
Infection of
HeLacells
with HRV-2 causes thesynthesis
ofcell-associated
viral RNA:single-stranded
viralRNA, double-stranded
replicative
form, and the putative
replicative intermediate.The single-stranded RNA from the infected cell has
the
samemigration rate on electrophoresis asthe
single-stranded
RNA isolated from the virion;they
aretherefore
assumed to be identical in size.We have no
evidence
that any cell-associatedsingle-stranded
RNA is larger than that of thevirion
(2).
Invitro
products of the polymerasecomplex when sedimented through a sucrose gradient appear to be more heterogeneous in size than the in vivo products extracts from the in-fected cell. The relative percentage of the
ribo-nuclease-resistant
RNA is higher in the in vitroproducts. Furthermore, the yield of virus and the total
synthesis
of viral RNA in vivo, as measured bythe
uptakeof
3H-uridine,
are lower at 37 C than at 34C; the incorporation of3H-GTP
into20 30
FRACTION NUMBER
FIG. 6. Sucrose gradient analysis ofRNA
synthe-sizedbycrudepolymerase
complex.
To2mlof
crudepolymerase, complex components
of
the assay were addedto give afinal
volumeof
4.0ml. The mixture wasincubatedat 34 Cfor30min then extractedwithphenolat room temperature.
One-half
of
the3HRNA was layered directly onto a sucrosegradient
and analyzedas inFig.3. The otherhalf
wastreated with pancreatic ribonuclease (40pg/ml
in 2.25SSC;
30min; 37 C) layeredonto a sucrose
gradient
andana-lyzedasinFig. 3.
(-*-)
Noribonucleasetreatment; (--0--) ribonuclease treatment.RNA in
vitro
by
thepolymerase
complex
isthe
same at
both
temperatures.If
the in vivo
effectof
temperature isdirectly
on RNAsynthesis,
then
weconclude
that
thein vitro
systemdoes
notaccurately reflect the in vivo
synthesis.
We
have
noevidence
of RNAchain
initiation
norof
release ofsingle-stranded
RNAfrom the
template.
Eitherpoint,
or someother
point,
could be sensitive
totemperature.
The
sedimentation
rateinasucrosegradient
ofthe
double-stranded
RNA is slower if thesedi-mentation is
performed
afterribonuclease
treat-mentrather than
sedimentation
beforeribonucle-ase treatment. A
similar
result isobtained
whenthe
double-stranded
RNA is labeled in vitro with3H-GTP
andtreated
withribonuclease
before and aftersedimentation.
We canspeculate
that the sizeor theconfiguration,
orboth,
of thedouble-stranded
RNA ischanged
after ribonucleasetreatment,
causing
ittosediment
ataslower
rate. The polymerasecomplex
has asedimentation
VOL. 10, 1972
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.493.139.429.65.316.2]FIG. 7. Sucrose gradient analysis of RNA synthe-sized by crude polymerase complex after treatment with sodiumdeoxycholate. Thecrudepolymerase com-plex (2 ml) was made 0.5% in deoxycholate. RNA
synthesis, phenol extraction, sucrosegradient analysis,
and ribonuclease treatment were performed as in
Fig. 3 and 6. (-*-) No ribonuclease treatment;
(- -a- -) ribonuclease treatment.
valueofapproximately120Saftertreatmentofthe membrane fraction with DOC. The polymerase activity is spread over a broad peak, suggesting
that the complexmay be attached to membrane pieces of various sizes; it may contain variable numbers of polymerase molecules and variable numbers ofnascent chains. Wehave no
experi-mentsinwhich theendogenousRNAtemplate isnot required. Clearly, the type of RNA labeled with IH-GTP by the polymerase complex after detergent treatment is different from the RNA
labeled by the complex before detergent
treat-ment. The detergent-treated complexsynthesizes
only double-stranded RNA. Similar results have been observed in other detergent-treated
polym-erasesystems(1). Since therearenucleasespresent
in detergent-treated as well as
non-detergent-treated cytoplasmicextractsofinfected HeLa cells (Yin, unpublisheddata),we canspeculatethatthe
integrity of the membranemay be necessary for
the synthesis of
single-stranded
RNAby somehow protecting it from theexposureofnucleases.
ACKNOWLEDGMENTS
We thank LynnMageeandDiana Faheyfortheir excellent
assistance.
LITERATURE CITED
1. Arlinghaus. R. B., and J. Polatnick. 1969. The isolation oftwo enzyme-ribonucleic acid complexes involved inthe synthe-sis offoot-and-mouth diseasevirusribonucleic acid. Proc.
Nat. Acad. Sci. U.S.A. 62:821-828.
2. Clements, J. B., and S. J. Martin. 1971. Evidence for large
strands of ribonucleic acidinduced bya bovine
entero-virus.J.Gen.Virol. 12:221-232.
3. Ehrenfeld,E., J. V. Maizel, and D. F. Summers. 1970.
Sol-uble RNApolymerase complexfrom polio-infected HeLa
cells. Virology 40:840-846.
4. Hamre, D. 1968. Morphology andChemistry of rhinoviruses,
p. 5-8. In J. L. Melnick (ed.), Monographs in
virol-ogy,vol. 1.S. Karger, New York.
5. Howe,C., L. T. Lee, and0.Ottersena 1969. Slicer foruse
in agar gel electrophoresis analysis of enzymes. Appl. Microbiol. 17:183-184.
6. Loening, U. E. 1967. The fractionation ofhighmolecular
weight ribonucleic acid by polyacrylamide-gel electro-phoresis.Biochem. J. 102:251-257.
7. McCoy, T.A.,M.Maxwell,andP. F. Kruse, Jr. 1959. Amino
acidrequirementsof theNovikoffhepatomaInvitro.Proc.
Soc.Expt.Biol. Med. 100:115-118.
8. Nair, C. N.,and K.K.Lonberg-Holm. 1971. Infectivity and
sedimentation of rhinovirus ribonucleic acid. J. Virol. 7:278-280.
9. Noble, J.,S. J.Kass,and L. Levintow. 1969. Analysis of
polio specific RNA ininfected HeLa cells by
polyacryl-amide gel electrophoresis. Virology 37:535-544.
10.Scherrer, K., and J. E. Darnell, Jr. 1962. Sedimentation characteristics ofrapidlylabeled RNAfrom HeLa ceUls. Biochem. Biophys. Res. Commun. 7:436-449.
11. Stott,E. J.,and G. F. Heath. 1970. Factorsaffecting the
growth ofrhinovirus 2 in suspensioncultures of L132
ceUs.J.Gen.Virol.6:15-24.
12. Tyrrell, D.A.J. 1968. Rhinoviruses,p.67-124. In S.Gard,
C. HaUauer, K F. Meyer (ed.), Virology monographs,
vol. 2,p.67-124. Springer-Verlag, New York.