Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Derivation and Functional Characterization of a Consensus
DNA Binding Sequence for the Tas Transcriptional
Activator of Simian Foamy Virus Type 1
YIBIN KANG1ANDBRYAN R. CULLEN1,2*Department of Genetics1and Howard Hughes Medical Institute,2Duke University
Medical Center, Durham, North Carolina 27710
Received 20 January 1998/Accepted 26 March 1998
Although DNA binding sites specific for the Bel-1 and Tas transcriptional activators, encoded, respectively, by the human and simian foamy viruses, have been mutationally defined, they show little evident sequence identity. As a result, the sequence determinants for DNA binding by both Bel-1 and Tas have remained unclear. Here, we report the use of a novel in vivo randomization and selection strategy to identify a Tas DNA binding site consensus. This approach takes advantage of the fact that Tas can effectively activate gene expression in yeast cells via a Tas DNA binding site derived from the simian foamy virus type 1 (SFV-1) internal promoter. The defined Tas DNA binding site consensus extends over approximately 25 bp and contains a critical core sequence of;5 bp. Positions adjacent to this core sequence, while clearly also subject to selection, show a significantly higher level of sequence variation. Surprisingly, the wild-type SFV-1 internal promoter Tas DNA binding site fails to conform to the consensus at several positions. Further analysis demonstrated that the consensus sequence bound Tas more effectively than did the wild-type sequence in vitro and could mediate an enhanced Tas response in vivo when substituted into the SFV-1 internal promoter context. These findings explain the limited sequence identity observed for mutationally defined Tas or Bel-1 response elements and should facilitate the identification of Tas DNA target sites located elsewhere in the SFV-1 genome.
Foamy viruses, including human foamy virus (HFV) and the simian foamy viruses (SFVs), are unusual among retroviruses in that they contain two distinct promoter elements (25). As in all retroviruses, a promoter element located in the viral long terminal repeat (LTR) directs the synthesis of a genome-length transcript that gives rise to the viral structural proteins Gag, Pol, and Env. The second foamy virus promoter, termed the internal promoter (IP), is located at the 39 end of the envelope gene (4, 16, 19). The IP is primarily responsible for the expression of two open reading frames located between env and the 39LTR, one of which encodes a transcriptional trans-activator termed Bel-1 in HFV and Tas in the SFVs (4, 15). Importantly, both of these promoter elements are strongly activated upon expression of the cognate Bel-1 or Tas regula-tory protein (4, 14, 15, 21, 26, 28). Mutational analysis has demonstrated that IP function is critical for foamy virus repli-cation in culture, most probably because loss of IP function results in a marked drop in the expression of Bel-1/Tas, which is known to be essential for viral replication (17, 18).
Analysis of the mechanism of action of Bel-1 in HFV and of Tas in SFV-1 has demonstrated that these virally encoded transcription factors are DNA binding proteins, a property which distinguishes Bel-1/Tas from the functionally equivalent human immunodeficiency virus type 1 Tat and human T-cell leukemia virus type 1 Tax proteins (12, 30). Binding sites for Bel-1 have been defined in both the IP and LTR promoter of HFV (12, 13), while binding sites for Tas have been identified in the SFV-1 IP and in the gag gene (3, 30), although the role and importance of this latter Tas-dependent enhancer element remains unclear. For HFV, it has been demonstrated that the
Bel-1 DNA binding site located in the IP displays a far higher affinity for Bel-1 than the major Bel-1 DNA binding site lo-cated in the LTR promoter (13). It has been hypothesized that this difference in affinity may, at least in part, explain the observation that the HFV IP is activated significantly earlier than the LTR promoter during the foamy virus life cycle (15, 19). This difference in affinity, if functionally important, could also explain the limited homology noted between the HFV LTR and IP Bel-1 binding sites (13). Similarly, the SFV-1 IP and Gag gene Tas binding sites also display only limited ho-mology to each other and to Tas-responsive DNA sequences present in the SFV-1 LTR (3, 20, 30). These findings, com-bined with the observation that Bel-1 and Tas are specific only for target sequences present in their cognate viral genome (4, 13), have meant that no consensus DNA binding site for either Bel-1 or Tas has been proposed, even though minimal DNA binding sites for both proteins have been defined (3, 12, 13, 30). In this study, we have used a novel in vivo randomization and selection procedure to define a consensus DNA binding site for the SFV-1 Tas protein. We demonstrate that this consensus DNA sequence binds Tas with a higher affinity than does the wild-type IP Tas binding site both in vivo and in vitro. In ad-dition, we have constructed a highly variant Tas DNA binding site that, while identical to the minimal IP Tas binding site at only 8 of 25 positions, can nevertheless respond effectively to Tas in vivo. The flexibility of the DNA binding specificity of Tas appears to explain the marked variability in the sequence of Tas responsive sequences previously identified in the SFV-1 genome.
MATERIALS AND METHODS
Construction of molecular clones.The yeast reporter plasmids pLGSD5 and pJLB have been described previously (8, 10). Both contain the lacZ indicator gene under the control of a basal yeast cyc promoter. However, pJLB differs from pLGSD5 in that the former lacks the yeast cyc promoter regulatory element
UASG, which contains negative regulatory elements (8). As a result, the pJLB
vector displays a higher basal activity but also a significantly stronger response to
* Corresponding author. Mailing address: Howard Hughes Medical Institute, Duke University Medical Center, Box 3025, Durham, NC 27710. Phone: (919) 684-3369. Fax: (919) 681-8979. E-mail: Culle002 @mc.duke.edu.
5502
on November 9, 2019 by guest
http://jvi.asm.org/
weak transactivators. A pJLB-based yeast indicator plasmid containing the
min-imal Tas DNA binding site (269 to245) from the SFV-1 IP has been described
previously (13). This wild-type Tas binding sequence, the minimal Bel-1 binding
site (2163 to2139) defined in the HFV IP, and various mutant Tas DNA
binding sites (UP, VA, HS1, and HS2 [see Fig. 3]), were synthesized as oligo-nucleotides with flanking XhoI sites, annealed to make them double stranded,
and then cloned into the XhoI site 59proximal to the cyc promoter present in the
indicator plasmids pLGSD5 and pJLB. In addition, wild-type and defective
(knockout [KO]) forms of a larger SFV-1 IP sequence, extending from2150 to
229 relative to the IP transcription start site, were PCR amplified with primers
that introduced flanking XhoI restriction sites, using the corresponding mamma-lian expression constructs (described below) as templates, and cloned into the
XhoI site of pJLB. Orientation of inserts was screened by PCR and confirmed by
DNA sequencing.
The chloramphenicol acetyltransferase (CAT) gene-based mammalian
indica-tor plasmid pS1IP-1/CAT, containing an SFV-1 IP sequence (2357 to195)
introduced between the Asp718 and BamHI sites of plasmid pCAT-22A2s (27), has been described previously (4). Shorter fragments of the SFV-1 IP, extending
from2159 to113 or from286 to113, were PCR amplified and cloned into the
same sites in pCAT-22A2s. A variant Tas target sequence predicted by the consensus Tas binding site (VA [see Fig. 3]), a putative up-mutant of the Tas DNA binding sequence (UP), and a defective Tas DNA binding sequence (KO) were each introduced into the SFV-1 IP in place of the wild-type Tas binding site by combinatorial PCR, and the resultant chimeric IP fragments were cloned into the Asp718-BamHI digested pCAT-22A2s plasmid. The mammalian expression plasmids pcTas and pBC12/CMV have been described previously (5, 13).
In vivo randomization selection of Tas response sequences.The randomized Tas binding site libraries R1 to R5, as well as R3A and R3B, were generated by
a single-step PCR procedure. The 59primers used were as follows:
59-GACCGCTCGAG TTGCA ATCAC TGGAA ATAGA AGTTA CAGATCCGCCAGGCGTGT-39 R1 R2 R3 R4 R5
The primer bears an XhoI site near the 59end, followed by the minimal Tas
binding sequence (269 to245) derived from the SFV-1 IP, and finally a short
sequence homologous to pLGSD5 vector sequences located immediately 39to
the unique XhoI site located adjacent to the cyc promoter. The 25-bp Tas binding sequence was divided into five regions (R1 to R5, as shown in the underlined sequence of the primer) and each region was replaced with five random nucle-otides in each respective primer (e.g., NNNNN in place of TTGCA in the R1 primer, where N refers to an equal mixture of all four bases). The R3 region was further subdivided into two subregions, R3A and R3B, with randomization of
either the two 59nucleotides (TG) or the three 39nucleotides (GAA). In each
case, the 39primer used was 59-TATGCTACAAAGGACCTAATG-39,
corre-sponding to a coding region in the lacZ gene in pLGSD5. PCRs were carried out with the parental pLGSD5 plasmid as the template and resulted in products with
a XhoI site at the 59end of the randomized Tas binding sequence and a unique
SphI site in the amplified pLGSD5 vector sequence. The PCR products were
then digested with XhoI and SphI and cloned between these two sites in the indicator plasmid pLGSD5. The resultant plasmids contained randomized Tas
binding sequences inserted 39 to the XhoI site present in the cyc promoter
59-flanking region of the pLGSD5 indicator plasmid.
The ligated randomized libraries were phenol-chloroform extracted, ethanol
precipitated, and then electroporated into Escherichia coli DH5a. After selection
for ampicillin-resistant transformants, each library was found to consist of more than 4,000 independent clones. DNA derived from each pooled library was then introduced into Saccharomyces cerevisiae PSY 316 (11) along with the Tas ex-pression plasmid pYCplacIII-Tas (9, 13). The transformed yeast cells were
plated on uracil- and leucine-deficient (Ura2Leu2) plate covered with a
Hy-bond-N nylon membrane (Amersham). After 3 days of growth, the nylon
mem-brane with yeast colonies was lifted from the plate, frozen at2140°C for 10 min,
and then thawed at room temperature. An in situb-galactosidase (b-gal) assay
was carried out by placing the nylon membrane on filter papers soaked in 0.53
Z buffer (1) with 0.3 mg of chlorophenolred-b-D-galactopyranoside (CPRG;
Boehringer Mannheim) per ml and 0.1% (vol/vol) 2-mercaptoethanol. After 1 h of incubation at room temperature, colonies that turned dark red (positives) or
those that remained white (negatives) were picked and recovered on Leu2Ura2
plates, and the yeast indicator plasmids harboring candidate Tas target se-quences were rescued after overnight culture. A second yeast transformation and
b-gal assay were then performed with the selected indicator plasmids together
with either pYCplacIII-Tas or the parental plasmid pYCplacIII (9) to quantify the level of transactivation by Tas and to identify any false (i.e., Tas-indepen-dent) positive clones. The Tas response sequences in the positive or true-negative clones were then obtained by ABI automatic cycle sequencing (PE Applied Biosystems).
Yeast transformation and analysis.The lacZ indicator plasmids containing various mutant Tas DNA target sequences and the single-copy, pYCplacIII-based Bel-1 or Tas yeast expression plasmid (9, 13) were cotransformed into the
yeast strain PSY316. After 3 days of growth selection on Ura2Leu2plates,
overnight cultures were prepared and cell extracts were assayed forb-gal activity
as described previously (1).
Mammalian cell culture and transfection.COS cell cultures (35-mm plates)
were transfected by the DEAE-dextran procedure (6) with 1mg of a CAT-based
indicator construct containing wild-type or mutant forms of the SFV-1 IP, and up
to 200 ng of the pcTas expression plasmid. To maintain a level of 1.2mg of DNA
per transfection in all experiments, transfection cocktails were also supple-mented with the parental, negative control plasmid pBC12/CMV where
neces-sary. Induced CAT activities were assayed at;50 h posttransfection as
previ-ously described (22).
Gel retardation analysis.The fusion protein glutathione S-transferase (GST)– Tas was expressed in the protease-deficient E. coli XA90 and purified as previ-ously described (13, 30). The SFV-1 IP DNA sequences used for gel retardation
and competition analysis were generated by PCR and extended from2159 to
113 in the SFV-1 IP. The wild-type DNA probe was labeled with [g-32P]ATP
with T4 polynucleotide kinase, and the total isotope incorporation was deter-mined by scintillation counting after column purification. The binding reaction
was carried out with;104cpm (;0.2 ng) of the probe and 50 ng of GST-Tas
fusion protein in 40ml of binding buffer (10 mM Tris-Cl [pH 7.5], 65 mM KCl,
2.5 mM MgCl21 mM dithiothreitol, 5% glycerol) containing 200 ng of
poly(dI-dC) and 40mg of bovine serum albumin. Binding was allowed to proceed for 30
min at 4°C, and the reaction products were resolved on a 5% native polyacryl-amide gel and visualized by autoradiography. Competitor DNA fragments were PCR amplified from the respective cat-based mammalian indicator plasmids described above. A 154-bp DNA fragment containing the Mason-Pfizer monkey virus constitutive transport element (7) was amplified by PCR and served as a nonspecific competitor DNA. For competition experiments, competitor DNAs were added at an 8- or 80-fold molar excess over the labeled probe fragment and were incubated with GST-Tas for 10 min before addition of the probe. The results of competition experiments were quantitated with a PhosphorImager and Image QuaNT software (Molecular Dynamics).
RESULTS
Previously, Zou and Luciw (30) defined a 25-bp Tas DNA binding site in the SFV-1 IP by using DNase I protection analysis and also showed by gel shift analysis that this DNA sequence was sufficient for specific Tas binding in vitro. Sub-sequently, we demonstrated that a single copy of this 25-bp sequence, which extends from 269 to245 in the SFV-1 IP relative to the transcription start site, is sufficient to render a yeast cyc promoter element highly responsive to Tas when introduced 59to this minimal promoter and analyzed in yeast cells (13).
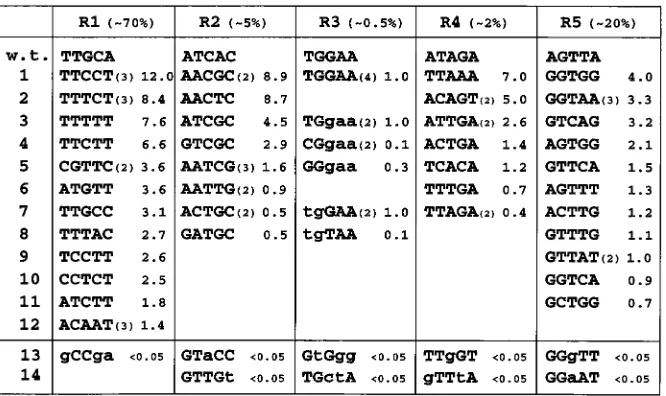
To define the consensus sequence for Tas DNA binding in yeast cells in vivo, we subdivided the minimal 25-bp SFV-1 IP Tas DNA binding site into five regions of 5 bp each, termed R1 through R5, as shown in Fig. 1. Each 5-bp segment was then individually randomized in the context of the lacZ-based indi-cator construct pLGSD5 to give five libraries, each containing 1,024 different candidate Tas target sequences (see Materials and Methods for details). Each library was cloned into E. coli and analyzed for integrity before being introduced into the yeast strain PSY316 together with a yeast Tas expression plas-mid. After 3 days of selection, yeast colonies containing DNA sequences that retained Tas responsiveness were identified by staining with theb-gal substrate CPRG. Approximately 4,000 to 5,000 colonies were screened per library. Individual respon-sive colonies were then picked, and the responrespon-sive plasmids were recovered. The Tas responsiveness of each indicator plas-mid was validated by retransformation into yeast in the pres-ence and abspres-ence of a Tas expression plasmid, and the precise level of b-gal activity induced by Tas, for each responsive indicator plasmid, was quantified as previously described. As shown in Fig. 1, a global analysis of the five libraries obtained during this research effort demonstrated that the central R3 library contained very few strongly positive clones (;0.5%). A higher but still modest percentage of positive colonies was obtained with the closely flanking libraries R2 and R4 (;5 and ;2%, respectively). In contrast, the R1 and R5 libraries, rep-resenting the 59and 39extremes of the SFV-1 IP Tas binding site, displayed high levels of positivity, with ;70 and;20%, respectively, showing readily detectableb-gal activity.
The small number of strongly positive clones observed with the R3 library, all of which displayed the wild-type sequence,
on November 9, 2019 by guest
http://jvi.asm.org/
prompted us to generate two additional libraries, termed R3A and R3B, in which only the 592 bp or the 393 bp of the R3 sequence were randomized. This allowed us to identify addi-tional, non-wild-type sequences covering this region that re-tained at least partial Tas responsiveness. Such clones were difficult to identify in the original R3 library since this partic-ular library generated a significant background of transcrip-tionally active but Tas-nonresponsive plasmids (data not shown). Consensus Tas DNA binding sequence.An analysis of the Tas-responsive sequences obtained in the above screen is shown in Fig. 1, and the data are summarized in Fig. 2, which also pre-sents the derived consensus Tas binding sequence. Based on these data, it is apparent that the core 59-TGGAA-39sequence covered by the R3 library is critical for Tas binding. Three of these positions were found to be invariant in this analysis, while variation at the other two positions was infrequent and, when observed, resulted in a marked drop in Tas responsiveness.
Analysis of the other four libraries, R1, R2, R4, and R5, gave the perhaps surprising result that many of the recovered plas-mids were more responsive to Tas than was the wild-type SFV-1 IP Tas binding site itself, in some cases by a factor of up to 10-fold. This result is remarkable in that it implies that the Tas binding site in the SFV IP has not been evolutionarily selected for maximal Tas binding affinity. Indeed, at several positions (e.g., position 5 in the R1 library and positions 2 and 4 in the R2 library), the nucleotide observed in the wild-type IP Tas binding site seems to be quite strongly selected against and is therefore lacking in the Tas binding consensus. For example, the adenine residue at position 4 in the R2 region of the wild-type Tas binding site was not detected in any of the 13 positive clones derived from the R2 library.
An additional interesting aspect of the sequences recovered from the R1, R2, R4, and R5 libraries is that although there is a clear selection at almost all of the 20 positions analyzed, strongly Tas-responsive clones with variant nucleotides at
ev-ery one of these positions were detected. Thus, while Tas clearly favors a particular nucleotide sequence and a consensus can therefore be readily derived, Tas DNA binding is highly plastic in that favorable nucleotides at certain positions appear able to compensate for less favorable nucleotides at others. The only clear exception to this is provided by the nucleotides in the R3 “core” sequence, which appear to comprise uniquely critical determinants of Tas binding.
To gain further insight into the requirements for Tas bind-ing, we also attempted to select several Tas-nonresponsive clones for each of the five libraries R1 through R5, and a small number of such negative sequences is shown in rows 13 and 14 of Fig. 1. Although truly Tas-nonresponsive clones were readily obtained for the R2 through R5 libraries, such clones proved difficult to identify in the R1 library, in that most such negative R1 clones proved to have deletions or rearrangements, in sev-eral cases elsewhere in the minimal Tas binding sequence. However, one nonresponsive sequence was obtained and is given in the R1 column of Fig. 1. As may be seen, this sequence includes residues at three sites that were not detected in any of
[image:3.612.134.467.69.267.2]FIG. 1. Selection of Tas-responsive sequences from randomized libraries in vivo. Introduction of a 25-bp Tas binding sequence, derived from the SFV-1 IP, adjacent to a minimal yeast cyc promoter element renders this promoter highly responsive to the Tas transactivator. To identify residues required for activation by Tas, this 25-bp sequence was subdivided into five regions of 5 bp each, termed R1 through R5, and each 5-bp sequence was independently randomized. Tas-responsive (rows 1 to 12) and Tas-nonresponsive (rows 13 and 14) DNA sequences were then selected after transformation into yeast cells and sequenced. Recovered strongly responsive sequences for each region R1 to R5 are aligned with the SFV-1 IP wild-type sequence (row w.t.). The first row also gives the approximate overall percentage of highly Tas responsive clones observed in each library by staining of yeast transformants on filters. The R3 library was further subdivided into an R3A library, involving randomization of only the 59two residues of R3, and an R3B library, involving randomization of the 39three residues. Residues held constant in these libraries are shown as lowercase letters in the upper section of column R3. Some sequences were recovered more than once, as indicated by a number in parentheses next to the relevant sequence. Rows 13 and 14 give Tas-nonresponsive sequences recovered during this screen, with residues not observed in any recovered positive sequence indicated in lowercase letters. The level of induction mediated by each selected sequence in yeast cells upon coexpression of Tas is given to the right of each sequence, as a multiple of the activity seen with the wild-type IP sequence, which is arbitrarily set at 1.0.
FIG. 2. Consensus Tas DNA binding sequence. This figure presents a com-pilation of the sequence data presented in Fig. 1, with the frequency of recovery of each base at each location in the randomized Tas-responsive sequence given as a percentage. The most prevalent base at each position is highlighted. At the bottom, these data are summarized to give a consensus Tas DNA binding site. The following abbreviations are used: W5A or T; Y5C or T; S5G or C; K5 G or T; and N5largely random.
on November 9, 2019 by guest
http://jvi.asm.org/
the 19 selected Tas-responsive clones and also has suboptimal residues at the other two positions. Overall, these data suggest that while almost all possible sequences in the R1 region will display at least some Tas response, it is nevertheless possible to significantly increase the efficiency of Tas binding by including an optimal sequence in R1.
As predicted from the smaller percentage of positive clones in the R2 through R5 libraries, it proved readily possible to isolate Tas-nonresponsive clones from each of these libraries. In each case, these sequences contained one or more residues that were entirely lacking in the selected positive clones. Res-idues that appear highly deleterious to Tas binding include an A residue at position 3 and a T residue at position 5 in R2, G residues at position 1 or 3 in R4, and a purine at position 3 in R5. This last result is of interest since the T residue predicted by the consensus sequence (Fig. 2) for position 3 in R5 is clearly the most highly selected residue in this library. Thus, selection for this residue may, on its own, explain the majority of the Tas-nonresponsive clones in the R5 library, given that these comprise only;80% of the total.
Biological activity of Tas binding site variants in yeast.The data presented in Fig. 1 and 2 suggest that it should be possible to derive highly variant forms of the Tas IP DNA binding site that retain good, or even enhanced, Tas binding ability. To test whether this was indeed the case, we designed two 25-bp oli-gonucleotides termed UP and VA (for variant). The UP se-quence differs at seven positions from the wild-type SFV-1 IP Tas binding site (Fig. 3A) and contains the optimal nucleotide sequence for Tas binding at every position, as predicted from the data shown in Fig. 1. In contrast, the 25-bp VA sequence was designed to be maximally different from the wild-type SFV-1 IP sequence yet to still largely conform to the Tas DNA binding site consensus given in Fig. 2. It was therefore antici-pated that the VA sequence, which differs from the wild-type sequence at 17 of 25 positions (Fig. 3A), would retain detect-able but not maximal Tas binding ability.
Both the UP and VA sequences were introduced into the pLGSD5 yeast indicator construct in both the sense and anti-sense orientation, and their Tas responsiveness was compared to that of the parental SFV-1 sequence (Table 1). As predicted, the UP sequence indeed proved to be 9- to 15-fold more responsive to Tas than was the parental SFV-1 sequence. Sim-ilarly, the VA sequence was found to retain 10 to 30% of the activity of the wild-type SFV-1 IP sequence. In contrast, intro-duced mutations that did not conform to the consensus
se-quence entirely abrogated Tas responsiveness (Fig. 1 and see below). Therefore, in the yeast system, the consensus sequence identified in Fig. 1 and 2 is highly predictive of the ability to respond to expression of the Tas transcriptional activator.
As previously shown (4, 13), and confirmed in Table 1, the Tas protein is unable to effectively interact with a 25-bp min-imal HFV IP-derived DNA sequence that serves as an effective DNA target site for Bel-1. Similarly, Bel-1 is unable to activate gene expression directed by a promoter containing the minimal SFV-1 IP Tas DNA binding site (Table 1) (4, 13). Comparison of the sequence of the HFV IP Bel-1 binding site with the consensus Tas binding site (Fig. 3B) suggested that it should be possible to render this 25-bp HFV sequence Tas responsive by changing five (HS1) or six (HS2) base pairs. The HS1 and HS2 variants of the HFV IP Bel-1 binding site were therefore in-troduced into the pLGSD5 yeast indicator construct. As shown in Table 1, the five mutated base pairs introduced into HS1 permitted a low level of response to Tas (;6%) while changing one additional base pair, in HS2, gave rise to a very substantial Tas response (;40% of the wild-type SFV-1 IP). However, both HS1 and HS2 were rendered nonresponsive to Bel-1 by these introduced mutations.
Previous work (4, 30) has suggested that all sequences in the SFV-1 IP required for response to Tas are contained within a 121-bp fragment, extending from2150 to229 relative to the IP transcription start site, and further suggested that this se-quence contained only a single Tas DNA binding site. To confirm that this was indeed the case, we compared the bio-logical activity in yeast cells of the extended2150 to229 IP sequence with the minimal 269 to 245 IP Tas binding
[image:4.612.54.289.68.228.2]se-FIG. 3. Tas DNA binding site variants. This figure gives the sequence of the UP, VA, and KO mutants of the269 to245 SFV-1 IP minimal Tas DNA binding site and of the HS1 and HS2 mutants of the2163 to2139 HFV IP minimal Bel-1 DNA binding site.
TABLE 1. Biological activity of Tas DNA binding-site mutants in yeast cells
Indicator plasmida b-Gal activity (mOD/ml) b
Negatived 1Tas 1Bel-1
pLG-SFV IP269/245(1) ,5 2,112 ,5 pLG-SFV IP269/245(2) ,5 632 ,5
pLG-UP(1) ,5 17,924 (8.5) NDc
pLG-UP(2) ,5 9,321 (15.0) ND
pLG-HFV IP2163/2139(1) ,5 ,5 425
pLG-HS1(1) ,5 128 (0.06) ,5
pLG-HS2(1) ,5 792 (0.38) ,5
pLG-VA(1) ,5 198 (0.09) ND
pLG-VA(2) ,5 173 (0.28) ND
pJLB-SFV IP269/245(1) ,50 52,440 ,50
pJLB-VA(1) ,50 9,760 (0.16) ND pJLB-VA(2) 200 17,860 (0.34) ND
pJLB-SFV IP2150/229(1) ,50 44,439 (0.85) ND pJLB-SFV IP2150/229(2) ,50 37,736 (0.72) ND
pJLB-KO2150/229(1) ,50 ,50 ND pJLB-KO2150/229(2) ,50 ,50 ND aSense orientation of the inserted Tas binding site is indicated by (1), and
antisense orientation is indicated by (2).
bNumbers in parentheses give the level of Tas-induced b-gal activity as a
multiple of the level seen with the269 to245 sequence. mOD, milli-optical density units (see reference 1 for assay description).
cND, not determined.
dParental pYCplacIII vector used to express Tas and Bel-1; see Materials and
Methods for details.
on November 9, 2019 by guest
http://jvi.asm.org/
quence after insertion into the pJLB yeast indicator construct (Table 1). An additional construct, containing the KO muta-tion predicted to block Tas binding (Fig. 3C) in the2150 to 229 sequence context, was also tested. If the2150 to229 IP sequence contains a second Tas DNA binding site, we would expect the 2150 to 229 sequence to retain significant Tas responsiveness after introduction of the KO mutation and would also expect it to give a stronger response to Tas than the minimal 269 to 245 sequence, given the known synergistic activity of Tas binding sites in yeast cells (13). In fact, the extended2150 to 229 indicator construct was not more re-sponsive to Tas than the minimal269 to245 construct, and this activity was entirely abrogated by the KO mutation. These data therefore serve to confirm the proposal, derived from an earlier in vitro analysis (30), that the SFV-1 IP contains only a single Tas binding site.
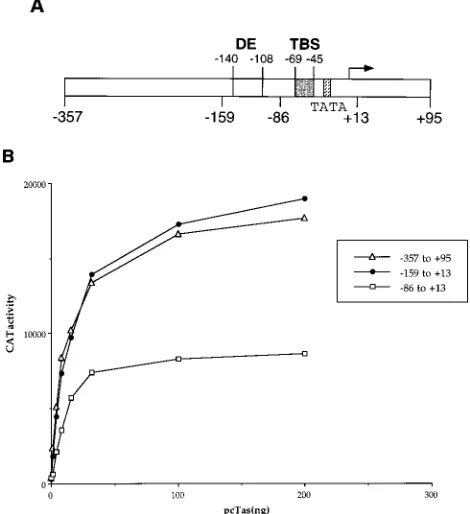
Biological activity of Tas binding site variants in mamma-lian cells.All the in vivo data on Tas DNA binding presented thus far were obtained with yeast cells, and we therefore wished to confirm these findings in the more physiologically relevant context of cultured simian cells. Previous mutational analysis of the SFV-1 IP (4, 30) has suggested that this pro-moter contains only three sequence elements required for maximal Tas response (Fig. 4A). These are a TATA box, seen in the majority of RNA polymerase II-specific promoters; the Tas binding site, located between residues245 and269
rela-tive to the transcription start site; and a third DNA sequence, termed the distal element, located between nucleotides2108 and2140 (30). While the distal element remains only poorly characterized, it does not bind Tas (30) (Table 1) and is there-fore thought to serve as a cellular factor binding site. Surpris-ingly, previous work (4, 30) has demonstrated that mutation of either the distal element or the Tas binding site sequence alone results in only a two- to fourfold drop in the Tas responsiveness of the IP; however, mutation of both sites results in the loss of Tas response (30). The observation that the single DNA target site for Tas present in the SFV-1 IP can be mutated with only a partial loss of Tas responsiveness is surprising and prompted us to reexamine the biological activity of the SFV-1 IP in mammalian cells and, in particular, to test the ability of wild-type and mutant SFV-1 IP derivatives to respond to a range of different Tas expression levels.
To perform this analysis, we used an SFV-1 IP-based indi-cator construct (pS1IP-1/CAT) containing the 2357 to 195 promoter sequence previously reported by Campbell et al. (4) to contain all the sequences required for full Tas responsive-ness (Fig. 4A). In addition, we constructed two IP deletion mutants. The first of these retains sequences extending from 2159 to113 in the SFV-1 IP and should represent a minimal, fully Tas-responsive IP. A second construct, extending from 286 to 113, has lost the distal-element sequence yet retains the complete Tas binding site (Fig. 4A). This promoter is predicted to retain partial Tas responsiveness.
As shown in Fig. 4B, both the2357 to195 and the2159 to 113 construct were highly Tas responsive when transfected into the simian COS cell line. Importantly, both promoters displayed an equivalent level of activity over the entire range of Tas expression tested. In contrast, the286 to113 IP sequence displayed a two- to threefold-lower level of Tas response than these more complete SFV-1 IP constructs over the entire range of Tas levels tested in this experiment. These data therefore confirm the earlier work of Luciw and coworkers (4, 30), ob-tained with only saturating levels of Tas expression, showing that the2159 to113 IP sequence contains all the elements required for full Tas responsiveness and that a sequence lo-cated between 2159 and286 in the SFV-1 IP modestly en-hances the Tas response of the IP.
Based on the data presented in Fig. 4, we next constructed derivatives of the2159 to113 and286 to113 forms of the SFV-1 IP containing the UP, VA, and KO mutations of the TBS described in Fig. 3. These mutant SFV-1 IP promoters were then tested for their ability to respond to Tas in trans-fected COS cells after transfection of 200 ng of the pcTas expression plasmid, which is predicted to give a maximal Tas response (Fig. 4B), or 8 ng of the pcTas plasmid, which is predicted to give a half-maximal response that is therefore in the linear range of this assay (Fig. 4B).
As shown in Fig. 5, introduction of these mutations into the complete2159 to113 SFV-1 IP sequence gave rise to only modest phenotypic changes. In particular, the KO mutation that completely abrogates Tas binding in yeast cells (Table 1) reduced Tas responsiveness by only two- to threefold, as in-deed predicted by the earlier data of Zou and Luciw (30). Introduction of the UP mutation, which is predicted to en-hance Tas binding (Table 1), resulted in a modest,;twofold increase in Tas responsiveness that was significant only at low levels of Tas expression (Fig. 5). Finally, the VA sequence, which differs at 17 of 25 positions from the wild-type Tas binding site, permitted the same level of activation by Tas as the wild-type IP sequence when introduced into the2159 to 113 IP sequence context and tested in mammalian cells.
[image:5.612.52.287.72.329.2]As noted above, it has previously been reported that the Tas
FIG. 4. Activity of the SFV-1 IP in mammalian cells. (A) Structure of the SFV-1 IP. Functionally important elements include a TATA box, the Tas DNA binding site (TBS) located between269 and245 relative to the transcription start site (arrow), and a distal element (DE), located between2108 and2140, that is believed to form a cellular Tas cofactor binding site. (B) Dose-response curves of SFV-1 IP derivatives with Tas in transfected COS cells. Indicator constructs, consisting of the indicated SFV-1 IP derivatives linked to the cat indicator gene, were transfected into COS cells together with increasing levels of the Tas expression plasmid pcTas, as indicated. The cells were transfected with 1mg of the relevant indicator construct and up to 200 ng of pcTas. A DNA level of 1.2mg per transfection was maintained by supplementation with the parental pBC12/CMV plasmid, which also serves as a negative control. Transfected cells were harvested;50 h after transfection, and CAT levels were determined as previously described (22).
on November 9, 2019 by guest
http://jvi.asm.org/
response of the SFV-1 IP is mediated not only by the Tas binding site but also by a sequence termed the distal element, which is thought to form a cellular factor binding site (30). As shown by the phenotype of the KO mutation in the2159 to 113 IP sequence context and also by the work of others (30), the distal-element sequence appears to be able to mediate activation by Tas even in the absence of a functional Tas binding site. However, in the context of the 286 to113 IP sequence lacking the distal element (Fig. 4A), the Tas response should be entirely dependent on the integrity of the Tas bind-ing site.
As shown in Fig. 5, this is indeed the case. Thus, the KO mutation that had only a modest (ca. two- to threefold) effect in the 2159 to113 sequence context dramatically inhibited the Tas response in the 286 to 113 IP sequence context. Further, the UP mutation of the Tas binding site had a clear positive phenotype in the 286 to113 sequence context, re-sulting in a level of Tas response that was equivalent to that seen with the wild-type form of the extended 2159 to 113 promoter element. Finally, the VA mutant again showed a level of Tas response in the286 to113 sequence context that was equivalent to that seen with the wild-type 286 to 113 sequence. Overall, these data obtained with mammalian cells therefore demonstrate that the DNA sequence requirements for Tas binding defined above in the yeast cell context are also valid in mammalian cells.
Tas DNA binding specificity in vitro.As a final confirmation of the validity of the Tas binding consensus given in Fig. 2, we examined the ability of the UP, VA, and KO mutants of this SFV-1 sequence to compete with the wild-type SFV-1 IP for binding to recombinant Tas protein by using a competitive electrophoretic mobility shift assay. As shown in Fig. 6, addi-tion of recombinant GST-Tas to a labeled SFV-1 IP DNA probe resulted in the detection of two specific DNA-protein complexes labeled C1 and C2. The origin of the C2 complex is unclear, but this could reflect dimerization of either the Tas
protein or GST, which is known to form dimers. Formation of both these complexes is effectively competed by an excess of the unlabeled wild-type SFV-1 IP DNA sequence (lanes 3 and 4) but not by an equivalent level of a nonspecific competitor DNA (lanes 11 and 12). Importantly, the addition of an SFV-1 IP-derived DNA sequence bearing the UP mutation resulted in a significantly enhanced level of competition (lanes 5 and 6), demonstrating that the UP mutation indeed enhances Tas bind-ing in vitro. In contrast, an SFV-1 IP DNA competitor bearbind-ing the KO mutation did not compete for binding any more effec-tively than the nonspecific DNA competitor did (compare lanes 9 and 10 with lanes 11 and 12). Finally, a sequence containing the VA mutation of the SFV-1 IP also competed for Tas DNA binding in vitro, although perhaps slightly less efficiently than the wild-type IP sequence did (lanes 7 and 8). This result ap-pears to mirror the in vivo biological activity of the VA se-quence observed in yeast cells (Table 1), which was also slightly lower than that seen with the wild-type IP sequence. In con-clusion, these in vitro Tas binding data are therefore in agree-ment with the in vivo data derived from the yeast (Table 1) and mammalian (Fig. 5) systems and serve to further validate the Tas DNA binding consensus given in Fig. 2.
DISCUSSION
The replication of primate foamy viruses is dependent on the biological activity of not only the viral LTR promoter but also a second promoter, the IP, located toward the 39end of the env gene (4, 16, 19). Both promoter elements are strongly activated by the virally encoded Bel-1/Tas transcriptional trans-activator (4, 14, 15, 21, 26, 28), and it is therefore the interplay of Bel-1/Tas with these two promoter elements that largely controls the level of foamy virus gene expression.
Analysis of the biological activity of HFV Bel-1 and of
FIG. 5. Activation of SFV-1 IP mutants by Tas. Indicator constructs consist-ing of the SFV-1 IP (2159 to113 or286 to113) linked to the cat indicator gene were mutated by introduction of the UP, VA, or KO mutations (Fig. 3) into the Tas DNA binding site. The resultant constructs were then transfected into COS cells in the presence of limiting (8 ng) or saturating (200 ng) levels of the pcTas indicator construct. The pBC12/CMV plasmid served as a negative control. Induced CAT expression levels were quantified at;50 h posttransfection. These data represent the mean for three experiments, with standard deviations indi-cated by error bars.
FIG. 6. DNA binding by SFV-1 Tas in vitro. The end-labeled DNA probe
used in this EMSA consists of the wild-type2159 to113 SFV-1 IP sequence.
Incubation with recombinant GST-Tas (50 ng) resulted in the detection of two retarded DNA-protein complexes labeled C1 and C2. Preincubation with an 8- or
80-fold excess of the unlabeled wild-type (WT)2159 to113 DNA fragment
inhibits complex formation (lanes 3 and 4), while the same level of a nonspecific
(NS) competitor has little or no effect. Competition by the2159 to113 IP DNA
sequence bearing the UP, VA, or KO mutation of the Tas binding site (Fig. 3) is also shown. The percent residual Tas DNA binding, compared to the binding with no added competitor (lane 2), was determined by using a PhosphorImager and is given at the bottom of the figure. Free Int. Pr. Probe, free internal promoter DNA probe.
on November 9, 2019 by guest
http://jvi.asm.org/
SFV-1 Tas has demonstrated that these viral transcription fac-tors are DNA binding proteins and has identified minimal, $25-bp binding sites for Bel-1 in the HFV LTR and IP and for Tas in the SFV-1 IP and gag gene (3, 12, 13, 30). However, comparison of these sequences with each other and with those of other phenotypically defined Bel-1 or Tas response elements has failed to identify a clear DNA binding consensus. The lack of such a consensus reflects two findings. First, Bel-1 and Tas interact only with sites present in their cognate virus (4, 13), thus making any comparison of the DNA binding sites of these two related proteins uninformative at present. Second, it ap-pears that the foamy virus IP has evolved to contain a signifi-cantly higher-affinity target sequence for Bel-1/Tas than has the LTR promoter (13), thus suggesting that Bel-1/Tas binding sites in the LTR are likely to diverge significantly from the optimal Bel-1/Tas DNA binding consensus.
In this paper, we report the use of a novel randomization and selection strategy with yeast cells to define the DNA bind-ing consensus for SFV-1 Tas. Yeast cells were chosen because these lower eukaryotes were previously shown to be permissive for transcriptional activation by both Bel-1 and Tas via their respective, mutationally defined DNA binding sites (1, 12, 13). We chose the SFV-1 IP Tas DNA binding site for this analysis because Tas gives a significantly higher level of transcriptional activation than Bel-1 in yeast cells (13), thus facilitating the identification of positive clones, and because the IP, as noted above, contains a particularly high-affinity DNA binding site for Tas (13). An alternative strategy, used for several DNA binding proteins (2, 23, 24, 29), would have been to identify a consensus Tas DNA binding site in vitro by using several cycles of PCR amplification and selection for Tas binding. An advan-tage of this latter, in vitro technique is that it would have permitted the simultaneous randomization of essentially the entire Tas binding site. In contrast, the in vivo randomization/ selection strategy described here is not able to analyze more than five or six nucleotide positions simultaneously because (i) more extensive randomization would require screening an un-wieldy number of yeast transformants and (ii) we have found that the percentage of false (i.e., Tas-independent)-positive clones, which largely represent yeast transcription factor bind-ing sites, increases when more nucleotides are randomized. The fact that in any given random library, much of the DNA binding site must therefore remain fixed could affect the range of positive sequences obtained by this approach. However, this concern is balanced by the anticipation that this in vivo ap-proach may be more likely to identify physiologically relevant DNA-protein interactions.
A series of Tas-responsive and nonresponsive sequences identified using by in vivo randomization and selection ap-proach are listed in Fig. 1 and summarized in Fig. 2, which also gives the derived consensus sequence for Tas DNA binding. Based on these data, we suggest that Tas DNA binding sites consist of a core recognition sequence that approximately co-incides with the R3 sequence 59-TGGAA-39. Flanking this, on either side, are sequences that are defined largely by the R2 and R4 libraries that, while critical for Tas binding, are never-theless more tolerant of sequence variation. Finally, while the R5 and, particularly, R1 regions contribute relatively little crit-ical sequence information to the Tas DNA binding site, DNA sequences present in these regions can significantly modulate the affinity of Tas for its target site (Fig. 1). Overall, these data indicate that the consensus DNA target site for Tas is some-what unusual in terms of both its large size and the relative plasticity of this sequence outside of the core R3 region. While it is interesting to speculate that the large size of this target DNA sequence could reflect binding by a Tas dimer or
multi-mer, we note that the Tas binding consensus (Fig. 2) does not give any evidence of being a direct or inverted repeat sequence. Perhaps the most surprising aspect of the Tas DNA binding consensus given in Fig. 2 is the implication that the wild-type SFV-1 IP Tas DNA binding site is suboptimal at several loca-tions, suggesting that this site has not been selected for maxi-mal Tas binding efficiency. Indeed, several of the recovered mutant sequences shown in Fig. 1 proved to be more effective target sites for Tas when tested in yeast cells.
To validate the relevance of the consensus Tas DNA binding sequence given in Fig. 2, we used this consensus to design a sequence, termed UP, that should be fully optimal for Tas binding and a second sequence, termed VA, that should be able to bind Tas effectively despite differing at 17 of 25 posi-tions from the wild-type SFV-1 IP sequence (Fig. 3A). Analysis of the UP and VA sequences in yeast cells (Table 1) and in vitro (Fig. 6) showed that the UP sequence indeed bound Tas more effectively than did the wild-type IP sequence while the VA sequence also bound Tas but somewhat less well than did the wild type. In contrast, an SFV-1 IP sequence mutated in the Tas binding-site core, termed KO, failed to detectably bind Tas in either assay system.
Demonstration of the relevance of the Tas DNA binding consensus in simian cells was complicated by the fact that the SFV-1 IP contains two partially redundant Tas response ele-ments (Fig. 4A). While the first of these coincides with the Tas DNA binding site, the second sequence, referred to as the distal element, fails to bind Tas both in vitro (30) and in vivo (Table 1). This finding, combined with the fact that the distal-element sequence can mediate Tas function in mammalian cells (30) (Fig. 4B) but not yeast cells (Table 1), strongly sug-gests that the distal element represents a DNA binding site for a cellular cofactor(s). Despite this complication, it nevertheless proved possible to also validate the Tas binding consensus in mammalian cells. In particular, in the context of a minimal SFV-1 IP promoter lacking the distal-element sequence, the KO mutation was found to block activation by Tas in mamma-lian cells whereas the UP mutation was found to increase the Tas response by two- to threefold. The maximally variant VA mutation was also observed to retain Tas responsiveness (Fig. 5). Together, these data demonstrate that Tas DNA binding in mammalian cells (Fig. 5), as in yeast cells (Table 1) and in vitro (Fig. 6), is effectively predicted by the Tas DNA binding con-sensus given in Fig. 2.
The data presented in Fig. 5 may also shed some light on the role of the putative cellular distal-element DNA binding fac-tor. The distal-element sequence is not a Tas binding site (30) (Table 1) and has no phenotype in the absence of Tas (Fig. 5), but it can mediate significant activation of the SFV-1 IP by Tas in the absence of an intact Tas DNA binding site (30) (Fig. 4B). This finding suggests that the cellular distal-element bind-ing protein acts by facilitatbind-ing the recruitment of Tas to the SFV-1 IP. Consistent with this hypothesis, while deletion of the distal element significantly reduces the activation of the SFV-1 IP by Tas, this reduction can be overcome by substitution of
FIG. 7. The SFV-1 Tas binding site in gag conforms to the Tas binding consensus. This figure shows an alignment of the Tas binding site in the SFV-1
gag gene, as defined by DNase I protection analysis (3), with the IP Tas binding
site and with the Tas binding consensus. A continuous line indicates tight con-servation, while a dashed line indicates that this residue was detected in the pool of Tas binding sequences listed in Fig. 1.
on November 9, 2019 by guest
http://jvi.asm.org/
the high-affinity UP Tas DNA binding site in place of the wild-type sequence (Fig. 5). In contrast, the UP mutant has only a minimal phenotype in mammalian cells in the context of an SFV-1 IP containing an intact distal element (Fig. 5), suggest-ing that Tas DNA bindsuggest-ing is already highly efficient in this environment. The hypothesis that a cellular cofactor specific for the distal-element sequence significantly enhances Tas bind-ing to the SFV-1 IP in mammalian cells thus may explain why the IP Tas DNA binding site is not more closely homologous to the optimal Tas DNA binding site (Fig. 2). However, it is also possible that the sequence of the IP Tas DNA binding site is simply constrained by the underlying envelope coding sequence.
A final point is that the proposed Tas binding site consensus should permit the identification of Tas binding sites located elsewhere in the SFV-1 genome. Indeed, a comparison of the SFV-1 gag gene Tas DNA binding site (3) with the consensus sequence shows extensive sequence homology (Fig. 7), partic-ularly around the core region. An analysis of regions within the SFV-1 LTR that have previously been implicated in mediating Tas transactivation of the SFV-1 LTR promoter (20) identified several regions with homology to the consensus, including par-ticularly between21052 and21028, between2375 and2351, and (in the antisense orientation) between2956 and2980 and between2163 to2187. The question whether these sequences are indeed important for Tas-mediated activation of the SFV-1 LTR promoter can now be experimentally addressed.
ACKNOWLEDGMENTS
We thank Paul Luciw for several SFV-1 clones used in this work and Hal Bogerd and Wade Blair for helpful discussions.
This research was funded by the Howard Hughes Medical Institute.
REFERENCES
1. Blair, W. S., H. Bogerd, and B. R. Cullen. 1994. Genetic analysis indicates that the human foamy virus Bel-1 protein contains a transcription activation domain of the acidic class. J. Virol. 68:3803–3808.
2. Blackwell, T. K., and H. Weintraub. 1990. Differences and similarities in DNA-binding preferences of MyoD and E2A protein complexes revealed by binding site selection. Science 250:1104–1110.
3. Campbell, M., C. Eng, and P. A. Luciw. 1996. The simian foamy virus type 1 transcriptional transactivator (Tas) binds and activates an enhancer ele-ment in the gag gene. J. Virol. 70:6847–6855.
4. Campbell, M., L. Renshaw-Gegg, R. Renne, and P. A. Luciw. 1994. Character-ization of the internal promoter of simian foamy viruses. J. Virol. 68:4811–4820. 5. Cullen, B. R. 1986. trans-activation of human immunodeficiency virus occurs
via a bimodal mechanism. Cell 46:973–982.
6. Cullen, B. R. 1987. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 152:684–704.
7. Ernst, R. K., M. Bray, D. Rekosh, and M. Hammarskjo¨ld. 1997. A structural retroviral RNA element that mediates nucleocytoplasmic export of intron-containing RNA. Mol. Cell. Biol. 17:135–144.
8. Finley, R. L., Jr., S. Chen, J. Ma, P. Byrne, and R. W. West, Jr. 1990.
Opposing regulatory functions of positive and negative elements in UASG
control transcription of the yeast GAL genes. Mol. Cell. Biol. 10:5663–5670. 9. Gietz, R. D., and A. Sugino. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair re-striction sites. Gene 74:527–534.
10. Guarente, L., R. Yocum, and P. Gifford. 1982. A GAL10-CYC1 hybrid yeast promoter identifies the GAL4 regulatory region as an upstream site. Proc. Natl. Acad. Sci. USA 79:7410–7414.
11. Harper, J. W., G. R. Adami, N. Wei, K. Keyomarsi, and S. J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816.
12. He, F., W. S. Blair, J. Fukushima, and B. R. Cullen. 1996. The human foamy virus Bel-1 transcription factor is a sequence-specific DNA binding protein. J. Virol. 70:3902–3908.
13. Kang, Y., W. S. Blair, and B. R. Cullen. 1998. Identification and functional characterization of a high-affinity Bel-1 DNA binding site located in the human foamy virus internal promoter. J. Virol. 72:504–511.
14. Keller, A., K. M. Partin, M. Lo¨chelt, H. Bannert, R. M. Flu¨gel, and B. R.
Cullen.1991. Characterization of the transcriptional trans activator of hu-man foamy retrovirus. J. Virol. 65:2589–2594.
15. Lo¨chelt, M., R. M. Flu¨gel, and M. Aboud. 1994. The human foamy virus internal promoter directs the expression of the functional Bel-1 transactiva-tor and Bet protein early after infection. J. Virol. 68:638–645.
16. Lo¨chelt, M., W. Muranyi, and R. M. Flu¨gel. 1993. Human foamy virus genome possesses an internal, Bel-1-dependent and functional promoter. Proc. Natl. Acad. Sci. USA 90:7317–7321.
17. Lo¨chelt, M., S. F. Yu, M. L. Linial, and R. M. Flu¨gel. 1995. The human foamy virus internal promoter is required for efficient gene expression and infec-tivity. Virology 206:601–610.
18. Lo¨chelt, M., H. Zentgraf, and R. M. Flu¨gel. 1991. Construction of an infec-tious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology 184:43–54.
19. Mergia, A. 1994. Simian foamy virus type 1 contains a second promoter
located at the 39end of the env gene. Virology 199:219–222.
20. Mergia, A., E. Pratt-Lowe, K. E. S. Shaw, L. W. Renshaw-Gegg, and P. A.
Luciw.1992. cis-acting regulatory regions in the long terminal repeat of simian foamy virus type 1. J. Virol. 66:251–257.
21. Mergia, A., K. E. S. Shaw, E. Pratt-Lowe, P. A. Barry, and P. A. Luciw. 1991. Identification of the simian foamy virus transcriptional transactivator gene (taf). J. Virol. 65:2903–2909.
22. Neumann, J. R., C. A. Morency, and K. O. Russian. 1987. A novel rapid assay for chloramphenicol acetyltransferase gene expression. BioTechniques 5:444– 447.
23. Oliphant, A. R., C. J. Brandl, and K. Struhl. 1989. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol. Cell. Biol.
9:2944–2949.
24. Rauscher, F. J., III, J. F. Morris, O. E. Tournay, D. M. Cook, and T. Curran. 1990. Binding of the Wilms’ tumor locus zinc finger protein to the EGR-1 consensus sequence. Science 250:1259–1262.
25. Rethwilm, A. 1995. Regulation of foamy virus gene expression. Curr. Top. Microbiol. Immunol. 193:1–24.
26. Rethwilm, A., O. Erlwein, G. Baunach, B. Maurer, and V. Ter Meulen. 1991. The transcriptional transactivator of human foamy virus maps to the bel 1 genomic region. Proc. Natl. Acad. Sci. USA 88:941–945.
27. Sparger, E. E., B. L. Shacklett, L. Renshaw-Gegg, P. A. Barry, N. C.
Ped-ersen, J. H. Elder, and P. A. Luciw.1992. Regulation of gene expression directed by the long terminal repeat of the feline immunodeficiency virus. Virology 187:165–177.
28. Venkatesh, L. K., P. A. Theodorakis, and G. Chinnadurai. 1991. Distinct
cis-acting regions in U3 regulate trans-activation of the human
spumaretro-virus long terminal repeat by the viral bel1 gene product. Nucleic Acids Res.
19:3661–3666.
29. Wright, W. E., M. Binder, and W. Funk. 1991. Cyclic amplification and selection of targets (CASTing) for the myogenin consensus binding site. Mol. Cell. Biol. 11:4104–4110.
30. Zou, J. X., and P. A. Luciw. 1996. The transcriptional transactivator of simian foamy virus 1 binds to a DNA target element in the viral internal promoter. Proc. Natl. Acad. Sci. USA 93:326–330.