Molecular characterisation of genes involved in iron homeostasis.
192
0
0
Full text
(2) Vellore-632004, April 21, 2014. DECLARATION. I hereby declare that the thesis entitled “Molecular characterisation of genes involved in iron homeostasis” is based on the results of the work carried out by me for the degree of DOCTOR OF PHILOSOPHY under the supervision of Dr.Poonkuzhali.B, Department of Haematology, Christian Medical College, Vellore. This work has not formed the basis of any associate ship, fellowship, degree or diploma of any other University.. Rekha.A Department of Haematology, Christian Medical College, Vellore Candidate. ii.
(3) Vellore-632004, April 21, 2014. CERTIFICATE This is to certify that the thesis entitled “Molecular characterisation of genes involved in iron homeostasis” is based on the results of the work carried out by Ms. Rekha.A for the degree of DOCTOR OF PHILOSOPHY under my supervision. This work has not formed the basis of any associate ship, fellowship, degree or diploma of any other University. This thesis represents independent research carried out by the candidate, under my supervision.. Dr.Poonkuzhali Balasubramanian Professor, Department of Haematology, Christian Medical College, Vellore Supervisor and Guide. iii.
(4) Vellore-632004, April 21, 2014. CERTIFICATE. This is to certify that Ms.Rekha.A has completed her requirements for the submission of her Ph.D. thesis entitled “Molecular characterisation of genes involved in iron homeostasis”. This thesis is a bonafide record of the work carried out by the candidate during the period of study under my guidance, and it has not formed the basis of any associate ship, fellowship, degree or diploma of any other University. This thesis represents independent work carried out by the candidate, under my supervision.. Dr. Eunice Sindhuvi Edison, Associate Professor, Department of Haematology, Christian Medical College Vellore Co-Guide.. iv.
(5) ACKNOWLEDGEMENTS. First and foremost, I thank almighty God for all his blessings, wisdom and knowledge to accomplish this endeavour in my life. I would like to express my sincere gratitude to my Guide Dr.Poonkuzhali Balasubramanian for her guidance and support all through the study. I am deeply indebted and thankful to my mentor and co-guide Dr.Eunice Sindhuvi.E for her tireless support, priceless care, encouragement and guidance which made this thesis possible. I would like to thank the head of the department Dr.Alok Srivastava for his extensive support and invaluable comments helped to improve and complete the study. I would like to acknowledge my sincere thanks to Dr. Mammen Chandy who initiated this project and supported tremendously. My special thanks to Dr.Shaji RV and Dr.G.Jayandharan for their valuable advice and necessary help. I would especially like to thank Dr. Vikram Mathews, Dr. Biju George, and Dr.Auro Viswabandya, Dr.Aby Abraham and all other clinicians, nurses in the department for their support for this study. I would like to express my appreciation to Dr. Vinod Abraham, Dr. Daisy Singh and Ms.Kalaiselvi (Research nurse) of Community Health centre for their support. I also owe a debt of gratitude to Dr. Victoria, Dr. Joe Fleming, Dr. Arun Jose and Mr.Joseph of Department of Clinical Biochemistry, Mr. Aaron Chapla of Department of Molecular Endocrinology and Dr. Mayka Sanchez of IMPPC, Spain for their support and help to complete the study successfully. I would like to extend my heartfelt warm thanks to my dear friends and lab mates for their help, support, inspiration and memorable moments to cherish always.My special thanks to Ms.Divya.G project student for her help in this study. I gratefully acknowledge Ms.Kavitha (Biostatistician), Mr. Sam, Mrs.Sankari ,Mrs. Rekha Singh, Mr.Benjamin, Mr. Joel and Mr. Srinivasan for their immense help over these years. I express my sincere thanks to CSIR for supporting me with fellowship and DBT for financial support for this study. Words are not enough to express my deep gratitude to my dear parents and sisters who unconditionally supported and inspired all my life.. v.
(6) The Great questions of the day will not be settled by means of speeches and majority decisions but by iron and blood Otto Von Bismarck. vi.
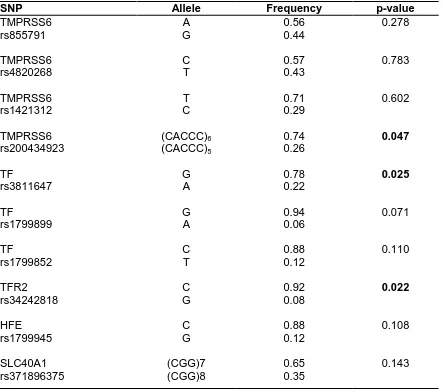
(7) TABLE OF CONTENTS 1. INTRODUCTION 2. REVIEW OF LITERATURE 2.1 Overview 2.2 Historical perspective of anemia and iron deficiency 2.3 Iron: Properties and Functions 2.4 Distribution and storage of iron 2.5 Iron absorption and transport 2.6 Placental iron transport 2.7 Iron and brain 2.8 Proteins involved in iron absorption and transport 2.8.1 Divalent metal transporter protein 1 (DMT1) 2.8.2 Duodenal cytochrome b reductase 1(DCYTB) 2.8.3 Heme carrier protein (HCP-1) 2.8.4 Hephaestin (HEPH) 2.8.5 Ferroportin (FPN1) 2.8.6 Transferrin (TF) 2.8.7 Ceruloplasmin (CP) 2.8.8 Six transmembrane epithelial antigen of prostate 3 (STEAP3) 2.9 Iron uptake and transferrin receptors 2.10 Intracellular iron storage: Ferritin and hemosiderin 2.11 Erythropoiesis and Iron 2.12 Mitochondria and heme synthesis 2.13 Reticuloendothelial system and iron recycling 2.14 Heme and its biological role 2.15 Importance of iron-sulphur cluster 2.16 Cytoprotective enzymes and antioxidants 2.17 Cellular iron homeostasis 2.18 Hepatic iron metabolism and proteins 2.19 Iron sensors and regulatory proteins 2.19.1 Hereditary hemochromatosis protein (HFE) 2.19.2 Hepcidin (HAMP) 2.19.3 Identification of hepcidin 2.19.4 Hepcidin Regulation 2.19.5 TMPRSS6 (Transmembrane Serine Protease 6) 2.19.6 Hemojuvelin (HJV) 2.19.7 Growth differentiation factor 15 (GDF15) 2.20 Systemic iron homeostasis 2.21 Dysregulation of iron homeostasis 2.21.1 Iron deficiency anemia (IDA) 2.21.1.1 Etiology of iron deficiency anemia. 2 5. 8. 12. 17 19. 27 30. 38 41. vii.
(8) 2.21.1.2 Stages of iron deficiency and diagnosis 2.21.1.3 Treatment 2.22 Inherited Disorders of Iron Metabolism 2.22.1 Iron Refractory Iron Deficiency Anaemia (IRIDA) 2.22.2 Hypotransferrinemia/Atransferrinemia 2.22.3 Aceruloplasminemia 2.22.4 Microcytic anemia with DMT1 mutation 2.22.5 Disorders of heme synthesis 2.22.6 Sideroblastic anemias 2.23 Genome wide association studies (GWAS) 2.24 Hereditary hemochromatosis (HH) 2.25 Secondary iron overload 2.25.1 Beta thalassemia 2.25.2 Pathophysiology and iron overload 2.25.3 Therapeutic options 2.26 International and National Status. 45. 50 51 53. 56. 3. AIMS AND OBJECTIVES. 59. 4. PATIENTS AND METHODS. 61. 4.1 Patients 4.1.1 Iron deficiency anemia (IDA) 4.1.2 Iron deficiency anemia in pregnancy (IDAP) 4.1.3 Hereditary hemochromatosis (HHC) 4.1.4 β-thalassemia major 4.1.5 Controls 4.1.6 Sample size calculation 4.2 Study material 4.3 Oral iron absorption test (OIAT) 4.4 Analysis of Haematological parameters 4.5 Analysis of Biochemical parameters 4.5.1 Measurement of serum Hepcidin 4.5.2 Measurement of serum GDF15 4.5.3 Measurement of transferrin by ELISA 4.6 Molecular Genetic Analysis 4.6.1 DNA extraction 4.6.2 RNA extraction 4.6.3 DNA and RNA Quality and Quantification 4.6.4 Reticulocyte isolation 4.6.5 Polymerase chain reaction (PCR) 4. 7 Genotyping 4.7.1 Restriction Fragment Length polymorphism (RFLP) 4.7.2 DNA Fragment analysis by capillary electrophoresis. 63. 64. 66. viii.
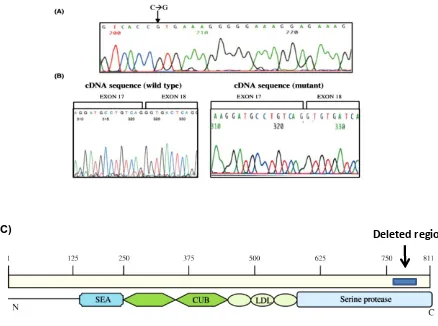
(9) 4.7.3 High resolution melting analysis (HRM) 4.7.4 Reverse dot blot (RDB) 4.7.5 Conformation sensitive gel electrophoresis (CSGE) 4.7.6 DNA sequencing by Sanger method 4.7.7 Targeted Resequencing by Ion Torrent™ Next-Generation Sequencing 4.7.8 Insilico analysis 4.8 Gene expression analysis 74 4.8.1 cDNA synthesis and RQ-PCR 4.8.2 Western blotting 4.8.3 Immunofluorescence 4.9 In vitro and ex-vivo experiments 76 4.9.1 Primary erythroid culture 4.9.2 Cell culture 4.9.3 Iron chelation 4.9.4 Expression of iron genes during erythroid differentiation 4.9.5 In vitro transferrin assay 4.9.6 siRNA transfection 4.10 Statistical analysis 78 5. RESULTS 80 5.1 Iron deficiency anaemia (IDA) 5.1.1 Characteristics of study group 5.1.2 Haematological and biochemical analysis in IDA 5.1.3 SNP genotyping 5.1.4 Identification of variants associated with poor iron absorption 5.2 Identification of mutations in patients with atypical microcytic anemia with normal or high ferritin 87 5.2.1 IRIDA (Iron Refractory Iron Deficiency Anemia) 5.2.1.1 Clinical and biochemical profile 5.2.1.2 Mutation analysis 5.2.1.3 Characterisation of mutation 5.2.2 Hypotransferrinemia/Atransferrinemia 5.2.2.1 Patients’ characteristics 5.2.2.2 Mutation screening 5.2.2.3 Insilico analysis 5.2.2.4 Structural modelling of transferrin protein 5.2.2.5 Invitro transferrin assay 5.2.3 Aceruloplasminemia 5.2.4 Sideroblastic anemia 5.2.4.1 Patients’ clinical profile 5.2.4.2 Mutation analysis 5.2.4.3 Characterisation of promoter mutation in ALAS2. ix.
(10) 5.3 Iron deficiency anemia in pregnancy (IDAP) 5.3.1 Study cohort details 5.3.2 Haematological and biochemical analysis 5.3.3 Response to oral iron supplements 5.3.4 Factors influencing response to oral iron supplements 5.3.5 Role of genetic variants in response to iron supplements 5.3.6 Identification of genetic variants in subjects with poor response 5.4 Hereditary hemochromatosis (HH) 5.4.1 Patients’ profile 5.4.2 Mutation analysis 5.4.3 Haplotype analysis 5.4.4 Invitro analysis of hepcidin induction 5.5 Secondary iron overload in β-thalassemia 5.5.1 Patients’ characteristics 5.5.2 Biochemical profile 5.5.3 Factors influencing ferritin levels 5.5.4 Effect of genetic variants on ferritin and hepcidin. 98. 106. 112. 5.5.5 Analysis of GDF15 polymorphisms in-thalassemia 5.6 Expression analysis of genes involved in iron metabolism and regulation 117 5.6.1 Effect of iron deficiency on iron uptake, efflux and regulatory genes in erythropoiesis 5.6.2 Effect of iron deficiency on iron transport and regulatory genes in pregnancy 5.6.2.1 Effect of iron deficiency on placental gene expression 5.6.3 Expression of iron related genes during ineffective erythropoiesis in β thalassemia 5.6.3.1 Exvivo erythropoiesis and analysis of iron related genes 5.6.3.2 Crosstalk of p53 and GDF15 in β thalassemia major 6. DISCUSSION 130 7. SUMMARY AND CONCLUSIONS. 147. 8. IMPACT OF THE STUDY. 150. 9. BIBLIOGRAPHY 10. APPENDIX 11. PUBLICATIONS. x.
(11) ABBREVIATIONS IDA IDAP HH IRIDA XLSA WHO Fe2+ Fe3+ Hb TF CP CRP TIBC GDF15 GWAS SNP OIAT NTBI DMT1 HCP1 FPN1 TFRC IRE IRP STEAP3 ALAS2 HFE HAMP TMPRSS6 HRM CSGE RDB SIFT Polyphen2 DFO. -. Iron deficiency anemia Iron deficiency anemia in pregnancy Hereditary hemochromatosis Iron refractory iron deficiency anemia X linked sideroblastic anemia World health organisation Ferrous iron Ferric iron Haemoglobin Transferrin Ceruloplasmin Cellular reactive protein Transferrin iron binding capacity Growth differentiation factor 15 Genome wide association studies Single nucletide polymorphism Oral iron absorption test Non transferrin bound iron Divalent metal transporter 1 Heme carrier protein 1 Ferroportin 1 Transferrin receptor 1 Iron responsive element Iron regulatory protein Six transmembrane epithelial antigen of prostate 3 Aminolevulinic acid synthase2 Hereditary hemochromatosis protein Hepatic antimicrobial peptide Transmembrane serine protease 6 High resolution melting analysis Conformation sensitive gel electrophoresis Reverse dot blot Sorting intolerant from tolerant Polymorphism phenotyping 2 Desferrioxamine. xi.
(12) INTRODUCTION. 1.
(13) CHAPTER 1 INTRODUCTION Iron is virtually an indispensable micronutrient required by all living organisms; it has a major role in fundamental biological processes such as haemoglobin synthesis, oxygen transport, cellular respiration and cell division. However, excess iron can cause tissue damage by catalysing the synthesis of free-radical ions that damage cellular membranes, proteins, lipids and DNA. Excess circulating iron in patients with primary and secondary iron overload leads to abnormal iron accumulation in vital organs such as liver, spleen and heart, leading ultimately to cirrhosis, cardiac dysfunction, arthropathy, gonadal insufficiency, and diabetes. On the other side iron deficiency induces anemia, reducedworking capacity and motor activity, reduced lymphocyte activity and immune dysfunction (1). Iron deficiency anemia (IDA) in pregnancy (IDAP) has been implicated as a risk factor for preterm delivery, low birth weight, perinatal mortality and morbidity. Therefore cellular and systemic iron homeostasis is crucial and has to be tightly regulated. Disruption of any of these regulatory mechanisms will lead to either iron deficiency or overload. Iron deficiency is one of the leading risk factor for anemia affecting an estimated 2 billion people worldwide with the highest prevalence in India constituting around 74.3% of the population. IDA affects nearly 58% of pregnant women, 50% of non-pregnant nonlactating women, 56% adolescent girls, 30% adolescent boys and around 80% in children below two years of age in India (2).The aetiology of IDA has been mainly attributed to socioeconomic factors in India. In spite of addressing these etiologic factors, there are still a fraction of patients in whom the cause for IDA is unknown. In pregnancy, a proportion of women remain anaemic even after iron supplementation. In 2008, Finberg et al., characterised the genetic basis of congenital hypochromic, microcytic anemia termed as IRIDA (iron refractory iron deficiency anemia) and recent advances in. genome wide. association studies and haplotype analysis have demonstrated the role of genetic components in determining the iron status and susceptibility to IDA (3–7). The available data from previous studies signify the possible association of genetic modifiers which probably influence iron deficiency in India. Even though India continues to be one of the countries with the highest prevalence of IDA, no genetic studies have been carried out to the best of our knowledge. Hereditary hemochromatosis (HH) is an autosomal recessive disorder of iron metabolism characterised by increased iron absorption and deposition in various organs. If left undiagnosed and untreated, iron overload can be fatal. Early detection and treatment of 2.
(14) iron overload and hemochromatosis can delay or prevent irreversible complications and prolong life. It is the best studied of the primary iron overload syndromes that have been attributed to genetic variants in genes of iron metabolism. Primary iron overload due to HFE (Hemochromatosis gene) mutation is rare in India and the molecular basis of HH has not been well characterised (8). Screening based on common HFE mutation alone cannot exclude the involvement of other genes and mutations in unexplained iron overload in our population. β- thalassaemia is a monogenic disease caused by absent or inadequate production of β globin. India carries a huge burden (10%) of world’s thalassaemia patients with a carrier frequency of 3.3% in the subcontinent. Transfusions remain the mainstay of treatment for thalassemia major. Iron overload due to frequent transfusions and ineffective erythropoiesis is a major cause of morbidity in patients with β-thalassemia. It is quite distinct from primary iron overload in various aspects. There is little understanding of the inter-individual variations and the molecular events that occur during transfusional iron overload. Genetic variations in iron regulating genes such as HFE, hepcidin, ferroportin have been identified as genetic modifiers of iron overload in thalassaemic patients (9–12). Hepcidin plays a central regulatory role in response to body iron store and it is not well studied in transfusion dependent thalassaemic patients. Elucidating the role of genetic variants and hepcidin influence on iron metabolism may help to explain heterogeneity in iron overload. Previous studies have provided new insights into molecular mechanisms of iron deficiency and iron overload through identification of new genes. Genome-wide and candidate gene studies have associated genetic variants with iron status. However there is lack of comprehensive analysis of genetic variants which are associated with IDA and response to iron supplements. Role of genetic variants in modulating secondary iron overload is also not well understood. Therefore the present study was designed to identify the possible role of modifiers and association of known genetic variants in disorders of iron homeostasis such as IDA and hemochromatosis in India. Identification of genetic profiles and functional characterization will definitely enhance our understanding of iron homeostasis as well as help in proper diagnosis and better patient management.. 3.
(15) REVIEW OF LITERATURE. 4.
(16) CHAPTER 2 REVIEW OF LITERATURE 2.1 Overview Iron is an essential micronutrient required by all living organisms for fundamental processes involved in growth and survival. Iron is a d-block transition element at 26th position in periodic table exhibits various oxidation states ranging from -2 to +6. Its unique physico-chemical properties permit it to act as an important catalyst and cofactor mediating key fundamental processes in our biological system. Iron is an essential component for large number of proteins and enzymes principally in haemoglobin, an important heme containing protein involved in oxygen transport. Heme and iron forms an integral part of many other enzymes such as cytochromes, NADH dehydrogenase, succinate dehydrogenase in electron transport and energy metabolism; catalases and peroxidases in antioxidant system, ribonucleotide reductase in DNA synthesis and prolyl hydroxylase in oxygen sensing. The amount of total body iron is exquisitely balanced to prevent its deficiency and excess or else it will lead to anemia or iron overload which adversely affects the quality of life. Iron deficiency anemia is a common nutritional health problem affecting around two billion people worldwide. The major causes of iron deficiency anemia include increased demand during growth and pregnancy, increased loss due to bleeding and reduced bioavailability associated with malabsorption. In early 19th century Ferro kinetic studies provided new insights into the iron homeostasis in in vivo systems. With the invent of advanced techniques in molecular biology and genetics, present knowledge of iron homeostasis is dramatically increased and helped us to understand its critical role in pathophysiology of iron related disorders. The importance of iron in biology and its functions, regulation, and impact on various pathophysiological states will be reviewed in subsequent chapters. 2.2 Historical perspective of anemia and iron deficiency First prescription of iron was dated back to 1500 B.C in an Egyptian Medicine book “Ebers papyrus”. This scroll consists of approximately 700 medicinal formulas and remedies since then iron supplements were used unwittingly to cure fatigue or weakness. In Greek history, iron was considered as a ‘miracle drug’ to cure anemia and its discovery was quite amusing. Iron deficiency anemia was common among girls after puberty and named this condition as ‘”green sickness” or “chlorosis” because of their pale appearance with greenish yellow tint (1). In the late 1600s Thomas Sydenham of London discovered iron as a remedy for chlorosis and named as steel tonic. In 1893 Ralph Stockman from School of Medicine 5.
(17) Edinburgh showed the raise of haemoglobin levels in chlorotic women after treatment with iron. He also demonstrated that chlorotic girls were getting 1.3 - 3.4 mg of iron per day against the normal requirement of 9mg/day which is not enough to replace their daily losses. Simple iron supplements were available for medical purposes soon after these discoveries and iron deficiency anemia was detected by measuring haemoglobin levels in blood. In 1932 William B Castle demonstrated the use of parenteral iron in patients with hypochromic anemia and apparently showed there is no distinct advantage of parenteral iron administration compared to oral iron (13). It was also shown that there is an influence and difference in the incidence of anemia in terms of age and sex (14). Understanding of iron absorption and utilisation was almost complete approximately 50years back; illustrated in a review by Hugh W Josephs, suggesting that iron metabolism was a long sought out story but still elusive in many aspects, driving researchers to explore expanding field of iron biology (15). 2.3 Iron-Properties and Functions Iron is fairly reactive metal due to its incompletely filled d orbitals (d6) which can exists in two predominant states of oxidation Fe (II) and Fe (III).Iron in the earth crust appears as different minerals such as hematite (Fe2O3), Magnetite (Fe3O4), siderite (FeCO3) and iron pyrite (FeS2).The preferred biologic ligands for iron are oxygen, nitrogen and sulphur atoms. Fe3+ behaves as a hard acid with preference to ligands like hydroxyl, carboxyl ions whereas Fe2+ acts both like a hard and soft acid and Fe2+ would be able to accommodate histidine, protoporphyrin, cysteine molecules. Role of iron is appreciable with respect to its versatility in biochemical functions. The most important one is haemoprotein (haemoglobin and myoglobin) in which iron protoporphyrin incorporated into different apo-proteins.Heme is basically a derivative of porphyrin. Porphyrins are cyclic compounds formed by the fusion of 4pyrrole rings linked by methenebridges (=CH-).Porphyrins contains side chains attached to each of the other four pyrrolerings (Figure 2.1). 6.
(18) Figure .2.1 Structure of heme (iron-protoporphyrin) moiety. (Source: Li Zhang, World Scientific Publishing Co.Pte.Ltd. 2011) Heme is tightly bound to protein through hydrophobic interactions and by single coordinate bond between imidazole of a proximal histidine and ferrous iron. Each haemoglobin molecule consists of tetramer of globin polypeptide chain containing four heme units (Figure.2.2). Heme possesses an unoccupied sixth coordination site within the hydrophobic pocket capable of binding reversibly with oxygen. Figure 2.2 Quaternary structure of haemoglobin. (Source: Royer JR. Journal of Molecular Biology .2003) Polypeptide subunits (2 alpha and 2 beta subunits) held together by various interactions and play a crucial role in binding of oxygen to haemoglobin. The size of the iron atom is the key factor determines the low (deoxy or T) and high affinity (oxy or R) state of haemoglobin. Ferrous atom in the heme gets oxidised to ferric form where it loses its high affinity to oxygen. 7.
(19) Fe2+ is extremely water soluble (Ksp=10−39 M) but Fe3+ is quite insoluble in water (Ksp =10−18 M).Iron complexes readily undergo electron transfer and acid-base reactions which emphasizes the importance of iron in biological system. The important functional aspect of iron is its ability to undergo redox cycling. It is approximately 110 years ago H.J.H Fenton discovered the oxidation capacity of Fe (II)–H2O2with some organic acids. This oxidation effect mainly depends on the concentration ratio of H2O2 and Fe (II) in Fenton reagent. Superoxide radical then reduce Fe3+ to Fe2+ and molecular oxygen. Fe2+ + H2O2→ Fe3+ + •OH + OH− Fe3++ O2− → Fe2+ + O2 Overall the reaction results in oxygen, hydroxyl radical and hydroxyl anion from superoxide radical and hydrogen peroxide known as Haber-Weiss reaction. O2− + H2O2→ O2+ •OH + OH− Reactive oxygen species (ROS) and nitrogen species (RNS) are produced endogenously by all aerobic organisms and are generally involved in various biological processes and essential for life. Excessive ROS production leads to oxidation of biomolecules resulting in stress and damage. It is also known to be involved. in aging. ,cancer, cardiovascular diseases Alzheimer’s and other neurodegenerative disorders (16). 2.4 Distribution and storage of iron The total body iron content is approximately 4g (40-50mg/kg) in adults where more than 50% (2.1mg) of iron present in haemoglobin of red blood cells and developing erythroid cells; about 25% of iron stored in liver as ferritin and 10-15% constitute myoglobin and numerous other iron containing proteins (17). The normal distribution of iron in the human body is shown in Figure.2.3. Mammals lose iron by various means such as sloughing of mucosal and skin cells and bleeding. However there is no other physiological means to excrete excess iron from the body. Hence the iron homeostasis is maintained and finely tuned at the site of duodenal iron absorption. One of the important checkpoints of iron homeostasis lies in epithelial layers of duodenum; whereby it senses the changes in body iron status and responds to its demand. The multipotent crypt precursor cells expresses various proteins involved in iron uptake and transport which is different from that of mature enterocytes and finally it differentiates into enterocytes capable to. transport iron (18).. Majority of iron is found in the form of haem in haemoglobin, myoglobin, other ironcontaining enzymes such as catalase and the cytochromes. The rest of the total body iron. 8.
(20) exists as a non-haem iron, which consists of plasma iron, iron bound to transferrin (TF), and stored iron in ferritin and hemosiderin. Approximately 3mg of iron circulates in the plasma bound to TF, an abundant protein with very high affinity for ferric iron (19). Both haem iron and non–haem iron absorption are influenced by extra-luminal factors; iron absorption is inversely correlated with body iron stores and directly correlated with the rate of erythropoiesis (20).. Figure 2.3 Physiology of iron metabolism. 1800mg 600mg. 300mg. 3mg. Transferrin. 300-1000mg 10-15mg/day. (Source: Antonello Pietrangelo, N Engl J Med. 2004). Iron is stored in the form of ferritin, a highly conserved protein which can store up to 4500 iron atoms present in all tissues including liver,spleen,bone marrow,skeletal muscle and brain.The plasma ferritin level well correlated with tissue iron store and is used as good marker for assessing body iron stores under normal physiological conditions (21).From bacteria through fungi to animal and plants iron is stored in an unique form within a protein. 9.
(21) shell composed of number of ferritin molecules protecting iron from toxic oxidation-reduction reactions but remains as a mobilising store for iron. 2.5 Iron absorption and transport Normally 1-2mg of iron is absorbed from the diet and same amount is lost daily by means of sloughed mucosal cells, desquamation and menstruation. Duodenal iron uptake is mainly regulated at two interfaces; apical and basolateral membrane. Apical membrane faces into intestinal lumen transports the ferrous iron into the cell. Three known proteins are involved in intestinal iron uptake; Divalent metal transporter 1 (DMT1), Heme carrier protein (HCP-1)and Mucin-integrin-mobiliferrin.Divalent metal transporter 1 (DMT1, Nramp2 or DCT1) transfers iron across the apical membrane into the cell through a proton-coupled process and it was extensively characterised.HCP1 (Heme carrier protein 1) is a candidate intestinal haem /folate transporter found in the proximal side of the intestine, where haem absorption is maximum.HCP1 expression and heme uptake was up regulated. with enhanced HO-1. (heme oxygenase1) expression and found to be an adaptive mechanism to heme degradation (22). The other pathway which is not much defined is mucin-integrin-mobiliferrin. Mucin binds to iron at acidic pH making it available for absorption at the duodenal level. This explains how achlorhydria associated with iron deficiency anemia in some cases. Integrin present on the apical membrane of duodenal enterocytes facilitates the transport of iron through microvillus membrane.Mobiliferrin seen in close association with integrin acts as shuttle protein for iron in the cytoplasm (23). Mobiliferrin is also known to be co-localised with transferrin vesicles helps in iron delivery for hemoglobinisation in K562 cell line (24). Dietary iron can be classified into two forms of iron haem and non-haem. Haem is easily absorbed because it is not influenced by the inhibitors present in the diet; furthermore, it is directly taken up by enterocytes through an absorption pathway different from that of non–haem iron. Low pH of gastric juice helps to dissolve ingested non-haem iron by providing a proton-rich milieu facilitating the reduction of ferric iron to ferrous by duodenal cytochrome b reductase followed by DMT1 transport. Absorbed iron either from haem or non haem source enters enterocytes and is stored as ferritin or exported across the membrane by ferroportin (SLC40A1 or IREG1) according to the body iron requirements. Hephaestin (HEPH) helps in oxidation of Fe2+ to Fe3+ and loading into circulating transferrin (25). Ceruloplasmin (CP) is copper containing ferroxidase helps in. Fe2+ oxidation and. essential for export of iron from cells to plasma.. 10.
(22) 2.6 Placental iron transport Iron requirements are greater during pregnancy and iron is transferred from mother to foetus across placenta. Iron deficiency and anemia is very common in pregnancy, hence routine supplementation is mandatory in clinical practice. Maternal iron deficiency anemia ultimately results in preterm delivery ,low birth weight and other developmental problems in new born (26). The function of placenta is to transport nutrients, oxygen and other factors to foetus to ensure its growth and development. Although iron requirement is as low as 0.8mg in first trimester it increases up to 4-6mg in second and third trimester, Studies with radioactive isotopes revealed progressive raise in iron absorption as pregnancy advances (27). Total iron requirement during pregnancy was estimated to be around 1040mg and human infant is born with an average iron content of 270mg at birth. Transferrin receptors present on the syncytiotrophoblast binds diferric transferrin and gets internalised. Inside the endosome low pH triggers its release from transferrin where DMT1 helps to transport iron into cytoplasm, the released ferrous iron is exported out of the syncytiotrophoblast through FPN1.Zyklopen a copper ferroxidase helps in oxidation of Fe2+ and foetal transferrin receives the diferric iron (28). Most of the transferrin receptors are seen on microvillar membrane of the placenta. The intracellular iron was found to be directly proportional to the expression of TFRC.TFRC expression was up regulated during iron deficiency. Maternal iron metabolism is determined by the foetal iron requirements.Foetal liver seems to regulate the expression of maternal TFRC and hepcidin during pregnancy (29). Millard et al. investigated the gene expression pattern in pregnant rat models on different gestational days and demonstrated high level of TFRC expression with low transferrin saturation and low maternal body iron stores. Duodenal expression of DMT1, DCYTB and IREG1 was found to be down regulated with reduction in hepatic hepcidin, HFE and TFR2 which is returned to normal 24-48hrs postpartum (30). Placenta is the main source of GDF15 a known negative regulator of hepcidin in ineffective erythropoiesis; however its role in and pregnancy iron metabolism has yet to be studied. Serum GDF15 was observed at very high concentration in pregnancy and found to be peaked at third trimester. Moore et al., indicated the high level of GDF15. in first trimester 6.3 (± 0.02) ng/mL. compared to controls 0.36 (± 0.04) ng/mL which elevated to 12.2 (± 0.5) ng/mL during 2 nd trimester (31). 2.7 Iron and brain Iron is an important cofactor in central nervous system essential for oxidative phosphorylation, myelin synthesis, neurotransmitters, nitric oxide metabolism and oxygen. 11.
(23) transport. Brain accounts for 2% of total body iron content and concentration vary with specific regions. Concentration of ferritin and neuromelanin is highest in substantianiagra with approximately 20ng/mg in first year of life and increases gradually up to 200ng/mg by the fourth decade of life. It was postulated that the uptake of iron into brain continues throughout the life (32). Even though iron is involved in diverse brain functions there was no difference observed in oxidative phosphorylation, enzyme activity or turnover rate of neurotransmitters in animals with iron deficiency compared to controls. Dopamine receptors density alone was lower in animals when iron drops to 15% of normal level (33). Endothelial cells form blood brain barrier express transferrin receptors on the luminal side of blood capillaries. The iron uptake is through TF mediated endocytosis and subsequent release of iron into the plasma. The absence of DMT1 in previous studies suggests iron transport from luminal to abluminal side through vesicles and iron released from transferrin with the help of ATP/citrate. Iron bound to citrate or ATP is taken up by the cells. Astrocytes play an important role in importing iron into the cell. Brain microvasculature contains receptor for H-ferritin implicating role of ferritin in iron transport. Brain iron dyshomeostasis is a key feature in aging and several neurodegenerative disorders such as Alzheimer's, Parkinson's and Huntington's disease.Measuring iron levels using MRI revealed iron accumulation at early onset of disease, strengthening the fact that iron overload is the cause for neurodegenerative disease (34). Men are more likely to develop age related brain disorders than women at earlier ages although brain iron increases with age in both the genders (35). Neurodegeneration with brain iron accumulation (NBIA) is a group of inherited neurological disorders with increased iron in basal ganglia and associated neurological abnormalities. The genes described in association with NBIA include PANK2, PLA2G6, C19orf12, FA2H, ATP13A2, WDR45, FTL, CP and DCAF17. Aceruloplaminemia and neuroferritinopathy are due to the mutation present in Ceruloplasmin (CP) and L-ferritin (FTL) respectively. A number of chelators and antioxidants are helpful in removing excess iron from brain and total body iron. 2.8 Proteins involved in iron absorption and transport. 2.8.1 Divalent metal transporter protein 1 (DMT1) Divalent metal transporter protein 1 also known as NRAMP2, DCT1 and SLC11A2 is an integral membrane protein with 12 transmembrane domains; plays a crucial role in intestinal iron absorption and endosomal iron transport. It mediates iron uptake with pH coupled fashion highly expressed at the brush border of dudodenum.DMT1 can transport other divalent cations like Mn2+,Zn2+,Cu2+, Ni2+, Co2+,Pb2+ and Cd2+ ions. DPGN (Asp–Pro–Gly–Asn) motif of transmembrane domain 1 is conserved in all SLC11 family members and essential 12.
(24) for its function (36). Mutations in these residues were found to be associated with loss of its transport activity. DMT1 is highly conserved in evolution and similar proteins are seen in plants,insects,microorganisms and vertebrates (37). Animal models like microcytic anaemic (mk) mouse and Belgrade (b) rat inherited with defective iron transport was shown to have missense mutation G185R in DMT1 demonstrating its importance in apical membrane iron uptake and endosomal iron transport in association with transferrin cycle in other cells. Recently it has been shown that DMT1 is not only associated with endosomal iron transport but also helps to import iron from cytoplasm into mitochondria (38). DMT1 mRNA isoform with iron responsive element (IRE) at 3’UTR maintains iron homeostasis in response to iron levels.DMT1-nonIRE (without IRE) and DMT1-IRE (with IRE) differ from each other in 18-25 amino acids at C-terminal and found to have differential expression and tissue specificity. DMT1-non IRE expressed in erythroid precursor suggesting its role in endosomal iron transport and DMT1-IRE mainly expressed in epithelial cell mediating apical iron transport (39). Mutations in DMT1 gene transmitted as an autosomal recessive trait resulting in microcytic anemia with hepatic iron overload. So far ,only four patients have been identified with DMT1 mutations (40,41). Identification and characterisation of mutated genes in inherited anemias will be of great interest for better understanding iron homeostasis. 2.8.2 Duodenal cytochrome b reductase 1(DCYTB) DCYTB,a member of cytochrome b561family plasma membrane protein containing 286 amino acids with six transmembrane domain exhibiting ferrireductase activity in duodenal brush border membrane (42). DCYTB mRNA is induced by iron deficiency indicating its importance in iron metabolism. Its cDNA encodes haem and ascorbate binding sites where ascorbate serve as an electron donor for transmembrane reduction of iron (43). DCYTB has an important physiological role in both ferric and cupric reduction through DMT1 and copper transporter 1 respectively. DCYTB expression is induced even in mild iron deficiency and shown to have a negative correlation with serum iron saturation (44). 2.8.3 Heme carrier protein (HCP-1) HCP1, known as human proton coupled folate transporter(hPCFT/SLC46A1) discovered by Shayegi et al as a candidate protein with low affinity to heme in mouse duodenum.It is now well established proton coupled folate transporter with high affinity to folate influx (45). It is also known to be involved in macrophage iron recycling and recently it has been elucidated that inflammation disrupts iron homeostasis in macrophages via HCP1 down regulation (46). Cancer cells uptake of porphyrins are also known to be mediated through HCP-1 (47).. 13.
(25) 2.8.4 Hephaestin (HEPH) Hephaestin is a membrane bound multicopper ferroxidase necessary for iron transit from enterocytes into circulation. Anderson et al.,discovered the gene responsible for defective iron efflux in sexlinked anemia mouse (sla) (48). The sla mouse harbours a mutation resulting in frame shift deletion of 582bp in the mRNA of HEPH gene producing truncated protein less than 192 aminoacids.Previous study has shown that truncated protein still retains partial oxidase activity and additional ferroxidase such as ceruloplasmin compensates its functional defect (49). It has also been shown that thereis two fold increase in Hephaestin protein under iron deficiency which in turn stimulates the iron absorption and export.Hephaestin is identical (50%) and similar (68%) to ceruloplasmin (ferroxidase) at the amino acid level. 2.8.5 Ferroportin (FPN1) Ferroportin (SLC40A1, IREG1, MTP1, and SLC11A3) is the sole cellular iron exporter present in duodenal enterocytes, placenta, and macrophages and hepatocytes. Ferroportin predicted to have 9-12 transmembrane domain and identified as a gene up-regulated during iron absorption and facilitating the iron efflux across the basolateral membrane (50). FPN1 gene is highly conserved, encoding a protein of 571aa in length with a mass of 62KDa. It is a multipass integral membrane protein with at least nine transmembrane alpha helices. It act as. hepcidin receptor where hepcidin binds and causes proteosomal degradation of. ferroportin thereby inhibiting the efflux of iron through ferroportin, in enterocytes, macrophages and hepatocytes (51). Iron responsive element present at 5’UTR of FPN1 confers translational regulation of ferroportin in response to intracellular iron. However epithelial and erythroid precursors utilise another isoform of FPN1 which lacks IRE at 5’UTR (FPN1B) (52). The presence of FPN-non IRE isoform reveals how the FPN1 expression eludes IRP dependent suppression and enhances iron uptake during iron deficiency. The identification of FPN1B transcript implicated its role in erythroid precursors and helped to understand the possible mechanism of normocytic and microcytic features in anemia of inflammation and iron deficiency anemia respectively (53). Transcriptional regulation of ferroportin was first demonstrated in bone marrow derived macrophages where haem derived from human and murine RBCs led to marked increase in FPN1 mRNA and HO-1 mRNA (54). Recent study has demonstrated the presence of MARE/ARE sequence (MAF recognition element/antioxidant responsive element) at -7007/-7016. position of the FPN 1 promoter (55). This finding suggests a. possible mechanism of FPN1 transcriptional regulation through Bach1 repressor. Heme released from senescent RBCs imposes an oxidative stimulus to inhibit Keap1. Keap1. 14.
(26) inhibition blocks Nrf2 degradation results in nuclear accumulation which in turn displace Bach1 repressor from ARE/MARE sequence and activates FPN1 transcription. Ferroportin mutations results in a type IV hemochromatosis also known as ferroportin disease. It has an autosomal dominant pattern of inheritance. Zohn et al has described flatiron mouse model (ffe) with FPN missense mutation (H32R) affecting its localisation and iron export activity (56). The presentation of disease is different with the type of mutations present. Classical ferroportin disease is characterised by hyperferritinemia with low or normal transferrin saturation and iron overload in reticuloendothelial macrophages with borderline anemia. On the other hand non classical hemochromatosis characterised by high transferrin saturation with hepatic iron overload to hyperferritinemia and macrophage iron overload. Recent meta-analysis on ferroportin mutations suggest that Y64N, V72F, Y501C render hepcidin resistance similar to non-classical disease C326Y mutation. Other mutations which impair the iron export includes D157N, D181N, G80V, Q182H, R489K and V162del either through incorrect folding or subcellular mislocalisation (57). 2.8.6 Transferrin (TF) Transferrins are monomeric glycoproteins predominantly synthesised in liver and has been detected in various body fluids like plasma, bile, amniotic, cerebrospinal, lymph and breast milk. It consists of 679 amino acid residues with a molecular weight of 79kDa.This protein is mainly stabilised by 19 intra-chain disulphide bonds and is protected by three carbohydrate side chains (two N-linked-ASP 413 &Asn-611 and one is O-linked-Ser 32) (58). Transferrin is a bilobular protein with N-lobe (336 amino acids) and C-lobe (343 amino acids) that is linked by a spacer sequence. N and C lobe domains interact to form a hydrophilic binding site for iron where it can bind with two iron atoms. The binding site consists of four conserved amino acids including two tyrosines, one aspartic acid and one histidine. Additionally one carbonate molecule donates two oxygen molecules that stabilize the iron atom. Atransferrinemia and animal models with similar phenotype emphasizes its significant role in erythropoiesis and iron metabolism. It’s not only in atransferrinemia but also in other conditions such as inflammation and iron overload transferrin is decreased (59,60). Aberrant levels of transferrin glycosylation has been indicated in chronic alcohol consumption and congenital disorders of glycosylation but it does not interfere with Fe or transferrin binding; however it impairs cellular iron uptake in vitro and in vivo(61). Huggenvik for the first time reported the hypotransferrinaemic mice (hpx) model harbouring a splicing defect in transferrin gene which produces less than 1% normal level of serum transferrin (62). Other cells of transferrin production includes sertolicells, brain capillary endothelial cells, ependymal cells in the choroid plexus and oligodendroglial cells in the brain and. 15.
(27) induced lymphocytes, implicating the importance of transferrin to the organism at all levels (63,64).Transferrin molecule has immense potential from therapeutic perspective where it can be used to sequester free iron, to deliver drugs to rapidly dividing cells and to activate immune cells (58).. 2.8.7 Ceruloplasmin (CP) Ceruloplasmin or the sky blue protein is an abundant serum glycoprotein and a multicopper oxidase enzyme synthesised in liver, promotes iron loading onto transferrin. It is a single chain polypeptide of 1046 amino acids .The physiological role of this enzyme was discovered after identification of a patient with aceruloplaminemia. Mutations in ceruloplasmin gene leads to rare genetic disease ‘aceruloplasminemia’ presenting with iron overload in brain, liver, pancreas and retina. Ceruloplasmin is an acute phase reactant elevated during inflammation, tissue injury and in some cancers. Ceruloplasmin carries 70% of total copper in human plasma and albumin carries about 15%.Mutations in p-type ATPase prevents incorporation of copper into ceruloplasmin and leads to an autosomal recessive disease known as Wilson disease. Copper depositions occur in various tissues such as hepatic parenchymal cells, brain periphery of iris and kidney. Like other plasma proteins CP levels also drop with liver synthesising capabilities. Copper availability doesn’t affect its translation but apoenzyme without copper is found. to be unstable (65). Activated. macrophages are the principal source of CP during inflammation and it was experimentally proven that CP was induced by IFN-γ both at protein and mRNA level (43). But ceruloplasmin translation was. inhibited by a cytosolic inhibitor complex IFN-γ activated. inhibitor of translation (GAIT) where it can binds to GAIT element present in 3’UTR of CP gene (67). Astrocytes produces an alternative GPI-anchored form of ceruloplasmin which functions as an antioxidant system of CNS (68). The insufficient synthesis of ceruloplasmin or its decreased activity with subsequent brain iron accumulation is thought to be one of the underlying mechanisms for neurodegenerative disorders like Parkinson’s and Alzheimer’s disease.. 2.8.8 Six transmembrane epithelial antigen of prostate 3 (STEAP3) Steap3/TSAP6 (Tumour Suppressor Activated Pathway Protein6) is direct transcriptional target of p53. It is involved in cell cycle regulation, apoptosis and exosomal mediated secretion of proteins. It is highly expressed in haematopoietic tissues and found to be colocalised with transferrin cycle endosome. STEAP2 and STEAP3 were shown to have ferric and cupric reductase activity. The importance of STEAP3 in iron metabolism was recognised in nm1054. hypochromic microcytic mutant mouse (69). Later on employing positional 16.
(28) cloning strategy it was identified that STEAP3 is responsible for iron deficiency anemia in mutant mouse (70). It is also involved in iron homeostasis and inflammatory response in macrophages. STEAP3 depletion impaired the induction of various cytokines in macrophages and its activity. Grand Champ et al.,reported 3 siblings with heterozygous p.C100X(-) stop codon STEAP3 mutation resulting in transfusion dependent anemia (71). The father was heterozygous for this mutation but had normal STEAP3 mRNA levels; indicating the difference in the expression of normal allele between the father and daughter.. 2.9 Iron uptake and transferrin receptors There are two transferrin receptors described till date; TFRC (CD71)and TFR2.TFRC is made up of two identical disulphide-linked 90,000 kD subunits, each with three asparaginelinked and one threonine-linked glycans.TFRC and TFR2 share moderate homology and 45 % identical at protein level. Transferrin receptor (TFRC) is a membrane glycoprotein with definitive function of mediating cellular iron uptake from plasma transferrin. It is expressed in almost all cells but expression varies greatly with the cell type. Transferrin receptors are expressed exceptionally high in immature. erythroid cells, placental tissue and rapidly. dividing cells(normal and malignant) (72). The level of transferrin receptor is highest in early erythroid precursors through intermediate normoblastic stage and decreases as it mature to reticulocytes (73,74). Transferrin receptors (TFRC) are homodimeric membrane glycoproteins which binds iron loaded diferric transferrin during iron uptake. At physiological pH Fe (III)-TF binds to its receptor (TFRC) present on the cell surface, the TF-TFRC complex is internalized in clathrincoated pit forming endocytic vesicles. The internalized complex in the endosome is acidified by a vacuolar H+-ATPase (V-ATPase) that lowers the luminal pH to about 5.5. This acidification process induces conformational changes in TF-TFRC complex with subsequent release of iron. An endosomal ferrireductase; STEAP3 is responsible for reduction of endosomal ferric iron to ferrous iron. The endosomal DMT1 transports the ferrous iron into cytosol. At acidic pH apotransferrin remains bound to TFRC, and the complex is recycled to the cell surface. At neutral pH of the plasma, apotransferrin dissociates from TFRC and accepts next cargo of iron (Figure 2.4). Another protein Sec15l1 is also known to be involved in the mammalian exocyst complex. Recently sorting nexin 3 (Snx3) was identified and known to facilitate transferrin receptor recycling and thus it is essential for proper delivery of iron to erythroid progenitors. TFRC consists of multiple IRE elements at 3’UTR of mRNA where iron regulatory proteins (IRPs) binds and regulates its expression by stabilising the mRNA .TFR2 is predominantly expressed in liver and it lacks iron dependent regulation. The role of TFR2 in 17.
(29) iron metabolism was affirmed after identifying mutations in patients with non-HFE hemochromatosis later termed as type III hemochromatosis. TFR2 plays a critical role in sensing and regulation of systemic iron homeostasis (75). TFR2 co-localises with HFE in duodenal crypts and in hepatocytes, although the direct interaction between these two proteins were excluded in vitro and still elusive (76–78). Reduction of TFR2 in hypotransferrinaemic mouse despite of iron overload implicates its regulation is transferrin saturation dependent .A. similar phenomenon was. also observed in beta thalassemia. mouse (79). 2.10 Intracellular iron storage: Ferritin and hemosiderin Ferritin is an intracellular storage form of iron which sequesters iron in a nontoxic bioavailable form. Historically hemosiderin was first identified as iron rich granules in tissue with an intense Prussian blue reaction when treated with potassium ferrocyanide. Ferritin and hemosiderin consist. of a central ferric oxy-hydroxide core where ferritin is well. organised by apoferritin protein shell (32). Hemosiderin consists of some apoferritin and degraded ferritin products thought to be derived from intralysosomal aggregation and degradation of ferritin. Around 25% of total body iron is stored in the form of ferritin and seen in all mammalian tissues. But hemosiderin is restricted to reticuloendothelialsystem. The existence of iron chaperone molecules remain elusive, but recently a protein known as poly (rC)- binding protein 1 has been identified as a chaperon helps to deliver iron into ferritin, plays an important role in this process (80). Ferritin is an oligomeric protein with 24 identical subunits forms a hollow protein shell capable of sequestering up to 4500 iron atoms. Cytosolic ferritin made of two subunit types L(predominant in liver) and H (predominant in heart) chains with 50% sequence identity. Mitochondrial ferritin is an intron less gene containing a mitochondrial localisation signal and expressed in the mitochondrial matrix and exhibits 75% sequence identity with Ferritin H gene (81). H chain ferritin was characterised by ferroxidase centre involved in the oxidation of ferrous to ferric iron. L-chain is involved in the nucleation of iron in the ferritin core. Depending on the tissue and physiological status of the cell, the ratio of H and L ferritin vary widely (82). The ferroxidase centre is highly conserved and mutations of His65 and Glu62 impaired ferroxidase activity both in human and mouse models (83) .. 18.
(30) Figure 2.4 Transferrin cycle. (Source: Walz.med.harvard.edu). Ferritin is post transcriptionally regulated in response to intracellular pool of iron. In the absence of iron, iron regulatory proteins (IRP1 & IRP2) binds to IRE element present at 5’UTR of mRNA and inhibit its translation. Thus ferritin synthesis is induced when iron is high and decreased when iron levels are low. Although ferritin is an acute phase reactant serum ferritin levels is used as a clinically useful parameter to assess the iron stores. It has also been shown that ferritin mRNA is induced in response to iron (84). Point mutations in IRE of L-ferritin mRNA lead to its constitutive activation and high serum ferritin in absence of iron excess causing hyperferritinemia–cataract syndrome. On the other hand recently discovered mutation in the IRE of H ferritin leads to increased affinity of IRP to IRE and decreased ferritin. Cytokines regulates ferritin post transcriptionally (85). Pro-inflammatory cytokines and TNF-alpha ,IL-1alpha have been found to induce ferritin, suggesting that inflammation and stress have a regulatory effect on ferritin synthesis (86). Oxidative stress induces ferritin by inactivating IRP1 through. the reversible oxidation of cysteine residues (87).. Recently ferritin was identified as a marker of early erythroid precursors and macrophages in bone marrow (88). During erythropoiesis intracellular ferritin serve an iron donor for heme synthesis (89). 2.11 Erythropoiesis and Iron Erythropoiesis governs iron metabolism. Erythropoiesis is a dynamic process of generation of red blood cells from hematopoietic stem cells (HSC) (Figure 2.5). During mammalian development erythropoiesis begins in the yolk sac followed by foetal liver and bone marrow.. 19.
(31) Daily production of erythrocytes was found to be approximately 2*1011 cells. BFU-es are the most immature precursor committed to erythroid lineage. BFU-es require stem cell factor (SCF) and other growth factors to proliferate and differentiate to CFU-es. The next phase of differentiation is EPO dependent where CFU-es differentiated through various stages of proerythroblast,. basophilic,. polychromatophilic. and. orthochromatophilic. erythroblast. ultimately into mature red cell. Erythropoietin is the key regulator of erythropoiesis. Vitamin B12, folate and iron are the three important nutrients required for the active erythropoiesis. Folate, vitamin B12 are required for DNA synthesis whereas approximately 30-40mg/day of iron is required for haemoglobin synthesis. The iron utilisation is maximum at late basophilic erythroblast where the synthesis of haemoglobin is maximum (90). Early erythroblasts store iron as ferritin, and utilises stored iron for haemoglobin synthesis as it proliferate and differentiate. Transferrin receptor (CD71) expression is high on erythroid cells to maximise iron uptake during erythropoiesis and regulated by intracellular iron concentration. Soluble transferrin receptor (sTfR) is a major determinant of erythropoietic activity and a marker of tissue iron deficiency.Soluble transferrin receptor is a truncated monomer of TFRC lacking first 100 amino acids seen in animal and human serum (91). Hepcidin mRNA was suppressed during anaemia and hypoxia and more pronounced during ineffective erythropoiesis with enhanced iron absorption. Schranzhofer et al. demonstrated a discrete regulatory mechanism of iron during terminal maturation of mouse erythroid progenitors different from other cell types. The expression of ferritin and TFRC were found to be independent of intracellular iron concentration and IRP activity. Ferritin translation was impaired in order to ensure successful hemoglobinisation preventing iron storage whereas TFRC and ALAS2 translation was activated to maximise the iron uptake and heme synthesis (92). 2.12 Mitochondria and haem synthesis Mitochondrion plays a critical role in iron metabolism. It is the sole site of haem synthesis and a major generator of iron sulphur clusters (ISC). Majority of haem synthesis takes place in developing red cells and nearly 15% of synthesis occurs in liver where it is utilised for haem containing enzymes. First and last three steps of haem pathway take place in mitochondria and remaining in cytosol (Figure 2.6).The heme biosynthetic ability and its regulation in erythroid cells mainly depend on the availability of iron but in liver heme synthesis occurs as per the metabolic requirements. Heme biosynthetic enzymes are extensively studied and characterised recently. Aminolevulinate synthase (ALAS) is the first enzyme in heme synthetic pathway, catalyses the condensation of Glycine and succinyl CoA.. 20.
(32) Figure.2.5 Stages of erythroid differentiation Maturation of red blood cells from hematopoietic stem cell (HSC). The expression of various surface markers is shown which is characteristic to each erythroid precursor. Arrows indicates specific factors required at different stages of erythroid differentiation. SCF EPO Iron. (Source: Handbook, European society of Haematology, 2009) There are two isoforms of ALAS - housekeeping ALAS1 and erythroid specific ALAS2. ALAS1 & ALAS2 are encoded by chromosome 2 & X-chromosome respectively. Both isoforms require PLP (Pyridoxal-5-phospahate) for its activity where it binds to specific lysine of ALAS. PLP-Schiff base complex then reacts with succinyl CoA producing aminolevulinate. The next three enzymes (ALD,PBGD,URO3S) in heme synthetic pathway exhibits dual promoter system representing both erythroid and non erythroid restricted expression (93). Mammalian ferrochelatase (FECH) is 2Fe-2S containing enzyme catalyses the insertion of iron into protoporphyrin IX. Ferrochelatase expression is. regulated by intracellular iron. levels via Fe-S cluster at the C-terminus and also regulated by hypoxia (94,95). ALAS1 & ALAS2 isoforms contain two heme binding motifs (HRM) which acts as mitochondrial targeting sequence whereby binding of heme blocks mitochondrial import of the enzyme (96). Transcriptional regulation of ALAS2 is mediated through the erythroid specific. transcriptional. regulator. GATA1.. ALAS2. contains. 5’UTR-IRE. and. post. transcriptionally regulated by iron regulatory protein. Interaction of IRP-IRE complex on 5’UTR of ALAS2 blocks its translation under iron deficient state suppressing heme synthesis. On the other hand Fe-S cluster abolishes IRP binding to IRE in iron replete conditions suggesting the crosstalk between red cell iron availability and mitochondrial function.ALAS1 transcription is up-regulated by peroxisome proliferation activated co-activator 1α (PGC1-α). Transcription of PGC1-α in turn gets activated under low glucose concentration. Therefore glucose infusion attenuates the severity of neurovisceral porphyrias (97). Inherited mutations in enzymes involved in heme synthetic pathway have been described in X-linked sideroblastic anemia and other porphyrias.. 21.
(33) Figure 2.6 Heme biosynthetic pathway. (Source: Richard .S.Ajioka, Biochimica et Biophysica Acta.2006) In developing red cells, iron. delivered to mitochondria through docking of TF. containing endosome ; bypassing cytosol known as kiss and run hypothesis (98). There are other proteins which help in iron transport across mitochondrial membrane for active haemoglobinisation. The role of mitoferrin (Mfrn1) / SLC25A37 has discovered from Mfrn1 knockout mouse model where it presented with impaired iron import and reduced heme synthesis (99). Mitoferrin was implicated in mitochondrial iron delivery. It was also demonstrated that ABCB10, a mitochondrial inner membrane ATP binding cassette transporter induced during erythroid differentiation and it stabilises Mfrn1 in haematopoietic tissues (100). ABCB6 helps in translocation of coproporphyrinogen III from cytoplasm to mitochondria. One of the possible mechanism by which heme is transported to its target proteins by is protein-protein interaction which was evident from localisation and close proximity of cytochrome P450 with FECH (101). HCP-1 and HRG-1(heme responsive gene-1) are two newly identified proteins involved in heme import into cells. It has been shown that overexpression of HRG-1 in MEL cell line markedly increased the uptake of heme analog. 22.
(34) Abkovitz et al demonstrated that Feline leukaemia virus subgroup c receptor (FLVCR) is essential to maintain cellular heme level and inevitable for erythroid differentiation and iron homeostasis. It is highly expressed in haematopoietic cells and mediates heme efflux thus coordinates the regulation of heme and globin synthesis during erythroid differentiation with balanced supply of heme for erythropoiesis. FLVCR null mice showed lack of definitive erythropoiesis with craniofacial and limb deformities (102). It has been shown recently that an isoform of FLVCR (FLVCR-1b) overexpression promotes heme synthesis and Invitro differentiation whereas knockdown of FLVCR-1b led to mitochondrial iron accumulation and defective erythropoiesis (103). ABCG2 also known as BCRP highly expressed in hematopoietic progenitor cells has also been implicated in heme efflux in mitochondria.. 2.13 Importance of iron-sulphur cluster Iron sulphur clusters are important cofactors of proteins involved in several biological activities such as electron transport and respiratory chain, photosynthesis and DNA repair. Generally it is attached to cysteine residue of the protein. Fe-S clusters which exist mostly in cubane form are chemically versatile and possess the ability to maintain low reduction potential facilitates an efficient transfer of energy from NADH through respiratory chain complexes (104). Fe-S cluster can directly bind to protein and controls its function as in iron regulatory proteins (IRP). In the absence of Fe-S cluster IRP interacts with iron responsive element (IRE) and regulates expression of target genes involved in iron metabolism. Cysteine desulfurase (NFS1) is a pyridoxal 5'-phosphate (PLP)-dependent enzyme that catalyses the conversion of L-cysteine to L-alanine. NFS1 supply sulphur for Fe-S biogenesis whereby ISCU provides scaffold system upon which iron covalently binds to sulphur atom (105). A dimer of NFS1 forms a core with ISCU (iron sulphur cluster assembly enzyme), in presence of PLP cofactor NFS1 is stabilised by another small protein called ISD11 found in mitochondrial matrix. Frataxin is a highly conserved protein found in mitochondrial matrix act as an iron chaperone and iron storage protein and mainly involved in Fe-S formation. Frataxin binds to the NFSI-ISCU complex causing conformational change and initiates Fe-S biogenesis. Assembly of Fe-S clusters relies on its availability of electrons to attain proper configuration observed in Fe-S cluster. It is facilitated by proteins such as GLRX5, ferredoxin helps in reductive coupling of 2Fe-2S to 4Fe-4S. HScA (HSC70) and HScB (HSC20) helps to transfer Fe-S cluster from ISCU to target proteins (106). Mutations in proteins involved in Fe-S biogenesis cause diseases such as Friedreich’s ataxia (FRDA), ISCU myopathy and certain types of sideroblastic anemia (107). Fe-S proteins are essential for the function of two proteins of the citric acid cycle, succinate dehydrogenase and aconitase; for respiratory chain complexes I–III; and for 23.
(35) numerous other proteins in the mitochondria, cytosol and nucleus of eukaryotic cells. Intrusion of Fe-S biogenesis leads to mitochondrial iron overload and cytosolic iron depletion associated with various disease pathologies. 2.14 Heme and its biological role Heme controls a variety of biological processes such as erythropoiesis, neurogenesis, cell growth and differentiation. HRI (heme regulated inhibitor of translation) shown to have heme regulated eIF2α kinase activity which is activated under heme deficiency by multiple autophosphorylation which in turn phosphorylates eIF2 and block the protein synthesis (Figure 2.7). When heme concentration is high HRI get inactivated permitting globin chain synthesis and haemoglobin synthesis to occur (108). HRI is essential to prevent α and β globin precipitation and erythroblast apoptosis during iron deficiency. The consequence of iron deficiency is more severe in absence of HRI than with HRI. Thus HRI act as a physiological regulator of adaptive gene expression and survival of erythroid precursors (109). Bach1 transcriptional activity is also modulated by heme. It’s a basic leucine Zipper protein and heme regulated transcriptional repressor found in mammals. Bach1-Maf heterodimer binds to Maf recognition element (MARE) in target gene such as stress responsive genes, globin gene, HO-1 and ALAS2. Bach1 is normally located in nucleus and it is exported into cytoplasm when concentration of heme is high. It possesses six HRM motifs essential for heme mediated regulation. Heme negatively regulates the transcriptional repressor function of Bach1 by inhibiting its DNA binding ability (110). Heme is also found to be critical for neuronal differentiation. Heme deficiency induced by succinyl acetone a potent inhibitor of ALAD reduced the number and length of NGF (Nerve Growth Factor) induced neurites. NGF induced Ras ERK1/2 signalling is inactivated during. heme deficiency and it alters the. expression of several other genes involved in NGF signalling such as MEKK1,P38 MAPK, p53 and c-myc .. 24.
(36) Figure 2. 7 Heme regulated activation of HRI. (Source: Chen et al, Blood.2007). 2.15 Cytoprotective enzymes and antioxidants Mitochondria, the power house in aerobic tissues are the main site of free radicals generation. Several sites of superoxide radical generation have been identified in mitochondrial respiratory chain that found to be varied with organ and mitochondrial activity. Aerobic organisms possess an intricate system of cytoprotection against oxidative damage. Superoxide dismutase (SOD) enzyme catalyses the reduction of two superoxide radicals to H2O2 and O2 (O2− + O2− + 2H+ → H2O2+ O2). There are three distinct SOD in mammalian cells. SOD1 is a Cu/Zn SOD homodimer exclusively found in cytosol and familial amyotrophic lateral sclerosis was found to be associated with mutations in SOD1. SOD2 is Mn/SOD found in mitochondria and SOD3 is Cu/Zn tetramer with protective role in blood vessel walls. Hydrogen peroxide is neutralised by enzyme catalase and glutathione peroxidase (H2O2+ H2O22H2O + O2). Catalase is a heme containing enzyme localised in peroxisomes. There are four members of selenium containing glutathione peroxidases (Gpx1-4) where first three forms are known to reduce H2O2 and last one GPx4 reduces phospholipid hydroperoxides and oxidises glutathione. Glutathione is tripeptide γglutamylcysteinylglycine, an important cellular antioxidant supplying electrons for the reduction of peroxides H2O2 + 2GSH →GSSH+ 2H2O. Heme oxygenases are evolutionarily conserved enzymes catabolize heme into carbon monoxide, biliverdin and iron. There are two isoforms HO-1 and HO-2 encoded by HMOX-1 and HMOX-2 respectively. HO-1 induced during oxidative stress whereas HO-2 is. 25.
(37) constitutively expressed. HO-1 is transcriptionally regulated by various stimuli like heme, oxidative stress, nerve growth factor, TNF-alpha, interleukin 1β, interferon γ and phenolic compounds. Three MAPK pathways extracellular signal-regulated kinase (ERK), c-Jun Nterminal kinase (JNK), and p38 are shown to be involved in up regulation of HO-1 in response to stress and other stimuli (111). Oxidative stress suppresses the activity of Bach1 a transcriptional repressor through blocking Keap1. Keap1 dissociates from Nrf2 (NF-E2 related factor-2) leads to its translocation from cytoplasm into nucleus where it dimerises with Maf proteins and activates transcription. of target gene such as HO-1(112).The. relevance of HO-1 system relies on its ability to protect cell from oxidative stress and damage. Polymorphisms in HO-1 promoter or its deficiency were associated with severity of coronary heart disease (113). Pharmacological induction of HO-1 or administration of end products of heme catabolism were shown to have therapeutic effect on many diseases(114). Akihiro et al., described the first case report of HO-1 deficiency due to a deletional mutation of exon2 and two dinucleotide deletion within exon3 of HO-1 gene . Decreased activity of HO-1 resulted in extreme susceptibility to stress and environmental toxic substances in this patient (115). Deficiency in NADPH P450 reductase enzyme also responsible for reduced HO-1 activity in some patients (116). Accumulating evidence suggests a significant role of HO-1 in detoxification and cytoprotection. 2.16 Reticulo-endothelial system and iron recycling Although the primary function of macrophage is in host defence macrophages of reticuloendothelial system is important and ensures the daily iron requirement for erythropoiesis. It participates in recycling of haemoglobin iron from senescent red blood cells through phagocytosis (Figure 2.8). Heme moiety is released into the lumen of phagolysosome where heme oxygenase-1(HO-1) catalyses degradation of heme into carbon monoxide, iron, biliverdin and its subsequent metabolite bilirubin by biliverdin reductase. The liberated iron is then exported out of the phagosome via DMT1 and Nramp1.Increased HO-1 activity can result in pro or antioxidant effect based on cellular redox potential and fate of heme iron (117). Nramp1 (natural resistance associated macrophage protein1) is an iron transporter localised in late phagolysosome and promotes efficient recycling of iron followed by eythrophagocytosis preventing its accumulation in liver and spleen (118). Heme and haemoglobin released into circulation during haemolysis. Free haemoglobin in the circulation. form a complex with plasma protein Haptoglobin and it. recognised by CD163 receptor on monocytes and macrophages cleared from the circulation (119). Similarly free heme cleared from circulation by a. heme binding protein hemopexin. Heme-hemopexin Complex binds to its receptor (CD91) low density lipoprotein receptor. 26.
(38) related protein present on hepatocytes,macrophages, neurons and syncytiotrophoblasts (120). Erythroblastic islands are essential microenvironment required for erythroid maturation where macrophages provide essential nutrients and factors for proliferation and survival (121). Disturbance in erythrophagocytosis plays pathophysiological role in anemia of chronic disease and hemochromatosis (122).. Figure .2.8 Macrophage iron recycling. (Source: Carole Beaumount, Haematologica.2010). 2.17 Cellular iron homeostasis Cellular iron homeostasis is maintained by the action of Iron Regulatory Proteins (IRP) through its interaction with iron responsive elements (IRE) present on target mRNAs (Figure 2.9). IRP mediates post transcriptional regulation of genes involved in iron uptake, storage and iron efflux thus maintains cellular iron balance under different conditions like iron deficiency and iron overload. IREs are evolutionarily conserved hairpin structures of 25–30 nts with a loop contains a conserved 5’CAGUGH-3’sequence (H denotes A, C or U), where the underlined C and G residues form a base pair. TFRC mRNA contains multiple IREs within its long 3’UTR, whereas the mRNAs encoding H- and L-ferritin contain a single IRE in their 5’UTRs. Iron 27.
(39) regulatory proteins 1 and 2 (IRP-1, IRP-2) interact with iron-responsive elements (IREs) present in the 5’ or 3’-untranslated regions (UTR) of several mRNAs coding for proteins in iron metabolism (TFRC, FT, FPN1, DMT1, ALAS2). IREs are also present in other genes involved in cell cycle, oxidative phosphorylation and cytoskeletal reorganisation.(MRCK αmyotonic dystrophy kinase related DC 42-binding kinase α ;Cdc14a-Cell division cycle 14a SD-Succinate dehydrogenase; mitochondrialaconitase-mACO). IRP-1 modulates its IREbinding activity in response to available cytoplasmic iron. In normal iron supply, due to the insertion of a [4Fe-4S] cluster, IRP-1 converts from an IRE-binding apoprotein to an enzymatic holoprotein with aconitase activity whereas iron deprivation, accumulates the apoprotein and binds to iron regulatory elements. A second cytoplasmic protein, IRP-2, shows extensive sequence homology to IRP-1 and an equal affinity for ferritin IREs. IRP-2 activity is induced by iron deprivation and decreased in iron-replete conditions (123). In ironstarved cells the IRE–IRP interactions stabilize TFRC mRNA and impose a steric hindrance to ferritin translation (Figure 2.10). As a result, increased TFRC levels stimulate the uptake of more iron from plasma transferrin to withstand iron deficiency. The inhibition ferritin synthesis leads to low tissue ferritin indication of storage iron depletion (124). Recent studies showed that not only IRP but miRNAs are also involved in regulation of iron genes. The overexpression of miR-485-3p repressed ferroportin and increased intracellular ferritin by targeting 3’UTR of FPN mRNA (125).. Figure 2.9 Iron responsive element sequence in different mRNAs. (Source: Paul Piccinelli & Tore Samuelsson, RNA, 2007). 28.
(40) The presence of IRE in 5’UTR of HIF2α (hypoxia inducible factor). suggests a. possible mechanism by how the erythropoiesis is regulated through modulating EPO levels under iron deficiency and replete conditions (126). HIF coordinates oxygen metabolism and iron availability, in the absence of iron; EPO dependent stimulation of erythropoiesis gets terminated to prevent immature RBC generation. HIF-2alpha translation is mainly regulated by IRP-1 which overrides PHD/VHL mediated HIF -2 alpha stability. IRP1-HIF2α regulatory axis furnishes new target to modulate cell metabolism depending on altered oxygen and iron status (127). Cells gauge intracellular iron and synergistically alter the activity of IRPs through a mechanism that depends on the protein called FBXL5 (F-box and leucine-rich repeat protein5). FBXL5 senses iron through an evolutionarily conserved hemerythrin domain related to a family of iron- and oxygen-binding proteins in bacteria and invertebrates. Because iron and oxygen stabilize FBXL5, it degrades IRPs. in cells that are iron-. replete(128).The physiological importance of IRP-IRE system is evident from hereditary hyperferritinemia cataract syndrome due to ferritin L chain IRE mutation. Progressive neurodegenerative disease with anemia was also observed in mice lacking IRP2 (129) indicates the significance of IRP-IRE in cellular iron regulation. 2.18 Hepatic iron metabolism The liver plays a major role in iron metabolism and regulation. Approximately 8% of plasma iron turnover is mediated by hepatocytes. Liver is the main storage repository for iron hence it is severely affected in iron overload conditions leads to cirrhosis and hepatocellular carcinoma. Liver regulates iron trafficking through hepcidin production and it is the main site of synthesis of many proteins involved iron metabolism such as transferrin, ceruloplasmin, haptoglobin and hemopexin etc. In liver, iron is distributed around periportal region of the liver with decreasing gradient towards centrilobularregion. Liver expresses a wide variety of proteins essential for iron homeostasis (130). The liver acquires iron through different receptors as shown in Figure 2.11. Hepatocytes can acquire iron from various compounds like non-transferrin-bound iron (NTBI), ferritin, hemoglobin-haptoglobin complexes and heme-haemopexin (receptor CD91) complexes. Apart from transferrin bound iron liver can uptake lactoferrin bound iron where lactoferrin binds to Low density lipoprotein receptor related protein (LRP) and is targeted to lysosomes for degradation. Diferric transferrin-TFRC mediated uptake of iron is the main source of iron under normal circumstances. But in pathological states such as hemochromatosis and transfusion dependent anemias the iron binding capacity of transferrin. 29.
(41) Figure 2.10 Regulation of IRP activities under iron deficient and replete states. exceeds NTBI impending oxidative stress and organ damage (131). NTBI uptake was found to be increased in cells where DMT1 mRNA and protein expression was up regulated (132). In vivo experiments have shown that NTBI was directed to hepatocytes and cleared rapidly from circulation. Serum NTBI was mainly seen as ferric citrate and smaller amount bound to albumin (132). It has also been well characterised that NTBI uptake in HFE–KO mouse model was twofold higher than controls which contributes to iron overload in hereditary hemochromatosis. It has postulated that NTBI can be used as a useful marker for iron overload and liver dysfunction than serum ferritin concentration (133). 2.19 Iron sensors and regulatory proteins 2.19.1 Hereditary hemochromatosis protein (HFE) Hereditary hemochromatosis protein (HFE) is homologous to class I MHC-like protein designated as HLA-H highly expressed in liver and small intestine. Association between HH and HLA-A3 was first suggested in 1976. It was in 1996 HFE was identified as a candidate gene responsible for HH.. 30.
Figure




+7




Outline
Hepcidin Regulation
Therapeutic options
Molecular Genetic Analysis 1 DNA extraction
Hypotransferrinemia/Atransferrinemia 1 Patients’ characteristics
Secondary iron overload in β-thalassemia
DISCUSSION
Rekha Athiyarath , Alok Srivastava, Eunice.S.Edison Molecular basis of primary iron overload in India and the role of serum-derived factors in hepcidin regulation Ann
Related documents
Most companies recruit for full-time and internship positions, but some indicate Co-Op as a recruiting priority, while not attending Professional Practice
• Storage node - node that runs Account, Container, and Object services • ring - a set of mappings of OpenStack Object Storage data to physical devices To increase reliability, you
• Our goal is to make Pittsburgh Public Schools First Choice by offering a portfolio of quality school options that promote high student achievement in the most equitable and
We nd that if individuals dier in initial wealth and if commodity taxes can be evaded at a uniform cost, preferences have to be weakly separable between consumption and labor
Comments This can be a real eye-opener to learn what team members believe are requirements to succeed on your team. Teams often incorporate things into their “perfect team
It is the (education that will empower biology graduates for the application of biology knowledge and skills acquired in solving the problem of unemployment for oneself and others
During the thesis work, I measured six different parameters: the number of emergency processes, hash table entry number, caching replacement policy, cache entry
investment advice (for the relevant information requirement, see Article 24(3) of the MiFID II draft). Only then is it actually possible for banks to offer this service without
