0022-538X/83/010036-11$02.00/0
Copyright©1983,American Society forMicrobiology
Mutants
Deleted
in
the Agnogene of Simian Virus 40
Define
a
New
Complementation Group
JANET E. MERTZ,* ANDREWMURPHY,t ANDALICE BARKAN
McArdle Laboratory for Cancer Research, University of Wisconsin, Madison, Wisconsin 53706
Received 16 July 1982/Accepted 13 September 1982
Analysis of
the DNA sequence of the late leader region of simian virus 40indicates
that it might encode a 61-amino acid, highly basic protein, LP-1. Mutantsdeleted in this region
areviable,
but they produce infectious progeny more slowlythan
wild-type virus in established
monkey cells. On the basis of the rates ofappearance
and the sizes of mixed
plaques formed after cotransfections with pairsof
mutants, wefound that
mutantsdefective in
thesynthesis
of LP-1complement-ed
mutantsin all
known complementation
groupsof simian virus
40.Complemen-tation
wasalso observed in infections with virions
and was bidirectional.Therefore,
these mutantsdefine
a new complementation group, group G. Inaddition,
aprotein of the appropriate
molecularweight
for LP-1 (approximately 8x
103)
wassynthesized by wild-type virus-infected cells but not by mock-infectedor group
G
genemutant-infected
cells.
This protein, whose identity
has beenestablished
definitively by
Jay et al. (Nature (London)291:346-349,
1981), wassynthesized
at ahigh
rateatlate
times after infection,
waspresent predominantly
in the
cytoplasmic fraction of
cells, possessed a fairly short half-life, and wasabsent
from
maturevirions. Once formed,
virionsof
group G gene mutantsbehaved
biologically
and
physically like virions of wild-type virus.
On thebasis ofthese
findings and other known properties of
LP-1and mutants defectivein
LP-1synthesis,
wehypothesize
that LP-1functions
to facilitate virion assembly,possibly by
serving
as anonreusable
scaffolding protein.
The late
leader
region
of the simian virus
40(SV40)
genomeencodes several
interesting
func-tions. These include
(i) the
promotersfor
early-strand
RNAsynthesis
(2, 7a, 8, 11;
L. A.Trimble and
J.E.
Mertz,unpublished data) and
late-strand
RNAsynthesis
(7a;J. E.
Mertz,L. A.
Trimble,
T.J.
Miller,
andG.
Z.Hertz,
manuscript in
preparation),
(ii) the 5' ends of the
late-strand mRNAs
(39), and
(iii) the signals
involved in splicing the 5'-terminal leader
re-gions
ontothe
"bodies" (i.e.,
themain
protein-encoding regions) of the late mRNAs (39). Inaddition, Dhar
etal.(7) noted
that the late leaderregion
alsocontains
an "agnogene" that mightencode a
61-amino acid,
highly
basic
protein
(see
Fig. 1).
The
first
reportsof
mutantsdefective
in thelateleader
region of
theSV40 genome were byMertz and Berg (25),
Carbon
et al. (4), andShenk
et al.(31).
Morerecently,
numerousadditional
mutantswith
deletions spanning
vari-oussegments
of this
region have been isolated in
several
laboratories
(7a; 11, 12, 16, 34; Trimbleand Mertz,
unpublished data). Surprisingly,
allt Present address:DepartmentofHumanGenetics, Colum-bia MedicalSchool,NewYork,NY 10032.
of these
mutants areviable unless
they
lack
sequences
essential for viral
RNAsynthesis
orviral DNA
replication.
These viable
mutants areall
somewhat defective in the
rateof
production
of infectious virions in established lines of
Afri-can green
monkey
kidney
(AGMK) cells,
asindicated
by
both
(i)
the
delayed
appearanceand
smaller sizes
of the
plaques
which
they
produce
(12,
25,
34) (see
Table
1)
and
(ii)
lower
ratesof
production
of PFU
during
single cycles
of
growth
(1,
31).
The
severity
of the defectiveness
varies
considerably
frommutant to mutant(25)
and does
notcorrelate in
aclearly
discernible
manner
with either
thesize
orposition
of
thedeletion
(1).
Inaddition,
since
thekinetics and
rates
of
synthesis
of viral
DNA insingle
cycles
of
growth
aresimilar
tothose
observed in
wild-typevirus-infected cells
(1),
thedefect(s)
in
thesemutantsoccurs
in
the late partof the
lytic
cycle.
There
have beenseveral
reportsconcerning
theeffects of
avariety
of mutations
in the lateleader
region
on the structuresof
the viralmRNAs
synthesized
ininfected
monkey
cells(10,
10a,12,
28,
40).
The mainconclusion of
these
studies
has been thatboth
the 5'ends
andthe patterns
of
splicing
of the late mRNAs are36
on November 10, 2019 by guest
http://jvi.asm.org/
altered in complex ways.
Unfortunately,
noclearly
discernible pattern hasyetemerged fromthe mass of data.
However,
aworking
modelconsistent with theseresults has beenproposed
recently(10a, 23).
The data presented here are concerned with
the existence andpossible
function(s)
of the61-amino acid protein encoded by the late leader
region of SV40. Usinga modification of a
previ-ouslydeveloped complementationtest(24;J. E.
Mertz,
Ph.D. thesis, StanfordUniversity,
Stan-ford, Calif.,
1975),
we found that mutantsde-leted in this region define a new
complementa-tion group, group G. The
polypeptide
thatmaybe
responsible
for the observedcomplementa-tion behavior of thesemutants wasidentifiedby
sodium
dodecyl
sulfate-polyacrylamide gel
elec-trophoresis
ofappropriately
radiolabeledpro-teins obtained from
monkey
cells infected withwild-type
virus and leaderregion
mutants ofSV40.
Basedupon the knownproperties
ofthe61-amino acid
protein
and mutants defective inits synthesis, we propose that this
protein
mayfunction to facilitate virion
assembly, possibly
by
serving
as anonreusablescaffolding
protein.
(Preliminary
accountsof thegenetic
andpro-tein aspects of this work were
presented
at theTumor Virus
Meetings
onSV40,
Polyoma,
andAdenoviruses
heldatColdSpring
HarborLabo-ratory,
ColdSpring
Harbor, N.Y.,
during
thesummers of 1978 and 1980.
Subsequently,
welearned that
Jay
etal.[16],
Jackson andChalkley
[15],
P.Southern
and P.Berg
[personal
commu-nication],
and R.Margolskee
and D. Nathans[personal
communication]
have alsoindepen-dently
identified thisvirus-encoded
protein
inSV40-infected
monkey
cells.)
MATERIALSANDMETHODS
Cell lines.CV-1P, MA-134,andCV-1 areestablished lines of AGMK cells. These cell lines were grown as described previously (24) in medium supplemented with 5% fetal bovine serum. Primary AGMK cells were purchased from Flow Laboratories and were grown in mediumcontaining
10o
fetal bovine serum. Viruses and viral DNAs. Wild-type strain 776 was obtained from S. Weissman. WT800 is a plaque-purified derivative of SV40 strain Rh-911 (24).The late leader region deletion mutants 802,
dl-805, dlG806,dl-809, anddlG810 arenaturally arising
mutantsofWT800.Theselection,propagation, growth
characteristics, andsequences of these mutants have
been described previously (1, 25). Altered restriction mutant ar1077, which has a single base change at nucleotideresidue348, wasgenerously provided by D. Nathans. Theisolation and growth characteristics of this mutant have been described previously (32). SV-L, which was obtained from H. Ozer, is a tempera-ture-sensitive, host range, large-plaque variant of SV40 (27, 35).
The temperature-sensitive mutants tsA58 (38) and
tsB201, tsC219, and tsD202 (5) were provided byP.
Tegtmeyer and R. G. Martin, respectively. Mutant
dlE1226(6)wasobtained from C. Cole.
Virus stocks of each of thesemutants wereprepared
andtitratedasdescribedpreviously(24).Unless indi-catedotherwise,viral DNAwasisolateddirectlyfrom infected cells by the procedure of Hirt (14). The
supercoiled DNA remaining in the supernatant was
purified bytwocyclesofequilibrium centrifugation in
CsCI-ethidium bromide gradients (29). After removal ofethidium bromidebyrepeated extraction with iso-propanol, the viral DNAwasprecipitated with NaCl andethanol andsuspendedin 10 mM Tris(pH
7.6)-i
mMEDTA-10 mM NaCl.
Enzymes. Restriction enzymes wereobtained from commercialsourcesandwereusedassuggestedby the manufacturers.
Complementationtestswith viral DNAs.Solutions of
Tris-buffered saline (TBS) (17) (0.2 ml/60-mm dish)
containing varying dilutions of DNA of the mutant
beingtested, 4 ng of DNA from a known
complemen-tation group (= helper), and 100 p.g of DEAE-dextran were used totransfect freshlyconfluent monolayers of CV-1P cellsaspreviously described (24). After trans-fection, the cell monolayers were overlaid with agar medium (24) containing 4% fetal bovine serum and incubatedat40.5°C, and 3 days later the cells were fed with an additional overlay of agar medium supple-mented with 1% fetal bovine serum. At 6 days after infection, agar medium containing 0.01% neutral red wasadded, and the dishes weretransferred to 39.5°C for subsequent incubation. Each day thereafter, the plaques visible to theunaided eye werecounted, and their diameters were measured. Two mutants were presumedtocomplement when the plaques observed inthe mixedinfection appeared sooner and were larger than the plaques observed when cells were infected with either mutant by itself.
Complementation tests with virions. When a condi-tionally lethal mutant could serve as the lawn of potentially complementing helper virus, tests were performed essentially as described previously (24; Mertz, Ph.D.thesis). Briefly, freshly confluent mono-layers of CV-1P cells (1 x 106 to 2 x 106 cells per 60-mm dish) were coinfected with 0.1-ml portions of serial dilutions of the mutant being tested and 0.1-ml portions of the mutant of known complementation properties (2 x 105 to1 x 106PFU/ml).After incuba-tion for 2 h at 37°C with occasional rocking of the dishes, the cells were overlaid with agar medium and treatedsubsequently as described above.
When both mutants were viable although poorly growing (e.g., dlG806 and dlG810), complementation tests with virions were performed essentially as de-scribedbyBrockman andNathans (3).Briefly, CV-1P cells were infected at a multiplicity of infection of approximately 8 PFU of each mutant per cell and incubated at 37°C in medium containing 2% fetal bovine serum. The next day, the infected cells were removed fromthe dish withtrypsin, serially diluted in liquid mediumcontaining 10% fetal bovine serum, and plated onto new dishes. After incubation at 37°C for 4 htoenable the cells toattach to the dish, uninfected CV-1P cells (5x
10'
cells per 60-mm dish) wereadded tocreate amonolayer of cells. Incubation was contin-uedinliquid medium at 37°C for 1 additional day. The monolayers of cells were then washed once with TBS,on November 10, 2019 by guest
http://jvi.asm.org/
overlaid with agar medium, and treated subsequently as described above.
Agarose gel electrophoresis. DNA samples were electrophoresed at room temperature in horizontal gels(thickness, 1 to 2 mm) of 1.0 to 1.5% agarose in 4x TAbuffer (0.16 M Tris-acetate, pH 8.3, 0.08 M sodium acetate, 8 mM EDTA) at 1.5 to 2 V/cm until the bromophenol blue dye had migrated 3 to 6 cm. The gels werestained by incubation for 20to40minin4x TAbuffer containing 0.5 ,ug of ethidium bromide per ml, destained by incubation for 15 to 30 min in water, and photographed by using Tri-X or Polaroid type 57 film, an orange filter, and a short-wavelength UV light box.
Isolation and purification of[3H[lysine-labeled SV40 virions. MA-134 cells were infected with 20 PFU of WT800virus per cell and incubated at 37°C in medium supplemented with2% fetal bovine serum. After 34 h themedium in each 100-mm dish was replaced with 5 mlof mediumcontaining one-fifth the normal concen-trationoflysine,2% dialyzed fetal bovine serum, and 0.1 mCi ofL-[4,5-3H]lysine (40Ci/mmol)perml; 12 h later, 5 ml of complete medium containing 2% fetal bovine serum was added to the medium already pres-ent ineach dish. Incubation was continued at 37°C. The cells were harvested 118 h after infection by
scrapingthem off the dishes into TBS witha rubber
policeman.Thecellsweredisrupted bythreecycles of
freezingandthawing, extraction withchloroform,and
sonication. After centrifugation at 5,000 rpm for 10 min at4°C,theresultingsupernatantwascollected and
made0.75%Nonidet P-40. SV40 virionswerepurified
from this extract by sedimentation (SW41 rotor, 35,000 rpm, 4h,15°C)through 1.5 ml of 15% (wt/vol) sucroseinto 3 ml ofCsCl(density,1.33g/cm3)in TBS
lacking calcium and magnesium (26), followed by centrifugation (SW50.1 rotor, 30,000 rpm, 40 h, 4°C) to equilibrium in a density gradient of CsCl (average density, 1.33 g/cm3). After 100 p.gof bovine serum albumin wasadded,thepurifiedvirionsweredialyzed
against 10 mMNaPO4(pH7.2)-0.1 MNaCI-0.1 mM EDTAand storedat4°C.
Determination of ratios ofvirion particles toPFU. SV40 virions were purified as described above from mutant-infected andwild-typevirus-infected cells. Af-ter CsClequilibrium density gradient centrifugation, each band ofpurifiedvirionswascollectedseparately.
A portion of each purified virion preparation was diluted 100-fold into TBS containing2% fetal bovine serumand was storedfrozen until it wastitrated to determine the number of PFU per milliliter as de-scribed previously (10). The number of virionparticles
permilliliterwasdeterminedbyspectroscopy, assum-ing1 Uof absorbanceat260nmofSV40virionswas equivalent to 6.5 x 1012physical particles per ml (41).
RESULTS
Biological and physical properties of late leader
region mutants. Some of the biological and
phys-ical properties of late leader region mutants
dl-801 through dl-810 have been described
previ-ously
(1, 10,
lOa, 25; Mertz, Ph.D. thesis). In
this paper we describe experiments performed
with these mutants that were directed
specifical-ly toward trying to define the function(s) of the
agnogene.
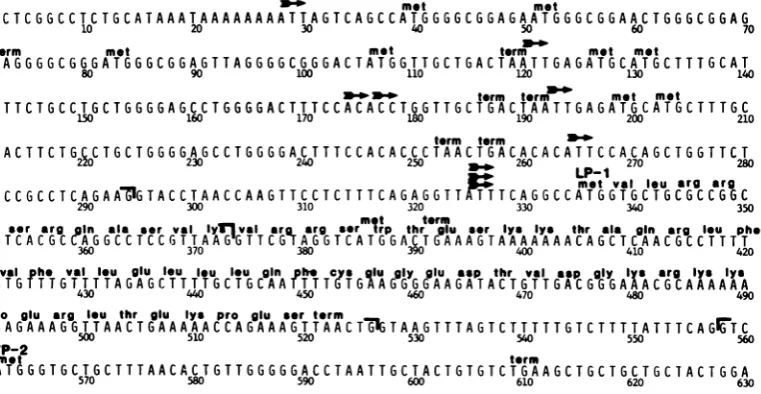
Figure
1shows the precise
maplocation of the61-amino acid protein that could be encoded by
moo- met met
GCCTCGGCCTCTGCATAAATAAAAAAAATTAGTCAGCCATGGGGCGGAGAATGGGCGGAACTGGGCGGAG
10 20 30 40 50 60 70
term met met term met met
TTAGGGGCGGGATGGGCGGAGTTAGGGGCGGGACTATGGTTGCTGACTAATTGAGATGCATGCTTTGCAT
80 90 100 110 120 130 140
no-wP.-Wterm term met met
ACTTCTGCCTGCTGGGGAGCCTGGGGACTTTCCACACCTLGGTTGCTGACTAATTGAGATGCATGCTTTGC
150 160 170 190 200 210
term term mo
ATACTTCTG2~0CTGCTGGGGAGCCTGGGGACTTTCCACACCCTAACTGACACACATTCCACAGCTGGTTCT
230 240 250 _ 260 270 280
-_
LP- 15s. met val Ieu erg arg
TTCCGCCTCAGAAnIGTACCTAACCAAGTTCCTCTTTCAGAGGTTNTTTTCAGGCCATGGTGCTGCGCCGGC
290 300 310 320 330 340 350
leu eer erg In ala *er vel lyTlval mr thrglu eer lye lys thr ala gin rg leu phe
T G T C ACGCCAG C C TC C G T TA A
GGaT
TCrGT AaG
6 TC A TGGA3C
T G AA A G T A AA A A AAACAGCTCAACGCCTTTT360 370 380 390 400 410 420
Vlhe
eal
leugIU
leuleu
leu gin h ugyguaptrvlap9Vepe cele @ E gnDph. glu glU9y glu Sep thr v.1 Se gIy ly*erg lye lye
TGTGTTTGTTTTAGAGCTTTTGCTGCAATTTTgTGAAGGGGAAGATACTGTTGACGGGAXACGCAAAAAAA
430 440 450 460 470 480 490
pro glu arg leu thr glu lye pro glu *er term
CCAGAAAGG6TTAACTGAAA AAACCAGAAAGGTTAACT3GTA A GTTTAGTCT T TTTGTCTTTTATTTCAGr'TC
500 510 520 530 540 550 560
VP-2met term
CATGGGTGCTGCTTTAACACTGTTGGGGGACCTAATTGCTACTGTGTCTGAAGCTGCTGCTGCTACTGGA
570 580 590 600 610 620 630
FIG. 1. Nucleotidesequenceof the late leaderregionoftheSV40genome.Nucleotide residuesarenumbered accordingtothesystem of Tooze(39), startingfrom thecenterof thepalindrome intheoriginof viralDNA replication. Thearrowsindicate thelocations of the5'endsofprominent speciesof latemRNAs,80to
90%o
of whichmaptoresidue 325(9). Thebrackets indicate the locations of donor andacceptorsplicesites used in theproductionof the 16S RNAsand the threesplicedclassesof19SRNAs(9).metandtermindicatethelocations of
triplet codons thatmayfunctionasinitiators andin-phaseterminatorsof translation.LP-1and VP-2indicate the AUG codons used in thesynthesisof the61-amino acidproductoftheagnogeneandVP-2,
respectively.
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
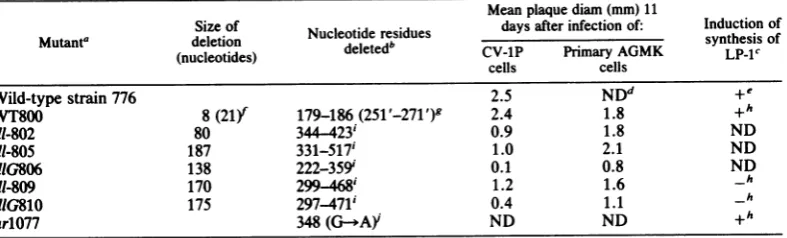
[image:3.489.64.445.408.605.2]TABLE 1. Summaryofphysical, biological, andgenetic propertiesof lateleaderregionmutants
Meanplaquediam (mm) 11
Sizeof Nucleotide residues daysafterinfection of: Inductionof
Mutant' deletion
deee'synthesis
of(nucleotides) deleted CV-1P Primary AGMK LP-1c
cells cells
Wild-type strain 776 2.5 NDd +e
WT800 8(21) 179-186 (251 '-271 ') 2.4 1.8 +h
dl-802 80 344 423' 0.9 1.8 ND
dl-805 187 331-517' 1.0 2.1 ND
dIG806
138222-359'
0.1 0.8 NDdI-809 170
299-468'
1.2 1.6 _hdlG810 175
297-471'
0.4 1.1_h
arlO77 348
(G-*AY
ND ND +ha Strain 776 and WT800 arenaturally occurringwild-type strains of SV40. G indicates that themutantbelongs
tocomplementationgroup G; a dash indicates that thecomplementationgroup has not beendeterminedyet.
bThenumberingsystem used here is the system shown inFig. 1 forwild-typestrain 776.Therefore, WT800is
indicated as having asubstitutioneven though it differs only in theprecise regionof the genome that isrepeated
in tandem. Wherethere isambiguityin theprecise endpointsof a deletion due to arepeatedsequence at itsends,
the largestpossible residuenumbers are given.
cDetermined as
described
in thelegend to Fig. 4. +, Presence; -, absence.dND, Notdetermined.
'Datafrom references 15 and 16.
fThenumberin parentheses indicates the size of an insertion.
8Trimbleand Mertz,unpublished data. The residue numbers in parentheses indicate the second copy of the
sequence that ispresentinplace of thedeletednucleotide residues. hData from this paper.
'These
mutants were derived from WT800; consequently, although not indicated, they all have the substitutionrelativetowild-typestrain776of residues 251' to 271' inplaceof residues 179 to186,aswellasthe deletions shown (1).iMutantar1077contains a single basepairchangeof G-C to A-T at nucleotide residue 348(32).
the late leader region of
theSV40 genome.
Based
uponthe nucleotide sequences of
theirgenomes
(1)-
(Table
1),mutants dl-801 through
dl-810 should all be defective in
thesynthesis
ofthis
protein;
whereas dl-802 contains
aframe-shift mutation
that
should result in premature
termination of
translation,
the
other
mutantsall
lack the
translation
initiation codon that
begins
atnucleotide residue
335.
Therefore,
someof
these
mutants wereselected for
further
study.
The
proposed
product
of the agnogene is
ahighly basic
protein containing
sevenlysine
resi-dues and
eight arginine
residues(Fig.
1).
Conse-quently,
this
histone-like
protein might
be
ex-pected
to function in association withSV40
minichromosomes
oras acomponentof virions.
SV40 virions
containing
mutantproteins
fre-quently exhibit
alteredphysical
orbiological
properties
(39).
Forexample,
incubation of
mu-tant
SV-L
virions
at50°C
for
2hin
TBS
contain-ing
serumreduces their
infectivity
104-fold
(36).
However,
when
wetreated virions
of
WT800and
mutantsdl-801
through
dl-810 in
ananalo-gous manner,
they
wereinactivated
lessthan
fivefold
(data
notshown).
Similarly,
mutants in theminor
capsid
proteins
VP-2and VP-3
pro-duce virions
with
verylow
specific
infectivities
because
they
aredefective in
adsorption
and
uncoating,
respectively (39). On the other hand,
the
ratios of
particles
to PFU
for 806 and
dl-810
virions
werewithin threefold of the ratio
observed with
WT800
virions
(data
notshown).
Therefore,
weconclude that the virions
pro-duced
by these late leader
region
mutants arelike the virions of wild-type virus with respect
tophysical integrity
and
adsorption, penetration,
and
uncoating.
Some
mutantsof
SV40 exhibit phenotypes
that
varywith the host
cell
type.For
example,
mutant
SV-L
forms
plaques in
primary
AGMK
cells
500-fold
moreefficiently
than in
established
AGMK cells (37). Therefore,
wealso
compared
the
abilities of wild-type virus and
mutantsdl-801
through dl-810
toform plaques on freshly
confluent monolayers of CV-1P and primary
AGMK cells that were infected in
parallel
withappropriately diluted virus stocks
andincubated
at37°C in
agarmedium.
Asexpected,
theleaderregion
mutantsproduced
plaques in CV-1P cellsthat were smaller than theplaques made by
wild-type
virus
(Table
1),but
atefficiencies
that wereequal
tothose observed
inprimary AGMK
cells(data
notshown). Surprising, therefore,
wasthefinding
thatall mutants except
dl-806 and dl-810,
the two poorest
growing mutants, formed
wild-type-sized plaques in primary AGMK
cells(Ta-VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
40 MERTZ, MURPHY, AND BARKAN
TABLE 2. Ratesof appearance of plaques in mixed infectionsa
Infecting DNAs %of maximumno.of
plaques
onrnfecting DNAs thefollowing days after infection:
Mutant Helper 7 8 9 10 11
dl-810 None 3 10 29 69 100
dl-810 tsA58 67 96 100 100
dl-810 tsB201 54 92 100 100
dl-810 tsC219 65 99 100 100 dl-810 tsD202 60 98 100 dl-810 dIE1226 69 100 100
tsB201 tsA58 60 96 99 100
tsB201 tsD202 86 100 100 tsB201 dlE1226 93 100 100
a
CV-IP
cells in 60-mm disheswere cotransfectedwith 0.04 to 0.2 ngof mutant DNA and 4 ng ofhelper DNAin 0.2 mlof TBScontaining 500 ,ugof DEAE-dextran per ml and treatedsubsequentlyasdescribed in the text. The valuespresentedaretheaverages from
duplicate sets of infected cell monolayers, each of
which contained 30 to 90 plaques by 11 days after infection.
ble 1). This result
indicates that, although
viri-onsof these
mutants areindistinguishable from
those
of wild
typewith
respect toadsorption,
penetration,
and
uncoating, these
mutants arenevertheless defective in
atleast
onefunction
that
is
notimportant for efficient growth
inprimary AGMK cells.
Thefinding
that mutantsdl-806 and dl-810
form,
evenin
primary AGMK
cells,
somewhat smaller
plaques than wild
typedoes
probably
indicates that these
two mutantspossess one or more
additional
defects that
arenot
compensated for in either primary AGMK
orCV-1P cells.
Mutants
dl-806
anddl-810 define
anewcomple-mentationgroup of SV40, group G.
If the
agno-gene
encodes
aprotein
that
is
synthesized in
SV40-infected
cells, these late leader region
mutantsmay
be
defective in
afunction
that canbe supplied in trans by a coinfecting helper
virus. Totestthishypothesis,
complementation
testswere
performed
by usingamodification ofa mixed-plaque assay that has been described
previously
(24; Mertz, Ph.D. thesis). Monolay-ersofCV-1P
cellswerecotransfected with serialdilutions of DNA ofalateleaderregionmutant
and 4 ngof DNA ofa mutantfromeach of the
known
complementation
groups(= helper). Thecells were incubated at 40.5°C, a temperature
that is
nonpermissive
for the growth of thetemperature-sensitive
helpers. In addition,al-though
mutantsdl-806, dl-810, and dlE1226
areviable,onaveragetheir plaquesappearlater and
growmuchmoreslowly than the plaques of wild
type (Table 1) (6).
Consequently,
most of theplaques that are visible within 8 days after
infectionarelikelytobe mixed plaquesresulting
fromtwo
complementing
mutants.Tables 2 and 3 show thetypeof data obtained
from one experiment of this kind. When cells
were
transfected
with mutant dl-810 DNA byitself, only 10% of the plaqueswerevisible with
anunaidedeyewithin 8days. On theother hand,
more than 90% of the plaques were visible by
this time when DNA from
complementation
groupsA,B, C, D,orEwasincluded.
Comple-mentation with mutants defective in small t
antigen was not tested because these group F
mutants make plaques that are only slightly
smallerthan those of wild type. Nevertheless,
we assume that late leader regionmutants can alsobe
complemented
bygroupFmutantssincethelatter are defective in afunction expressed
early ratherthan lateinthe lytic cycle (39).
Thetotal numbers ofplaques that eventually
appeared with and without helper DNA were
similar(datanotshown). This resultwas
expect-edsince4ngof helper DNAper60-mm dish of
cells isenoughtoinfect allof the
approximately
2%of the cells thatare susceptible to
[image:5.489.51.449.523.643.2]transfec-tion by the DEAE-dextran technique whichwe
TABLE 3. Ratesofgrowthofplaques resultingfrom mixedinfectionsa
Infecting DNAs Mean sizes(mm) of visible plaques on the following days after infection:
Mutant Helper 8 9 10 11
dl-810 None 0.3
(0.1-0.7)b
0.3(0.1-0.7) 0.5(0.1-1.8) 0.6(0.1-1.5) dl-810 tsA58 1.0(0.2-2.0) 1.6(0.4-2.7) 1.8(1.2-2.8)dl-810 tsB201 0.8 (0.3-1.6) 1.4(0.4-2.8) 1.9(1.1-3.0) dl-810 tsC219 0.6(0.2-1.2) 1.3(0.5-2.2) 1.5(1.0-2.2) dl-810 tsD202 1.0(0.2-1.5) 1.6(0.5-3.2)
dl-810 dlE1226 0.8(0.2-1.7) 1.3(0.6-2.1)
tsB201 tsAS8 1.1(0.3-2.2) 2.0(1.3-3.0) 2.4(1.8-3.5) tsB201 tsD202 1.2(0.3-2.0) 1.4(0.8-2.3)
tsB201
dIE1226
1.9(0.8-3.0)aThe valuespresentedwereobtained from thesame setof infections describedinTable2, footnote a; each number is the average of the diameters of6 to12plaques selectedatrandom formeasurement.
bThe numbers inparenthesesareranges.
on November 10, 2019 by guest
http://jvi.asm.org/
used (24).
Consequently,
few, if
any,of the
plaques
observed in the
cotransfections should
have
arisen from cells
receiving only dl-810.
This expectation
wasconfirmed
by thefinding
that,
exceptfor the data
at8
days, the
rangesof
plaque
sizes observed
inthe
cotransfections
were always
outside
the mean plaquesizes
ob-served in the absence of helper (Table
3). Eventhe
one apparentexception actually followed
this rule
since,
given that only 10% of
thedl-810
plaques
werevisible
at8
days (Table 2), the real,
asopposed
toobserved,
meanplaque size
in the
absence
of
helper
wasless than 0.1
mm.There-fore,
asmeasured
both
by the
ratesof
appear-anceand by the sizes of the plaques,
mutantdl-810
complemented
mutantsin all of
theother
known complementation
groupsof SV40.
Several
typesof experiments
wereperformed
toconfirm that these results
wereinterpreted
correctly and that the
apparentcomplementa-tion did
nothave
trivial, uninteresting
explana-tions.
First,
complementation
tests wereper-formed
in parallel with tsB201. The resulting
data
(Tables
2and
3)
were verysimilar
to thedata
obtained with
dl-810, although
the
meansand
rangesof
plaque sizes
mayhave been
slight-ly larger with tsB201.
Therefore,
weconclude
that
dl-810 complements the
mutantsin other
complementation
groupsalmost
aswell
as orequally
aswell
astsB201,
which is known
tocomplement other
mutantsexcellently
asdeter-mined
by
avariety of complementation
assays,including the classical
ones(39).Second, data similar
tothe data obtained with
dl-810
werealso
obtained in
complementation
tests
performed
in
ananalogous
fashion with
mutant
dl-806 (data
notshown).
The oneappar-ent
difference observed in these
two setsof
experiments
wasthat whereas the
helpers
in-creased the
ratesof
appearanceand sizes of
dl-810
plaques
by
3
to 4days (Tables
2and
3),
they increased those of dl-806
by
only
2 to3
days. This latter
finding
indicates that the
help-erscompensated for
only
someof the defects of
dl-806.
Third, complementation
testsbetween dl-806
and
dl-810
werealso
performed. However,
be-cause
these
mutants areviable,
the
monolayer of
cells would
havebeen
destroyed
by transfection
with 4 ng
of
DNAof
either oneof
them per60-mm
dish.
Consequently,
the tests wereper-formed
withvirions
by coinfecting
cells at amultiplicity of infection of approximately
8 PFUof
each mutant per cell and thendiluting
andreplating
theinfected cells
withfresh
monolay-ersof uninfected
cells. As acontrol, mixed
infections were also
done
with stocksof dl-810
and tsA58 virus.
Coinfection with dl-810
andtsA58 virus
indicated
that mixedplaques
ap-peared
and grew at ratessimilar
to the ratesobserved in the DNA
transfections
performed
with
the same mutants(data
notshown).
Never-theless,
the
plaques from the
coinfection with
dl-806 and dl-810
weresimilar
tothose observed
when cells were
infected with
either
mutantby
itself.
Therefore,
weconclude that
(i)
mutantsdl-806 and
dl-810
aredefective in the
sametrans-complementable function, and (ii)
asopposed
towhat
is observed with
group D mutants(39),
these
late leader
region
mutantscomplement
asefficiently when they
arepackaged
into virions
aswhen
they
arein the
form
of
purified
DNA,
thus
confirming
the
finding
that
they
are notdefective in
adsorption, penetration,
oruncoat-ing.
Theoretically, the plaques observed in the
experiments
described above could have been
produced by
(i) wild-type recombinants that
arose
in the
mixedly
infected cells
or(ii)
unidi-rectional
complementation in which the late
leader
region
mutants,being
viable, supplied
the
helpers with all
of the viral
products needed for
efficient
multiplication,
but the
helpers
wereunable
toimprove the slow
rateof growth of the
viable leader
mutants.If either of these
possibili-ties had
actually occurred,
wewould have
ex-pected the virus that
grew outof the
cotransfec-tions
tocontain either
(i)
atleast
somewild-type
SV40
genome or(ii) only
asmall
percentageof
leader
mutantgenomes.To
rule
outthese
possibilities, plaques
ob-served in
experiments similar
tothose
summa-rized in Tables
2and 3
werepicked and used
toproduce small stocks of viral
DNA. Aportion
ofeach of these DNA stocks
wasincubated with
HpaII
only
orHpaII
plus
HaeII
and then
elec-trophoresed in
agarosegels. Since
ourlate
lead-erregion
mutants areresistant
tocleavage by
HpaII (25), whereas dlE1226 is
resistant
to
cleavage by
HaeII(6), these reactions enabled
ustodistinguish
amongleader
mutant,dlE1226,
and
wild-type
genomes.The data obtained
from
oneexperiment of this
type are
shown
in
Fig.
2.Since these
DNAstocks
were grownfrom the
small amountsof
virus
presentin
single plaques,
anywild-type
recombinants produced in the initial infections
should have had
sufficient opportunity
tooutreplicate
the
mutants andbecome
thepre-dominant
species. Nevertheless,
nowild-type
DNA was detected in the DNAs grown
from
plaques
originating from
theeotransfections.
Instead,
these DNAstocks consisted of
approxi-mately
equimolar
amounts of the two mutants.Similar results
werealso obtained with
plaques
produced from cotransfections
withdl-810
anddlE1226 (data
notshown).
Therefore,
wecon-clude
that theplaques observed
in thesecomple-mentation
tests wereindeed
mixedplaques.
To
eliminate
definitively
thepossibility
thaton November 10, 2019 by guest
http://jvi.asm.org/
A30 E1226 E1226
G806 W"'WT800 £1226 G806 G806 FMa.{
!- O.9
1-I-0
[image:7.489.52.244.69.223.2]1 2 3 4 5 6 7 8 9 10
FIG. 2. Analysis of the viral genomes present in DNAstocks grown from mixed plaques. Plaques ob-tained in experiments similar to those described in Tables 2 and 3 were picked with a Pasteur pipette and placed in TBScontaining 2% fetal bovine serum. The virus wasreleased from the infected cells present in eachplaqueby three cycles of freezing and thawing and was used to infectconfluentmonolayersof CV-1 cells at a multiplicity of infection of 10-2 to 10-3
PFU/cell. The cells were then incubated at 37°C in medium supplemented with 2% fetal bovine serum. When more than50% of the cells exhibited cytopathic
effect,viral DNA was harvestedby theprocedure of
Hirt(14) andpurified as described in the text. All of the DNA samples were incubated with restriction endonucleaseHpaII.One-half of each sample was also incubated withrestriction endonucleaseHaeII(lanes 2, 4, 6, 8, and 10). The patterns of digestion were determinedbyelectrophoresis in a1.0%agarosegel.I, II,and L indicate thepositionsof supercoiled, nicked
circular, and unit length linearSV40DNAs,
respec-tively; 0.91 and 0.09 mark thepositions of the DNA
fragments that resulted from digestion of wild-type
DNA withbothHpaIIandHaeII. Lanes 1 and 2 and lanes 3 and 4,DNA samplesfrom plaques picked from cells infected as controls with d1G806 and WT800, respectively; lanes 5 and 6 and lanes 7 to 10, DNA samples fromplaquesobtained from mixed infections
with dlE1226 plus tsA30 and
dIE1226
plus dlG806,respectively.
complementation
wasunidirectional,
we alsolooked
for
changes either
intracellularly
or invirions in
therelative molar ratios
of
coinfecting
viruses
during
single cycles of growth.
Figure
3shows that
in cells
mixedly
infected
with dl-806and
wild
type(i)
the percentageof
theintracellu-lar
viral
DNA that was mutant remainedcon-stant
during
the courseof the infection
and(ii)
the
dl-806
genomes werepackaged
into virions asefficiently
as thewild-type
genomes were.The
first
of
theseconclusions
wasconfirmed
by
a
Southern blot
analysis (33)
of the
relativemolar ratios
of
thetwogenomesatearlier
timesafter
infection;
wefound
(data
notshown)
thateven at21 h the percentage of the intracellular
viral
DNA that wasdl-806
wasapproximately
70%.Results similartothesewerealso obtained
in experiments involving cells coinfected
with
dl-810 and wild-type virus (data not shown).
Therefore, we conclude that these late leader
region mutants (i) do not possess a cis-acting
defect in either viral DNAreplication orvirion
assembly and (ii) trulybenefit from thepresence
ofacomplementing helper.
In summary, the findings described above
clearly indicate that mutants dl-806 and dl-810
define a new complementation group of SV40.
We propose that this new complementation
groupbe named group G.
Synthesis of the agnogene protein in monkey
cellsinfected with wildtypeand late leaderregion
44 h
G) 0
=0
68 h
_ CZ
-D
1'-s
L-l
| _
Hpall-cut
PBR322
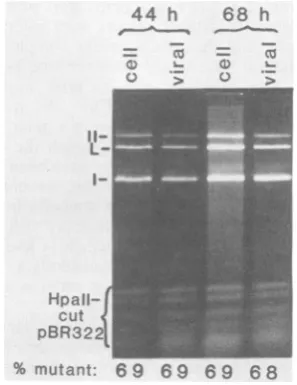
% mutant: 6 9 69 6 9 68 FIG. 3. Analysis of the viral genomes present
intra-cellularly (cell)andin virions(viral)atdifferent times
aftercoinfection withdIG806and WT800.CV-1P cells were mixedly infected with approximately 10 PFU each ofdIG806and WT800per cellandincubatedat
37°C.Atdifferent timesthereafter,batchesof infected
cells were harvested both by the method of Hirt(14)
andbythevirion isolationproceduredescribed in the text. Intracellular viral DNA was purified from the
Hirtlysatesby extraction withphenol andchloroform,
treatment with pancreatic RNase, and precipitation
with ethanol. Viral DNA was obtained from purified virionsby incubation at65Cfor 0.5 h in50mMTris
(pH 7.6)-S50 mM NaCl-10 mM EDTA, followed by
treatmentfor 2 h at37°Cwith 100,ug ofproteinaseK perml,extraction withphenol-CHCl3-isoamylalcohol (25:25:1), and precipitation with ethanol. Theresulting
viral DNA samples were incubated with restriction endonuclease HpaII and electrophoresed in a 1.3% agarosegel. Tocheck that thedigestionshadgoneto completion, pBR322 DNA was included in each reac-tion.The gel was photographedbyusingTri-Xfilm,
andthe relative amount of DNA in each band was quantified by densitometry. The fraction of the viral DNAthatwasdlG806wasdeterminedasfollows:(I+
II)/(I +II + L).
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.489.277.426.252.445.2]mutants of
SV40.
Tolook directly for synthesis
of
a61-amino acid leader-encoded protein,
which
we propose to name LP-1 (LPstanding
for leader-encoded protein), monkey cells
wereinfected with dl-809, dlG810, ar1077,
orwild
type,
radiolabeled
40 to 48 h laterwith
[3H]leu-cine,
[3H]lysine,
or[35S]methionine
for 0.5 h,
and then
fractionated into cytoplasmic and
nu-clear
components.The
resulting
extracts wereelectrophoresed in
gradient
polyacrylamide-so-dium dodecyl sulfate gels and examined by
fluorography.
The data
from
onesuch
experiment
(Fig. 4)
indicate that
(i)
aprotein of the
appropriate
molecular
weight for LP-1
(approximately 8
x103)
wassynthesized
at ahigh
rateby
WT800-infected cells but
notby
mock-infected
ordl-810-infected cells and (ii) whereas
a0.5 h
of
pulse-labeling
wassufficiently long for
mostof
the
newly
synthesized VP-1, VP-3, and cellular
histone proteins
tohave
been
transported
tothe
nucleus,
mostof the radiolabeled 8-kilodalton
virus-induced
protein
wasfound in the
cytoplas-mic
fraction.
In
addition, the fact that the
dl-810-infected cells
synthesized VP-1 and VP-3
atrates
comparable
tothe
ratesobserved in
WT800-infected cells indicates that the failure of
these
cells
tohave made the
8-kilodalton
protein
could be attributed
specifically
tothe mutation
in the late leader
region.
Other
experiments
(data
notshown) indicated
that
(i) whereas
mutantar1077,
which
theoreti-cally
contains
amissense mutation in LP-1,
induced the synthesis of the 8-kilodalton
pro-tein,
mutantdl-809
did
notand
(ii) the
8-kilodal-tonprotein
wasradiolabeled
by
[3H]lysine
and
by [3H]leucine,
but
notby
[35S]methionine.
This
latter
finding
is
consistent
with the
amino acid
composition
of the
protein
aspredicted from
the
DNA
sequenceand
indicates that the
only
me-thionine
of
the
protein,
which is located
atthe
amino terminus, is removed
intracellularly.
Finally,
preliminary
data
(S.
A.
Sedman and
J.
E.Mertz,unpublished data) indicated that
an8-kilodalton
protein
wasalso
synthesized
in
large
amountswhen
wild-type
SV40-specific
RNA
selected by
hybridization
toSV40 DNA
wasincluded in
amicrococcal nuclease-treated
rabbit
reticulocyte lysate in vitro translation
system.Together with the data
presented above,
this
finding leads
us toconclude that the
8-kilodalton
protein
is
probably LP-1, the product
of the
agnogene.Analysis of virion proteins. To
determine
whether the
8-kilodalton
virus-induced protein
wasincorporated
into virions, SV40 virions
werepurified from
[3H]lysine-labeled,
WT800-infected
monkey cells and electrophoresed in
asodium
dodecyl
sulfate-polyacrylamide tube gel.
The presence
and relative
amountsof the
vari-nuclei
0 0
0
-c o
co Xc v
kdaltons (5 H
VPI-2YP-1
-25.7-histones P Ia
LP-1
-1 2 3
-14.3-
-12.3--L P-i
-3
cytoplasm
o 0
-v
co co U,
(3
0 0_6 t: E
-mwquo wo
4 5 6
FIG. 4. Electrophoretic analysis of the
[3H]leu-cine-labeled proteins found in the nuclei and cyto-plasmofdlG810-and WT800-infected monkey cells. CV-1 cells in 100-mm dishes wereinfected with
ap-proximately20 PFU ofdlG810orWT800 percell and
incubated at 37°C in medium containing 4% fetal bovine serum. After 48 h, the cellswerewashedonce withTBS and once withmedium lacking leucine and then were incubated at 37°C for 30 min in 1 ml of leucine-free medium containing4% dialyzed fetal bo-vine serum and 300
plCi
ofL-[4,5-3H]leucine
(57 Ci/ mmol) per ml. The cells were then washed twice withTBS at0°Cand scrapedoff the dish in 1 mlof TBS
containing 0.5%Nonidet P-40 and 300 1Lgof
phenyl-methylsulfonylfluoride per ml. After incubationat0°C
for 5 min, the nucleiwerepelleted bycentrifugationat 2,000 x g for 10 min, washedtwice with TBS,
sus-pended in 2 ml of sample loading buffer (21), and
boiled for 5 min withfrequent blendingwithaVortex mixer. The supernatant from the first centrifugation
(cytoplasm)wasdiluteddirectlyinto 2x sample
load-ing buffer; 100,000 cpm of each samplewas electro-phoresed for 3 h at 125 V in a 15 to 20% gradient
acrylamide-sodium dodecylsulfategel preparedbythe
method of Laemmli (21). The gel was stained with Coomassie brilliant blue to visualize the molecular weight markers (chymotrypsinogen A, 25,700; lyso-zyme, 14,300;cytochrome c, 12,300; insulin, -3,000) andthenprocessed forfluorography (22). Lanes 3and 6contained nuclear andcytoplasmicextracts, respec-tively, of cells processed in parallel as controls but without virus present in the infection. (The slightly fastermobility of the VP-1synthesized in the dlG810-infectedcellsthantheVP-1synthesized in the WT800-infectedcellswasprobably dueto asecond-site muta-tionmapping intheVP-1gene thataroseduring serial passage of the mutant inmonkey cells
[Barkan
and Mertz,manuscriptinpreparation].)on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.489.250.446.91.314.2]44 AND BARKAN
ous
radiolabeled proteins
weredetermined by
scintillation spectroscopy after
slicing
thegel
into 125 sections, each 2
mmthick. This
experi-ment(data
notshown) indicated
that,
whereas
1,570 cpm was
presentin the fraction
containing
the
largest
amountof cellular histone protein
H4,
fewer
than 30
cpm was presentin any
of the
fractions expected
tocontain
LP-1.
Since
7of
the
61amino
acid residues in LP-1
arelysine
(Fig.
1),whereas
H4is also
composed
of
ap-proximately
11%
lysine
residues and is present
at alevel
of 20
to25
copies
pervirion
(39),
wecalculated that
onaverage, the
SV40 virions
contained
fewer than
twocopies
of
LP-1.The
conclusion that LP-1 is
probably
absent from
mature vinons agrees
both with the failure of
LP-1
toaccumulate in
large
amountsin the
nucleus
(Fig.
4) and with the
finding
that
onceformed,
virions of the late leader
region
mutantsbehave
biologically
and
physically
like virions of
wild-type SV40.
DISCUSSION
The main
findings of this work
arethat
mu-tants
deleted in the
major late leader
region
both
define
a newcomplementation group and fail
toinduce the synthesis of
an8-kilodalton protein
that
is made in
wild-type
SV40-infected cells.
After
wecompleted these
studies,
Jay
etal.
(16)
demonstrated
directly
by amino-terminal
se-quence
analysis that this
protein
is
indeed the
61-amino
acid
protein
LP-1
whose
sequenceis
shown in
Fig.
1.Other
workers
(15; Margolskee
and
Nathans, personal
communication;
South-ern
and Berg, personal
communication)
have
also
independently
found
an8-kilodalton
protein
in
SV40-infected cells and have identified it
by
avariety of
techniques,
including
mutational
anal-ysis
and determination of which amino acids
canand
cannotbe
used
toradiolabel it.
Therefore,
we
conclude that
ourlate
leader
region
mutants-fail
tomake
the8-kilodalton virus-encoded
pro-tein
LP-1because
they
lack the nucleotide
resi-dues required for its
synthesis.
What
is the
reasonfor
thecomplementation
properties of
mutantsdl-806 and dl-810?
Theconclusion
statedabove
provides
alikely
expla-nation. However,
these mutantsarealso
defec-tive
in"attenuation" of late-strand
RNAsyn-thesis
(13, 25a), the efficiencies of
splicing
andprecise locations of
the 5'ends of
thelate-strand
mRNAs
(10, 10a, 12,
28, 40;
A. Barkanand J. E.Mertz, manuscript
inpreparation),
and thepre-cise
ratesof
synthesis
invirus-infected
cells ofthe virion
proteins
VP-1, VP-2,
and VP-3(Bar-kan
andMertz,
manuscript
inpreparation) and,
possibly,
other leader-encoded
proteins
(see
be-low).
Consequently,
experiments
with mutantscontaining
single
nucleotidepair changes
ratherthan
deletions
will need to bedone
to answerthis
question
definitively.
Properties
andfunction(s)
of LP-1.The
data
presented
here and
elsewhere (13, 15,
16)indi-cate that LP-1
(i)
is
a61-amino
acid,
highly
basic,
acid-soluble, phosphorylated
protein
whose
amino terminus is
anunblocked
valine
residue;
(ii)
is
synthesized
from 16S
mRNAatahigh
rate atlate
times in the
lytic
cycle, but has
ahalf-life of
only
2 to 3h;
(iii)
binds
tosingle-stranded
anddouble-stranded DNA in
0.1 MNaCl; (iv)
ishelpful,
but
notessential, for
alate
step in the
viral
lytic
cycle; (v)
canfunction in
trans to aid
the
growth of mutants defective in
its
synthesis; (vi)
is
also
encoded, although
with
a somewhat
different sequence,
by
BK
virus,
arelated human
papovavirus;
(vii)
may
exist in
association with
SV40
minichromosomes
orin-termediates
invirion
assembly,
but
is
notpres-ent in mature
virions; (viii)
is
found
predomi-nantly
in the
cytoplasmic
fraction of cell
extracts,
although possibly
because of
leakage
from the nucleus
during
the
fractionation
proce-dure;
and
(ix)
may enable
VP-1 toperform
oneof its
functions
morereadily (Margolskee and
Nathans, personal
communication; Barkan and
Mertz, manuscript
inpreparation).
One
hypothesis consistent with all of these
findings
andthe
known properties of
mutantsdefective in the
synthesis of LP-1 is that
afunction of
LP-1may
be to facilitate
virion
assembly.
Possibly, this could be accomplished
by
LP-1serving
as anonreusable
scaffolding
protein which displaces HI from
theminichro-mosomes and
then promotes the
condensation
of the
virion
proteins
ontotheviralDNA.
IfLP-1
molecules
weredegraded during
or as aconse-quence
ofthisreaction, this hypothesis would
explain both their short half-life and their
pre-dominantly cytoplasmic location, i.e., the
cyto-plasmic molecules
arethe
onesthat have not yet
participated
inthereaction.
Consistent
with thisproposed role
forLP-1
is thefact that the
cellular
histone
protein
Hi,
although present
intracellularly
onSV40
minichromosomes,
isprobably missing from virions
(39).
Ifour
hypothesis
is true, it wouldprovide
agood rationalization
as towhy SV40
encodesLP-1 onthe same mRNAs used in the
synthesis
of
VP-1; by making polygenic
mRNAs,SV40
mayhave
developed
asimple
mechanismfor
guaranteeing production of these two
proteins
atthe constant
relative
rates at which it utilizesthem. In
addition,
other plausiblefunctions
ofLP-1 that should be considered
include roles
in(i)
control ofinitiation oflate-strand
RNAsyn-thesis,
(ii) attenuation of late-strand RNAsyn-thesis (13), and (iii) regulation of
translation
startcodon
selection on thesepolygenic
viralmRNAs.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Are other proteins also encoded by the late
leader region of SV40? The findings discussed
above demonstrate definitively that the AUG
codonat nucleotide residues 335 to 337 can be
utilized in vivo for initiation of translation. Since
30 to 50% of the
19S
viral mRNAs present inwild-type SV40-infected monkey cells lack
nu-cleotide residues 374 to 557 (9), it is likely that
nucleotide residues 335 to 337canalso function
for the synthesis ofa30-amino acid polypeptide.
Eight additional AUG codons are present
be-tweennucleotide residues 39 and 256 of the late
leader region(Fig. 1). Preliminary findings
(Bar-kanand Mertz, unpublished data) have indicated
thatsomeof these codonsmaybe utilized in the
synthesis of small (i.e., 11- to 20-amino acid)
polypeptides. Experiments are in progress to
testthis hypothesis. Ifcorrect,it would provide
anexplanation for the finding that the 5' ends of
the late SV40 mRNAs are situated throughout
this region. In addition, it would indicate that attenuation of late-strand SV40 RNA synthesis
(13, 25a) may function as a mechanism for
enabling these possibly functional late-strand
leader region polypeptides to be synthesized
early in the lytic cycleorathigherratesthan the
capsid proteins are.
Implications for the mechanism of translation
start codon selection in eucaryotic mRNAs.
To-gether with what is presently known about the
primarystructuresof theSV40 late mRNAs (9),
the conclusion that LP-1 is synthesized in vivo
indicates that VP-1, VP-2, and VP-3 are made
startingfrom the second, third,orevenfourth of
severalfunctional AUG codons presentin these
polygenic mRNAs. We have recently found that
VP-2, VP-3, and,possibly, VP-1 canbe
synthe-sized in vivo from the same spliced species of
19S
mRNAs (Barkan and Mertz, manuscript inpreparation). These findings are clearly
incon-sistent with the original "scanning model" for
translation start codon selection in eucaryotic
mRNAs ofKozak (18, 19). Recently, Kozak has
modified the modeltoallow ribosomestoinitiate
at a downstream site provided that all of the
upstream AUGs areflanked by unfavorable
se-quences suchthat some 40S ribosomes canget
through (20). Whether synthesis of the proteins
encodedby the polygenicSV40 mRNAs actually
occurs by thisor some other mechanism, such
as direct internal translational initiation or
se-quential synthesis oftwoor moreproteins by the
same ribosome, remainstobe determined.
ACKNOWLEDGMENTS
WeareindebtedtoChuckCole, Harvey Ozer, Bob Martin, DanNathans, Peter Tegtmeyer,and ShermanWeissman for supplyingmutants ofSV40, toCharles McLean andRoland Rueckertfor advice and assistance inanalyzing proteins in polyacrylamide gels, andtoPeterSouthern,Paul Berg, Bob Margolskee, and Dan Nathans for communicating results
before publication. We also thank Shi-Da Yu for technical assistance andWilliam Dove, Bill Sugden, and Howard Temin for helpfulcomments on themanuscript.
Thiswork was supported by Public Health Service research grants CA-07175 and CA-22443 from the National Cancer Institute. A.B. was supported by a fellowship from the Wis-consin Alumni Research Foundation and by Public Health Servicetraining grant T32CA-09135from the National Insti-tutes of Health.
LITERATURECITED
1. Barkan, A., and J. E. Mertz. 1981. DNA sequence analy-sis of simian virus 40 mutants with deletions mapping in the leader region of the late viral mRNA's: mutants with deletionssimilar in size and position exhibit varied pheno-types. J. Virol. 37:730-737.
2. Benoist, C., and P. Chambon. 1981. In vivo sequence requirements of theSV40 early promoter region. Nature (London)290:304-310.
3. Brockman, W. W., and D. Nathans. 1974. The isolation of simianvirus 40 variants with specifically altered genomes. Proc. Natl. Acad. Sci. U.S.A. 71:942-946.
4. Carbon, J., T. E.Shenk, and P. Berg. 1975. Biochemical procedure forproduction of small deletions in simian virus 40 DNA. Proc.Natl. Acad. Sci. U.S.A. 72:1392-13%. 5. Chou, J. Y., and R. G. Martin. 1974. Complementation
analysis of simian virus 40 mutants. J. Virol. 13:1101-1109.
6. Cole, C. N., T.Landers, S. P. Goff, S.Manteuil-Brutlag, and P. Berg. 1977. Physical and geneticcharacterization ofdeletion mutants of simian virus 40 constructed in vitro. J. Virol. 24:277-294.
7. Dhar, R., K. N. Subramanian, J. Pan, and S. M. Weiss-man.1977. Structure of a large segment of the genomeof simian virus 40 that does not encode known proteins. Proc.Natl. Acad. Sci. U.S.A. 74:827-831.
7a.Fromm, M., and P. Berg. 1982. Deletion mapping of DNA regionsrequired forSV40 early region promoter function in vivo. J. Mol.Appl. Genet. 1:457-481.
8. Ghosh, P. K., P.Lebowitz, R. J. Frisque, and Y. Gluzman. 1981.Identification of a promotercomponent involved in positioning the 5' termini of simian virus 40 early mRNAs. Proc. Natl. Acad. Sci. U.S.A. 78:100-104.
9. Ghosh, P. K., V. B. Reddy, J. Swinscoe, P.Lebowitz, and S. M. Weissman. 1978. Heterogeneity and S'-terminal structures of the late RNAs of simian virus 40. J. Mol. Biol. 126:813-846.
10. Ghosh, P. K., P. Roy, A. Barkan, J. E. Mertz, S.M. Weissman, and P. Lebowitz. 1981. Unspliced functional late 19S mRNAs containing intervening sequences are produced by a lateleader mutant of simian virus 40. Proc. Natl.Acad. Sci. U.S.A. 78:1386-1390.
10a.Ghosh, P. K., M. Piatak, J. E. Mertz, S. M.Welssman, and P.Lebowitz. 1982. Altered utilization of splice sites and 5' termini in late RNAs produced by leader region mutants of simianvirus 40. J. Virol. 44:610-624. 11. Gruss,P., R. Dhar, and G. Khoury. 1981.Simian virus 40
tandem repeated sequences as an element of the early promoter. Proc.Natl. Acad. Sci. U.S.A. 78:943-947. 12. Haegenan,G., H.vanHeuverswyn, D. Gheysen, and W.
Flers. 1979. Heterogeneity of the 5' terminus of late mRNA induced by a viable simian virus 40 deletion mutant. J. Virol.31:484-493.
13. Hay, N., H.Skolnik-David,and Y.Aloni. 1982. Attenua-tion inthe control ofSV40 gene expression. Cell 29:183-193.
14. Hirt, B.1%7. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol.26:365-369. 15. Jackson, V., and R. Chalkley. 1981. Use ofwhole-cell
fixation to visualize replicating and maturing simian virus 40:identification of new viral gene product. Proc. Natl. Acad.Sci. U.S.A. 78:6081-6085.
16. Jay, G., S. Nomura, C. W. Anderson, and G. Khoury. VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
46 MERTZ, MURPHY, AND BARKAN
1981. Identification of the SV40 agnogene product: a DNA bindingprotein. Nature(London)291:346-349. 17. Kimura, G., and R.Dulbecco. 1972. Isolation and
charac-terizationoftemperature-sensitivemutants of simian vi-rus40. Virology 49:394-403.
18. Kozak, M. 1978. How do eucaryotic ribosomes select initiationregions in messenger RNA? Cell 15:1109-1123. 19. Kozak, M. 1980.Evaluation of the"scanningmodel"for initiation of proteinsynthesis in eucaryotes. Cell 22:7-8. 20. Kozak, M. 1981.Possible roleofflankingnucleotides in recognition of the AUG initiator codon by eukaryotic ribosomes.NucleicAcidsRes. 9:5233-5252.
21. Laenmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head ofbacteriophage T4. Nature(London) 227:680-685.
22. Laskey, R. A., and A. D. Mills. 1975. Quantitativefilm detection of3Hand14Cinpolyacrylamidegels by fluorog-raphy. Eur. J.Biochem.56:335-341.
23.Mertz, J. E. 1982. Amodel forregulationof SV40 late geneexpression.J. Cell. Biochem.6(Suppl.):313. 24. Mertz, J. E., and P. Berg. 1974.Defective simian virus 40
genomes:isolation andgrowthof individual clones. Virol-ogy62:112-124.
25. Mertz, J. E., and P. Berg. 1974.Viable deletion mutants of simianvirus40:selective isolationbymeansofa restric-tion endonuclease from Hemophilus parainfluenzae. Proc. Natl. Acad.Sci.U.S.A. 71:4879-4883.
25a.Miller, T. J., D. L. Stephens, and J. E. Mertz. 1982. Kineticsofaccumulationandprocessingof simian virus 40RNA inXenopus laevis oocytesinjected withsimian virus 40 DNA. Mol. Cell. Biol. 2:1581-1594.
26. Ozer, H. L. 1972.Synthesis andassembly of simian virus 40. I. Differential synthesisof intact virions and empty shells. J. Virol. 9:41-51.
27. Ozer, H. L., and K. K. Takemoto. 1969. Site of host restriction ofsimian virus 40 mutants in anestablished African greenmonkey kidneycell line. J.Virol. 4:408-415.
28. Piatak, M., K. N. Subramanian,P.Roy, and S. M. Weiss-man. 1981. Late mRNAproduction byviable SV40 mu-tants with deletions in the leaderregion. J. Mol. Biol. 153:589-618.
29. Radloff, R., W. Bauer, and J. Vinograd. 1967. A dye-buoyant-densitymethodfor the detection and isolation of
closed circularduplexDNA:the closed circular DNA in HeLa cells. Proc. Natl. Acad. Sci. U.S.A. 57:1514-1521. 30. Reddy, V. B., B. Thimmappaya, R. Dhar, K. N. Subra-manian,B.S.Zain, J. Pan,P. K. Ghosh,M. L.Celma,
andS. M.Weissman. 1978. The genome of simian virus 40. Science200:494-502.
31. Shenk, T. E., J. Carbon, and P. Berg. 1976. Construction andanalysisofviable deletionmutantsof simian virus 40. J.Virol. 18:664-671.
32. Shortle, D., and D. Nathans. 1978. Local mutagenesis: a method for generating viral mutants with base substitu-tions inpreselected regions of the viral genome. Proc. Natl. Acad. Sci.U.S.A. 75:2170-2174.
33. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separated by gelelectrophoresis. J.Mol. Biol. 98:503-517.
34. Subramanian, K. N. 1979. Segments of simian virus 40 DNAspanningmostof theleader sequence of themajor late viral messenger RNA are dispensable. Proc. Natl. Acad. Sci.U.S.A.76:2556-2560.
35. Takemoto, K. K., R. L.Kirschstein, and K. Habel. 1966. Mutants of simian virus 40differing in plaque size, onco-genicity, and heat sensitivity. J. Bacteriol. 92:990-994. 36. Takemoto, K. K., and M. A. Martin. 1970. SV40
thermo-sensitive mutant: synthesis of viral DNA and virus-in-duced proteins atnonpermissive temperature. Virology 42:938-945.
37. Takemoto, K. K., G. J. Todaro, and K. Habel. 1968. Recovery of SV40 virus withgenetic markers of original inducing virus fromSV40-transformed mouse cells. Virol-ogy 35:1-8.
38. Tegtmeyer, P. 1975. Function ofsimian virus 40 gene A in transforming infection. J. Virol. 15:613-618.
39. Tooze, J. (ed.). 1981.Molecular biology of tumor viruses, part2, 2nd ed., revised. DNA tumor viruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. 40. Villarreal, L. P., R. T. White, and P. Berg. 1979.
Muta-tional alterations within the simian virus 40 leader seg-ment generate altered 16S and 19S mRNA's. J. Virol. 29:209-219.
41. Yoshiike, K. 1968. Studies on DNA from low-density particles of SV40. I. Heterogeneous defective virions produced by successive undiluted passages. Virology 34:391-401.