Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Triggering of Human Parainfluenza Virus 3 Fusion Protein (F) by the
Hemagglutinin-Neuraminidase (HN) Protein: an HN Mutation
Diminishes the Rate of F Activation and Fusion
Matteo Porotto, Matthew Murrell, Olga Greengard, and Anne Moscona*
Department of Pediatrics, Mount Sinai School of Medicine, New York, New York 10029
Received 24 October 2002/Accepted 13 December 2002
For human parainfluenza virus type 3 and many other paramyxoviruses, membrane fusion mediated by the fusion protein (F) has a stringent requirement for the presence of the homotypic hemagglutinin-neuraminidase protein (HN). With the goal of gaining further insight into the role of HN in the fusion process, we developed a simple method for quantitative comparison of the ability of wild-type and variant HNs to activate F. In this method, HN/F-coexpressing cells with red blood cells (RBC) bound to them at 4°C are transferred to 22°C, and at different times after transfer 4-guanidino-neu5Ac2en (4-GU-DANA) is added; this inhibitor of the HN-receptor interaction then releases all reversibly bound RBC but not those in which F insertion in the target membrane or fusion has occurred. Thus, the amount of irreversibly bound (nonreleased) RBC provides a measure of F activation, and the use of fluorescently labeled RBC permits microscopic assessment of the extent to which F insertion has progressed to fusion. We studied two neuraminidase-deficient HN variants, C28a, which has two mutations, P111S and D216N, and C28, which possesses the D216N mutation only. C28a but not C28 exhibits a slow fusion phenotype, although determination of the HNs’ receptor-binding avidity (with our sensitive method, employing RBC with different degrees of receptor depletion) showed that the receptor-binding avidity of C28a or C28 HN was not lower than that of the wild type. The F activation assay, however, revealed fusion-triggering defects in C28a HN. After 10 and also 20 min at 22°C, irreversible RBC binding was significantly less for cells coexpressing wild-type F with C28a HN than for cells coexpressing wild-type F with wild-type HN. In addition, F insertion progressed to fusion more slowly in the case of C28a HN-expressing cells than of wild-type HN-expressing cells. Identical defects were found for P111S HN, whereas for C28 HN, representing the 216 mutation of C28a, F activation and fusion were as rapid as for wild-type HN. The diminished fusion promotion capacity of C28a HN is therefore attributable to P111S, a mutation in the stalk region of the molecule that causes no decrease in receptor-binding avidity. C28a HN is the first parainfluenza virus variant found so far to be specifically defective in HNⴕs F-triggering and fusion promotion functions and may contribute to our understanding of transmission of the activating signal from HN to F.
Attachment of human parainfluenza virus type 3 (HPF3) to the host cell is mediated by the hemagglutinin-neuraminidase envelope protein (HN), which recognizes and binds to sialic acid-containing receptors of the cell surface. The ensuing fu-sion of the viral envelope and the cell membrane, with the consequent release of the nucleocapsid to the cytoplasm, is mediated by the other envelope protein, F (fusion protein). However, HN also plays an essential role here, since for fusion mediated by HPF3 F, there is a stringent requirement for the presence of homotypic HN. The third function of HN in the infection process comes into play after the production of new virions: HN⬘s receptor-cleaving neuraminidase activity ensures the release of these progeny virions and thus the infection of additional cells.
Although the necessity for homotypic HN for fusion medi-ated by the F protein of HPF3 and many other paramyxovi-ruses is well established, the underlying mechanism is not fully understood. One model posits that upon binding to the sialo-side receptors on the cell surface, HN undergoes a conforma-tional change which allows it to interact with F, converting it to
a fusion-active conformational state (11, 22). According to another model, an HN-F complex is formed during trafficking to the cell surface, and upon HN⬘s binding to the receptor, both proteins undergo a conformational change, with the re-sulting disruption of HN-F interaction and release of the fu-sion peptide into the target cell membrane (24, 25). The ability of HN and F to form a protein-protein complex has been demonstrated in several laboratories (6, 24, 25, 27), and it has been reported that HNs with mutations that inhibit complexing with F fail to trigger the F-mediated fusion of the viral enve-lope with the target cell membrane (6). Also, while HN-recep-tor binding is a precondition for fusion (14, 15), it is not clear whether the altered fusion promotion potential of variant HNs stems from quantitative changes in their receptor-binding abil-ity or from the effects of the mutations on other properties of the molecule.
The goal of our current studies is to obtain further insight into the relationship between HN⬘s receptor-binding and fu-sion promotion capacities. We have developed specific tools to allow direct analysis of sequential steps in the fusion process and for quantification of the relative receptor-binding avidity of wild-type and variant HNs. Many of the fundamental as-pects of this process remain to be understood; in particular, how does HN transmit an activating signal to F?
* Corresponding author. Mailing address: Department of Pediatrics, Mount Sinai School of Medicine, 1 Gustave L. Levy Place, New York, NY 10029. Phone: (212) 241-9709. Fax: (212) 860-3316. E-mail: Anne .moscona@mssm.edu.
3647
on November 8, 2019 by guest
http://jvi.asm.org/
The present study focuses on the previously characterized neuraminidase-deficient HPF3 variant C28a, which has wild-type F and two mutations in HN, resulting in a variant virus with undetectable neuraminidase activity (20). The first muta-tion, at residue 216, is partially responsible for the neuramin-idase-deficient phenotype because this is the same (single) mutation in another variant, C28, with neuraminidase activity which is decreased to approximately 30% of the wild-type neuraminidase activity level (9). The second mutation in C28a (P111S) further reduces the neuraminidase activity, resulting in essentially complete loss of neuraminidase activity (20). Al-though C28a is competent in receptor binding, the phenotypic behavior of C28a is indicative of diminished fusion capacity. We have now obtained evidence that the receptor-binding avidity of C28a HN is not lower but somewhat higher than that of wild-type HN but requires more time for F activation and fusion than does wild-type HN. This defect in fusion promo-tion is a direct consequence of the P111S mutapromo-tion, which causes a delay in triggering F.
MATERIALS AND METHODS
Cells and virus.CV-1 (African green monkey kidney) cells were grown in Eagle’s minimal essential medium (Mediatech Cellgro) supplemented withL -glutamine, antibiotics, and 10% fetal bovine serum (Sigma) in 5% CO2. 293T
(human kidney epithelial) cells were grown in Dulbecco’s modified Eagle’s me-dium (DMEM) (Mediatech Cellgro) supplemented with 10% fetal bovine serum and antibiotics. HPF3 wild-type virus stock was prepared by infecting CV-1 cell monolayers at a multiplicity of infection of 0.1 as described previously (12). Variant virus stocks were made in CV-1 cell monolayers from virus that was plaque purified three times (20). Viral titers were determined by plaque assay in CV-1 cells as described previously (12).
Chemical.4-guanidino-neu5Ac2en (4-GU-DANA) was prepared from Re-lenza Rotadisks (5 mg of zanamivir with lactose). A 50 mM stock solution was prepared by dissolving each 5-mg blister capsule in 285l of serum-free medium. Stock solutions were stored at⫺20°C.
Plaque assays and plaque size assessment.Supernatant fluid from infected or mock-infected cells was serially diluted in serum-free medium, and 200l of each serial dilution was added per well to confluent CV-1 cell monolayers in 24-well plates. Cells were incubated at 37°C with intermittent rocking. After 90 min, minimum essential medium containing 0.5% agarose was added to the dishes, and incubation was continued for different time periods (12 to 48 h) at 37°C. After removing the agarose overlay, the cells were fixed with phosphate-buffered saline–2% formaldehyde for 15 min and immunostained for plaque detection as described previously (12). For the purpose of determining plaque area, plaque diameters were measured at 7⫻to 45⫻magnification with a zoom stereo mi-croscope equipped with a micrometer.
HN and F constructs.Mutagenized HN cDNAs were digested withEcoRI and
BamHI and ligated into the digested pEGFP-C3 mammalian expression vector. The pUC19 vector (BD Biosciences Clontech, Palo Alto, Calif.) containing a wild-type F cDNA insert was used as the template DNA for PCR amplification of the wild-type F cDNA with primersXhoI/F/pCAGGS.MCS forward (5⬘-CCC TCGAGGACCATGCCAACCTCAATACTGC) andBamHI/F/pCAGGS.MCS reverse (5⬘-CCCGGATCCTTTGTTTGTTAATACATATGG), designed for li-gation into the pCAGGS.MCS expression vector (18). Positive clones were sent for sequencing to verify the mutations and to ensure that no additional alter-ations had been introduced, as described previously (20).
Transient expression of F and HN genes.Transfections were performed ac-cording to the PolyFect transfection reagent protocol (Qiagen, Valencia, Calif.). Briefly, 293T cell monolayers were seeded into T75 culture flasks (2.4⫻106
cells/flask) 24 h prior to transfection. Medium was removed from the cell mono-layers (40 to 80% confluent) and replaced with 7 ml of fresh 293T cell medium. A transfection mixture containing 8g of HN DNA, 1.3 ml of DMEM, and 80 l of PolyFect reagent was then added to the culture flask and incubated at 37°C. The following day, the cells were trypsinized and lifted from the cell culture flask. Cells were then seeded into 24-well Biocoat plates (Becton Dickson Labware, Bedford, Mass.) at a density of 5⫻105cells/well in 293T medium. Eight hours
later, a transfection mixture containing 0.5g of F DNA, 100l of DMEM, and 20l of PolyFect reagent was added to each well. Two hours later, 10 mU of
neuraminidase was added per well to prevent fusion of the monolayers (16) and to allow all the variant HN molecules, including that of C28a, to bind red blood cells (RBC) at 4°C (20). The cells were used for experiments 18 to 48 h later.
Quantification of cell surface expression of HN and F by ELISA.To quantify the amount of HN expressed on the cell surface of 293T cells, enzyme-linked immunosorbent assay (ELISA) was performed essentially as described previ-ously (2, 4) with the following modifications. Briefly, transfected 293T cells were washed with phosphate-buffered saline after incubation at 37°C and reacted with a mixture of anti-HPF3 HN or F monoclonal antibodies (1:1,000), supplied by Judy Beeler from the World Health Organization repository (in 293T cell com-plete medium.) The cells were left at room temperature for 30 min before being washed three times with medium. Horseradish peroxidase-conjugated anti-mouse immunoglobulin G (IgG) (Pierce) was then added to the cells in phos-phate-buffered saline–1% bovine serum albumin (1:10,000 dilution) and incu-bated for 30 min at room temperature. The cells were washed three times with phosphate-buffered saline-bovine serum albumin before incubation with sub-strate (3,3⬘,5,5⬘-tetramethylbenzidine; Pierce). Absorbance measurements on 200-l aliquots from each culture well were taken at 405 nm on an ELISA reader after adding 50l of H2SO4(Kinetics Reader model EL312e; Bio-Tek
Instru-ments, Winooski, Vt.).
Before each experiment, the surface expression of proteins was determined on all preparations of cells expressing HN to ensure that the expression levels of the proteins in these preparations were within 10% of one another. For the fusion studies shown in Fig. 5A, there was up to 10% variation in the expression levels of the different HNs as well as of the coexpressed F, and the results were therefore normalized to correct for these variations.
RBC release assays.RBC release assays were performed on 293T cells tran-siently expressing various constructs as described above. Cell monolayers were washed three times with serum-free medium and placed at 4°C with 300l of 1% RBC in DMEM for 90 min. The monolayers were then rinsed once with cold DMEM (without phenol red) to remove unbound RBC, and 200l of DMEM (without phenol red), warmed to 37°C, was added to the wells. The plates were then placed at 37°C for various amounts of time. At each time point, the plates were rocked to resuspend released RBC, and the medium was collected into a V-bottomed 96-well plate. The unreleased (bound/fused) RBC were lysed with 180l of RBC lysis solution (0.145 M NH4Cl, 17 mM Tris-HCl) and placed in
a flat-bottomed 96-well plate. Released RBC in 96-well V-bottomed plates were pelleted at 2,500 rpm for 10 min in a Beckman GS-6R centrifuge with a GH 3.8 rotor equipped with a MicroPlus carrier. The supernatant fluid was aspirated, and the RBC were lysed in 50l of distilled water and transferred to a 96-well flat-bottom plate.
Quantitation of RBC was performed as previously described (20). The absor-bance values of the released and bound RBC lysates were read at 540 nm (Kinetics Reader model EL312e; Bio-Tek Instruments, Winooski, Vt.) The two RBC lysates (released and bound) were then combined, and the absorbance values of the total bound RBC were read at 540 nm.
Partial removal of sialic acid receptors from RBC.Partial receptor depletion of RBC was achieved by treatment of 2 ml of a 10% RBC solution in serum-free medium for 2 h at 37°C with 0 to 50 mU ofClostridium perfringensneuraminidase (type X fromC. perfringens, catalog no. N-2133; Sigma Scientific, St. Louis, Mo.). Neuraminidase was then removed by pelleting the RBC, after which the super-natant fluid was aspirated and replaced with serum-free medium. This washing was repeated three times. Each set of RBC were then resuspended in serum-free medium to make final 2% RBC stocks.
Relative receptor-binding avidity of wild-type and variant HNs.RBC with different degrees of receptor depletion were prepared by treatment with various amounts of bacterial neuraminidase as described above. Aliquots of these and control (nondepleted) RBC preparations were added to monolayers of cells expressing wild-type or variant HNs at 4°C, and hemadsorption (HAD) was quantified as described previously (17). Curves obtained by plotting HAD against the degree of RBC depletion served to determine the relative receptor-binding avidity of the different HNs.
Fluorescent labeling of RBC.RBC were labeled with the fluorescent lipid-soluble dye R18, as described previously (3), to assess lipid mixing with HN/F-expressing cells.
RESULTS
Variant that fuses more slowly than wild-type HPF3.In the course of plaquing the neuraminidase-deficient C28a variant of HPF3 (20), we noted that plaques appeared somewhat later and grew more slowly than wild-type HPF3 plaques. This
on November 8, 2019 by guest
http://jvi.asm.org/
nomenon was not exhibited by the C28 variant (10), which has only one of the two mutations harbored by C28a. Quantitative confirmation of these observations is presented in Fig. 1: it may be seen that the area of C28a plaques increased at a slower rate than that of wild-type plaques. On the other hand, C28 plaque growth was similar to or slightly faster than that of the wild type. Since HPF3 plaque formation involves cell-cell fusion and does not depend on the release of progeny virions from the infected cell to neighboring cells, a failure of virion release (resulting from neuraminidase deficiency) was unlikely to ex-plain the slow growth rate of C28a plaques. We therefore examined the question of whether the slow fusion phenotype of C28a was attributable to a defect in HN⬘s ability to trigger F and whether this defect was the direct consequence of the P111S mutation.
[image:3.603.136.453.74.266.2]Release of wild-type and variant HNs from receptor.Before analyzing the ability of wild-type and variant HNs to trigger F (see below), we first needed to study the properties of wild-type and variant HNs in terms of their ability to release from and attach to RBC receptors. The method that we established for comparing the efficiency of the different HNs to release from the receptor is based on the finding that RBC bound to wild-type HN-expressing cells at 4°C can be released upon transfer to 37°C due to the receptor-cleaving function of wild-type HN⬘s neuraminidase activity (17). In the experiment shown in Fig. 2, RBC were allowed to bind to C28a, C28, P111S, and wild-type HN-expressing cells at 4°C, and after washing, the cells were transferred to 37°C; released RBC were then quantified as a percentage of total bound RBC (see the ordinate) at sequential times (abscissa).
FIG. 1. Plaque enlargement of C28a, C28, and wild-type (wt) viruses. The generation of plaques and determination of their areas at the indicated times (abscissa) after infection were done as described in Materials and Methods. The values for plaque areas (ordinate) are means⫾ standard deviation of results for 10 to 20 plaques.
FIG. 2. Release of wild-type, C28a, C28, and P111S HN from receptor. RBC were bound to cells expressing wild-type (wt), C28a, C28, or P111S HN at 4°C. The cells were then transferred to 37°C, and RBC release was quantified as described in Materials and Methods at the indicated times (minutes) (abscissa). The amount of released RBC is given as a percentage of total bound RBC (ordinate).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.603.131.454.516.693.2]It may be seen from Fig. 2 that wild-type HN-expressing cells released virtually all bound RBC between 30 and 120 min, whereas cells expressing the neuraminidase-dead C28a HN showed minimal RBC release throughout the experimental period. However, the curve for C28 HN, with the single-amino-acid change D216N, which is one of the two mutations in C28a, was similar to that for wild-type HN. It thus appears that the partial loss of neuraminidase activity of C28 HN does not cause detectable loss of RBC-releasing ability. On the other hand, the delay in RBC release from cells expressing HN with P111S (the second mutation in C28a) is evident in Fig. 2 from the significantly lower values at both 60 and 90 min for RBC release from P111S HN-expressing cells than from wild-type HN-expressing cells. The results in Fig. 2 thus suggest that the severe loss of C28a HN⬘s RBC-releasing efficiency is due to the synergistic effect of the two mutations in this HN variant. In previous studies, we found that release from the receptor de-pends on HN⬘s receptor-binding avidity as well as on its neur-aminidase activity (17). For this reason as well as because of the postulated role of receptor binding in F triggering, we compared the receptor-binding avidity of the wild-type and variant HNs.
Comparison of receptor-binding avidity of wild-type and variant HNs. Our previous observation that alterations in HN⬘s receptor-binding avidity can be revealed with the use of RBC partially depleted of their surface receptors led to the development of a sensitive method for determining the relative binding avidity of different HNs (17). This method, in which RBC treated with different amounts of bacterial neuramini-dase are used to quantify HAD (for details, see Materials and Methods), was now applied to cells expressing the different HNs studied here. For comparison, the experiments also
in-cluded cells expressing the HN of the ZM1 variant of HPF3, which has previously been shown to exhibit higher receptor-binding avidity than wild-type HN (17).
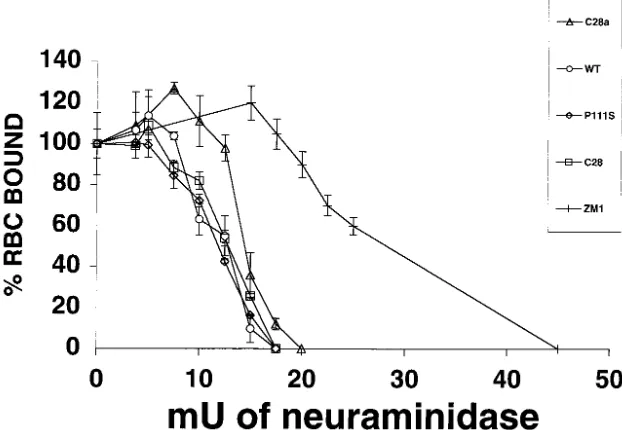
Figure 3 shows the relationship of HAD (ordinate) to the degree of RBC receptor depletion (abscissa). The curve for wild-type HN underwent a much steeper decline than that of ZM1 HN; even at a degrees of RBC receptor depletion at which RBC binding to ZM1 HN remained maximal (abscissa points 17.5 and 20 mU of neuraminidase), wild-type HN RBC binding was already abolished. In contrast, the curves for C28 HN as well as for C28a HN and P111S HN were very similar to the wild-type curve, demonstrating that these variants do not have diminished receptor-binding avidity. In fact, at RBC de-pletions of 10 and 12.5 mU of neuraminidase, the binding values for C28a HN were slightly higher than those for wild-type HN. It thus seems clear that the slow fusion phenowild-type of the C28a variant cannot be attributed to decreased receptor avidity of its HN.
4-GU-DANA releases RBC bound to HN-expressing cells.
[image:4.603.140.451.76.291.2]Our previous studies showed that in cells infected with C28a, the defective release of progeny virions could be overcome not only by neuraminidase supplementation, which destroys the receptors whereby virions remain attached to the host cells’ surface, but also by the addition of 4-GU-DANA, which, by competing with HN for its receptor, prevents attachment itself (20). We thus postulated that the lack of RBC release at 37°C from C28a HN-expressing cells (Fig. 2) could also be over-come by the addition of 4-GU-DANA. The releasing effect of 4-GU-DANA was also tested at 22°C, a temperature at which wild-type neuraminidase activity is low and therefore RBC are retained by cells expressing wild-type as well as neuraminidase-deficient HN.
FIG. 3. Quantitation of HN receptor binding for wild-type and variants. RBC with different degrees of receptor depletion were prepared by treatment with various amounts of bacterial neuraminidase (see abscissa) as described in Materials and Methods. Aliquots of these and control (undepleted) RBC preparations were used to quantify HAD on cell monolayers transfected with constructs expressing wild-type (WT, open circles), C28 (open squares), C28a (open triangles), P111S (open diamonds), or ZM1 (hash mark) HN. The extent of binding of each of the “depleted” RBC is expressed (ordinate) as a percentage of the control (i.e., of the amount of untreated, nondepleted RBC bound on cells expressing the corresponding HN). The points are means of results for triplicate monolayers, with bars denoting standard deviation.
on November 8, 2019 by guest
http://jvi.asm.org/
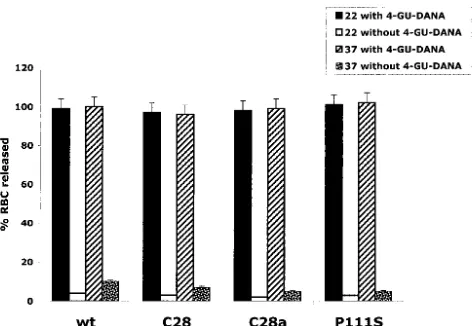
In the experiments of Fig. 4, RBC were allowed to bind to cells expressing wild-type HN, C28a HN, C28 HN, or P111S HN at 4°C, and after washing, the cells were transferred to 22°C or 37°C in the absence or presence of 4-GU-DANA. Fifteen minutes after this transfer, released RBC were quan-tified as in Fig. 2 and expressed as a percentage of total RBC. The results in Fig. 4 show that during these 15 min in the absence of 4-GU-DANA, wild-type HN and C28 express-ing cells released less than 10% and C28a HN or P111S HN-expressing cells released 5% of their bound RBC at 37°C, while at 22°C, at which neuraminidase activity is very low, no appre-ciable RBC release occurred from any of the cells. However, in the presence of 4-GU-DANA, all RBC were released in 15 min from cells expressing wild-type or any variant HN at both 22°C and 37°C.
These results indicate that 4-GU-DANA provides a useful tool for the study (next section) of cells coexpressing F with the different HNs because, while 4-GU-DANA releases all revers-ibly bound RBC (i.e., RBC bound via HN interaction with sialic acid receptors on RBC), we will show that it does not release those in which F insertion or fusion has occurred. Thus, the extent to which RBC remain bound to HN/F-coexpressing cells in the presence of 4-GU-DANA provides a measure of the F-activating potential of the different HNs irrespective of the extent to which their neuraminidase or receptor-binding activity affects RBC release.
Strategy to quantify F activation as a consequence of HN-F interaction.Taking advantage of the findings discussed in the previous section, we developed a simple strategy for quantify-ing F activation that results from HN-F interaction. This strat-egy uses the irreversible attachment of RBC to HN/F-coex-pressing cells as evidence that F has been effectively triggered by HN. Cells expressing HN are transfected with F, and HAD is performed at 4°C. The dishes are then transferred to a temperature, 22°C, high enough for F activation, which may lead to irreversible binding of or fusion with the RBC. 4-GU-DANA, added at various times after transfer to 22°C, will release all reversibly bound RBC, i.e., those bound only via
HN, but not those RBC for which F insertion or fusion has occurred. The quantity of RBC not released by 4-GU-DANA thus provides a measure of F insertion or fusion as a function of time. In order to also assess the extent to which F insertion progressed to fusion, RBC fluorescently labeled with R18 were used, and cells were examined microscopically.
It should be noted that for the experiments described above, neuraminidase was added to the coexpressing cell monolayers (see Materials and Methods) because without this addition, cells coexpressing F with wild-type or C28 HN tend to fuse with one another (thus precluding study of their interaction with RBC) (7). However, while C28a HN is capable of promoting fusion during virus infection, we observed that cells coexpress-ing C28a HN or P111S HN with F showed no or very little fusion even without neuraminidase addition. This observation, as well as the slow fusion phenotype of C28a (Fig. 1), suggested that C28a HN may be less effective or slower in fusion promo-tion rather than incapable of this funcpromo-tion. Examinapromo-tion of this hypothesis necessitated the quantitative method described above for assessing the rate of F triggering and fusion promo-tion by different HNs.
Comparison of F activation mediated by wild-type and vari-ant HNs.With the above-described strategy, we compared F activation mediated by wild-type HN, C28a HN, C28 HN, and P111S HN. Cells expressing one or another of these HNs were transiently transfected with wild-type F, and HAD was per-formed at 4°C. The monolayers were then transferred to 22°C, and 10, 20, or 60 min after transfer, 4-GU-DANA was added. The quantity of RBC that remained irreversibly bound was assayed 15 min later because (see Fig. 4) all reversibly bound RBC are released within 15 min. In Fig. 5A, the amount of irreversibly bound RBC is shown as a function of time at 22°C prior to the addition of 4-GU-DANA. F activation and inser-tion were complete by 20 min in the presence of wild-type HN or C28 HN. However, the values for C28a HN- and P111S HN-expressing cells were much lower at this time. During the shorter period of 10 min, when F activation by wild-type HN and C28 HN was about 50%, the values for C28a HN and P111S HN were again significantly lower. After 60 min at 22°C, the extent of irreversible binding (ordinate) was the same for cells coexpressing type F with any of the variant or wild-type HNs. The data show that C28a HN achieves F activation and irreversible RBC binding at a slower rate than wild-type HN or C28 HN and that this defect is attributable to the P111S mutation in C28a HN.
The RBC in the experiment in Fig. 5 were labeled with the fluorescent dye R18 (see Materials and Methods) so that we could ascertain whether F activation progressed to fusion of the cell membranes. The results of fluorescence microscopy (carried out at the same time as the measurements in Fig. 5A) are shown in Fig. 5B. By 20 min, redistribution of dye into the wild-type HN- and C28 HN-coexpressing cells was evident, indicating fusion. Far less RBC fusion was seen, however, in the C28a and P111S HN-coexpressing cells. Interestingly, cells coexpressing F with wild-type HN or C28 HN showed more fusion after 60 than after 20 min, indicating that even though irreversible binding was already maximal at 20 min (see Fig. 5A), fusion progressed further in the ensuing 40 min. By 60 min, the cells coexpressing C28a HN and P111S HN had at-FIG. 4. Release by 4-GU-DANA of RBC bound to HN-expressing
cells. RBC were bound to cells expressing wild-type (wt), C28a, C28, or P111S HN at 4°C. The cells were then placed at 22°C or 37°C in the presence of 10 mM 4-GU-DANA; released RBC, quantitated 15 min later, are expressed as a percentage of total bound RBC.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.603.47.283.68.231.2]FIG. 5. F activation mediated by wild-type (wt) HN compared with that mediated by C28a HN, C28 HN, and P111S HN. The experimen-tal strategy is described in the text. Cells expressing the indicated HNs and transfected with F were allowed to adsorb RBC at 4°C. They were then transferred to 22°C, and 4-GU-DANA was added either before (0 min) or 10, 20, or 60 min after transfer to 22°C. (A) The amount of RBC that remained bound 15 min after the addition of 4-GU-DANA was determined. These amounts, expressed as a percentage of total bound RBC (ordinate), are shown as a function of time at 22°C prior to the addition of 4-GU-DANA (abscissa). (B) Photographs were taken under a fluorescence microscope of the R18-labeled RBC on cell monolayers at the same time points as those shown in panel A.
on November 8, 2019 by guest
http://jvi.asm.org/
tained wild-type F activation levels (Fig. 5A), and there was significant fusion in these cells as well.
Thus, the data in Fig. 5A and 5B indicate that C28a HN, due to its P111S mutation, is slower than wild-type HN in both F activation and fusion. The results also show the potential of our method for separate examination of two sequential steps, i.e., quantification of F triggering and microscopic assessment of fusion. It is not possible to quantitatively compare the rate of F-triggering in transfected cells (shown in Fig. 5) with the rate of plaque growth in virus-infected cells (shown in Fig. 1) because plaque formation is a multistep process with variations in HN and F expression during viral entry, replication, cell fusion, and plaque enlargement. However, it is apparent that a slowing of the triggering process at the stage of each fusion event could, over time, act to delay plaque enlargement of C28a.
In the case of both simian virus 5 (19) and Newcastle disease virus (23), certain F molecules can mediate fusion in the ab-sence of HN, leading to the proposal that there may be a separate receptor for F (23); however, in both systems, HN enhances F-mediated fusion. It has been shown that, for simian virus 5, the presence of HN serves to decrease the energy requirement for F activation (19, 21). It is thus possible that HPF3 HNs that are more efficient at activation of F would do so at a lower temperature. Since the mutation in C28a-HN is less efficient in activating F at 22°C, we hypothesize that it will increase the temperature requirement for F activation. Future studies will test this hypothesis by comparing the temperature curve for F activation by C28a HN and P111S HN with that of wild-type HN.
DISCUSSION
With the goal of advancing the possibility of dissecting steps in the process whereby HN promotes F-mediated fusion, we developed a simple procedure for quantitatively comparing the F-triggering efficacy of wild-type and variant HNs. Applying this method and one we recently described for obtaining a relative measure of the HNs’ receptor-binding avidity (17), we found that C28a HN is slow in triggering even though its receptor-binding avidity is at least as high as that of the wild type. This triggering defect alone explains the slow fusion phe-notype of the HN variant C28a. While the neuraminidase de-ficiency of C28a HN is a composite result of its two mutations (20), the fusion defect that we found in this study is due entirely to one of these mutations, P111S.
One step in the fusion assay applied here, the release of all reversibly bound RBC by 4-GU-DANA, traces back to our past finding that sialic acid-derived neuraminidase inhibitors like 4-GU-DANA also interfere with the receptor-binding function of HN (8, 12). This dual action was consistent with subsequent crystallographic studies of Newcastle disease virus HN, indicating that a single site provides both the binding and the hydrolytic functions (5). Indeed, both functions are dimin-ished in many Newcastle disease virus HN mutants obtained by site-specific mutations directed to conserved sialic acid-binding site residues (4); the finding that some neuraminidase-deficient mutants had wild-type HAD capability was proposed to be due to the fact that binding operates at lower affinity and does not require as many precise interactions as does catalysis.
As a possible explanation for the enhanced syncytium-form-ing ability of two Newcastle disease virus HN mutants with minimal HAD activity, Connaris et al. (4) suggest the interest-ing possibility that these mutations evoked structural changes in HN that mimicked those which occur upon receptor binding and enable HN to activate F. In other studies, site-specific mutations directed to heptad repeat residues in the stalk re-gion (6, 22, 25), the transmembrane rere-gion (13), or the hydro-phobic surface region (26) of Newcastle disease virus HN yielded some mutants which had no HAD or neuraminidase defect but altered ability to promote fusion, as judged by syn-cytium formation. However, the assays of synsyn-cytium formation suffer from low resolving power and quantification difficulties (1). In addition, the standard HAD assay (with intact RBC) reveals only major differences in receptor binding, so one can-not exclude the possibility that the formation of syncytia larger or smaller than wild-type syncytia is the consequence of in-creased or dein-creased receptor-binding avidity. The method we now used permits, for HPF3 at least, quantitative comparison of the fusion promotion efficacy of HN as well as the receptor-binding avidity of different HNs. Thus, C28a HN, which causes F triggering and fusion with RBC at a slower rate than the wild type, is demonstrated here to be a paramyxovirus HN variant whose fusion promotion defect is clearly not due to decreased receptor-binding avidity.
For Newcastle disease virus, the diminished receptor-bind-ing function of some neuraminidase-deficient HN variants has been attributed to the attachment of oligosaccharides to the HN molecules, and their removal by exogenous neuraminidase treatment or site-directed mutagenesis was found to restore the receptor-binding function (10). Oligosaccharide attach-ment to HN of the HPF3 variant C28a may conceivably un-derlie our observation that cells expressing C28a HN fail to adsorb RBC at 4°C unless pretreated with neuraminidase (20). If so, then oligosaccharides become detached at 22°C or 37°C, since C28a-HN requires no neuraminidase pretreatment for HAD at these temperatures (20). However, our fusion assay requires that HN/F-coexpressing cells bind RBC at 4°C, and this was one of the reasons for pretreating the cells with bac-terial neuraminidase. Second, HN-expressing cells transfected with F can fuse with one another (thus precluding study of their fusion with RBC), and we found that this difficulty can also be overcome by the addition of neuraminidase prior to transfer of the cells to the temperatures required for fusion.
While considerable advances have been made in recent years in the understanding of structural changes in Fp required to initiate fusion (21), the question of how HN transmits an activating signal to F is largely unanswered. Insight into this process may be obtained with the aid of the procedure that we developed for quantitative assessment of F triggering by dif-ferent HNs and for distinguishing the triggering efficiency of different HNs from their receptor-binding and neuraminidase activities. This method, the irreversible attachment of RBC to HN/F-expressing cells, was made feasible by the fact that RBC in which F insertion or membrane fusion has occurred remain attached after the addition of 4-GU-DANA, while all other RBC are rapidly released by this agent irrespective of the different HNs’ receptor binding and neuraminidase activities. We have here identified a mutation in HN that can lead directly to a defect in triggering F. Previous studies, mentioned
on November 8, 2019 by guest
http://jvi.asm.org/
above (4, 6, 13, 22, 25, 26), identified mutations that alter syncytium formation without affecting HAD or neuraminidase, suggesting that fusion promotion can be separated from the other properties of paramyxovirus HNs. By assessing F trig-gering specifically, we now show that the defect caused by the P111S mutation is an F triggering deficiency and can, under the experimental conditions that we used, be definitively separated from other related properties of HN.
The delay in triggering F may be due to slower induction of the conformational change required to mediate fusion. If HN-F interaction occurs only at the cell surface following bind-ing of HN to sialic acid and this interaction induces F’s active state (21), then the triggering-defective P111S HN may have diminished interaction with F and thereby require a longer period of contact with its receptor to accomplish the necessary change. If, alternatively, constitutive HN-F interaction occurs intracellularly and inhibits F and binding of HN to sialic acid disrupts HN-F interaction (24), then P111S HN may interact more strongly with F or be less effective at releasing F. If HPF3 HN serves to decrease the energy requirement for F activation, as for simian virus 5 (19), then the P111S HN may require more energy for F activation and thereby require a higher temperature than wild-type HN to trigger F, and this question is under investigation.
Future studies to look at the physical interaction of the P111S HN with F (compared to the interaction of wild-type HN and C28 HN with F) should help us to address the ques-tion of whether HN⬘s receptor attachment at the cell surface leads to HN-F interaction, thus triggering F, or, conversely, whether HN and F interact during transit to the cell surface and HN⬘s receptor attachment at the cell surface leads to the release of F from an HN-F complex. If direct interaction be-tween HN and F in coexpressing cells occurs at the cell surface and it is HN⬘s receptor attachment that triggers F (21), then the HN-F interaction will be positive only upon addition of receptor donor cells. Observing a diminished interaction of P111S HN with F (compared to wild-type HN) upon addition of receptor donor cells to HN/F-expressing cells would support this model. On the other hand, if intracellular HN-F interac-tion inhibits F, then adding receptor donor cells should disrupt the HN-F interaction. Finding that the P111S HN interacts as well as or more strongly with F than wild-type HN would support the model in which HN-receptor binding leads to release of F from a complex (24).
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI 31971 to A.M. from the National Institutes of Health.
We thank Natalya Kanovich for excellent technical assistance and Thomas Weber for helpful discussions.
REFERENCES
1. Bagai, S., and R. Lamb.1995. Quantitative measurement of paramyxovirus fusion: differences in requirements of glycoproteins between simian virus 5 and human parainfluenza virus 3 or Newcastle disease virus. J. Virol.69: 6712–6719.
2. Bousse, T., T. Takimoto, W. Gorman, T. Takahashi, and A. Portner.1994. Regions on the hemagglutinin-neuraminidase proteins of human parainflu-enza virus type-1 and Sendai virus important for membrane fusion. Virology 204:506–514.
3. Chernomordik, L. V., V. A. Frolov, E. Leikina, P. Bronk, and J. Zimmerberg. 1998. The pathway of membrane fusion catalyzed by influenza hemaggluti-nin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J. Cell Biol.140:1369–1382.
4. Connaris, H., T. Takimoto, R. Russell, S. Crennell, I. Moustafa, A. Portner, and G. Taylor.2002. Probing the sialic acid binding site of the hemaggluti-nin-neuraminidase of Newcastle disease virus: identification of key amino acids involved in cell binding, catalysis, and fusion. J. Virol.76:1816–1824. 5. Crennell, S., T. Takimoto, A. Portner, and G. Taylor.2000. Crystal structure
of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol.7:1068–1074.
6. Deng, R., Z. Wang, P. J. Mahon, M. Marinello, A. Mirza, and R. M. Iorio. 1999. Mutations in the Newcastle disease virus hemagglutinin-neuramini-dase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion. Virology253:43–54.
7. Dutch, R., S. Joshi, and R. Lamb.1998. Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: com-parison of fusion reactions mediated by simian virus 5 F, human parainflu-enza virus type 3 F, and influparainflu-enza virus HA. J. Virol.72:7745–7753. 8. Greengard, O., N. Poltoratskaia, E. Leikina, J. Zimmerberg, and A.
Moscona.2000. The anti-influenza virus agent 4-GU-DANA (zanamivir) inhibits cell fusion mediated by human parainfluenza virus and influenza virus HA. J. Virol.74:11108–11114.
9. Huberman, K., R. Peluso, and A. Moscona.1995. The hemagglutinin-neura-minidase of human parainfluenza virus type 3: role of the neurahemagglutinin-neura-minidase in the viral life cycle. Virology214:294–300.
10. Iorio, R. M., G. M. Field, J. M. Sauvron, A. M. Mirza, R. Deng, P. J. Mahon, and J. P. Langedijk.2001. Structural and functional relationship between the receptor recognition and neuraminidase activities of the Newcastle disease virus hemagglutinin-neuraminidase protein: receptor recognition is depen-dent on neuraminidase activity. J. Virol.75:1918–1927.
11. Lamb, R.1993. Paramyxovirus fusion: a hypothesis for changes. Virology 197:1–11.
12. Levin Perlman, S., M. Jordan, R. Brossmer, O. Greengard, and A. Moscona. 1999. The use of a quantitative fusion assay to evaluate HN-receptor inter-action for human parainfluenza virus type 3. Virology265:57–65. 13. McGinnes, L., T. Sergel, and T. Morrison.1993. Mutations in the
transmem-brane domain of the HN protein of Newcastle disease virus affect the struc-ture and activity of the protein. Virology196:101–110.
14. Moscona, A., and R. W. Peluso.1991. Fusion properties of cells persistently infected with human parainfluenza virus type 3: participation of hemagglu-tinin-neuraminidase in membrane fusion. J. Virol.65:2773–2777. 15. Moscona, A., and R. W. Peluso.1993. Relative affinity of the human
para-influenza virus 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J. Virol.67:6463–6468.
16. Moscona, A., and R. W. Peluso.1992. Fusion properties of cells infected with human parainfluenza virus type 3: receptor requirements for viral spread and virus-mediated membrane fusion. J. Virol.66:6280–6287.
17. Murrell, M., M. Porotto, T. Weber, O. Greengard, and A. Moscona.2003. Mutations in human parainfluenza virus type 3 HN causing increased recep-tor binding activity and resistance to the transition state sialic acid analog 4-GU-DANA (zanamivir). J. Virol.77:309–317.
18. Niwa, H., K. Yamamura, and J. Miyazaki. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene108:193– 199.
19. Paterson, R. G., C. J. Russell, and R. A. Lamb.2000. Fusion protein of the paramyxovirus simian virus 5: destabilizing and stabilizing mutants of fusion activation. Virology270:17–30.
20. Porotto, M., O. Greengard, N. Poltoratskaia, M.-A. Horga, and A. Moscona. 2001. Human parainfluenza virus type 3 HN-receptor interaction: the effect of 4-GU-DANA on a neuraminidase-deficient variant. J. Virol.76:7481– 7488.
21. Russell, C. J., T. S. Jardetzky, and R. A. Lamb.2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 20:4024–4034.
22. Sergel, T., L. W. McGinnes, M. E. Peeples, and T. G. Morrison.1993. The attachment function of the Newcastle disease virus hemagglutinin-neuramin-idase protein can be separated from fusion promotion by mutation. Virology 193:717–726.
23. Sergel, T. A., L. W. McGinnes, and T. G. Morrison.2000. A single amino acid change in the Newcastle disease virus fusion protein alters the require-ment for HN protein in fusion. J. Virol.74:5101–5107.
24. Stone-Hulslander, J., and T. G. Morrison.1997. Detection of an interaction between the HN and F proteins in Newcastle disease virus-infected cells. J. Virol.71:6287–6295.
25. Stone-Hulslander, J., and T. G. Morrison.1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol.73:3630–3637.
26. Takimoto, T., G. L. Taylor, H. C. Connaris, S. J. Crennell, and A. Portner. 2002. Role of the hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J. Virol.76:13028–13033.
27. Yao, Q., X. Hu, and R. W. Compans.1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol.71:650–656.