Copyright © 1998, American Society for Microbiology. All Rights Reserved.
The Herpes Simplex Virus gE-gI Complex Facilitates Cell-to-Cell
Spread and Binds to Components of Cell Junctions
KEVIN S. DINGWELL1,2ANDDAVID C. JOHNSON2*
Department of Biology, McMaster University, Hamilton, Ontario, Canada L8N 3Z5,1and Department of
Molecular Immunology, and Microbiology, Oregon Health Sciences University, Portland, Oregon 972012
Received 22 June 1998/Accepted 5 August 1998
The herpes simplex virus (HSV) glycoprotein complex gE-gI mediates the spread of viruses between adjacent cells, and this property is especially evident for cells that form extensive cell junctions, e.g., epithelial cells, fibroblasts, and neurons. Mutants lacking gE or gI are not compromised in their ability to enter cells as extracellular viruses. Therefore, gE-gI functions specifically in the movement of virus across cell-cell contacts and, as such, provides a molecular handle on this poorly understood process. We expressed gE-gI in human epithelial cells by using replication-defective adenovirus (Ad) vectors. gE-gI accumulated at lateral surfaces of the epithelial cells, colocalizing with the adherens junction proteinb-catenin but was not found on either the apical or basal plasma membranes and did not colocalize with ZO-1, a component of tight junctions. In subconfluent monolayers, gE-gI was found at cell junctions but was absent from those lateral surfaces not in contact with another cell, as was the case forb-catenin. Similar localization of gE-gI to cell junctions was observed in HSV-infected epithelial cells. By contrast, HSV glycoprotein gD, expressed using a recombinant Ad vectors, was found primarily along the apical surfaces of cells, with little or no protein found on the basal or lateral surfaces. Expression of gE-gI without other HSV polypeptides did not cause redistribution of either ZO-1 orb-catenin or alter tight-junction functions. Together these results support a model in which gE-gI accumulates at sites of cell-cell contact by interacting with junctional components. We hypothesize that gE-gI mediates transfer of HSV across cell junctions by virtue of these interactions with cell junction components.
Herpes simplex virus (HSV) replicates in tissues of epithelial origin such as the oral and genital mucosae and corneal epi-thelium. HSV spreads efficiently through these tissues, gaining access to sensory neurons, which eventually become the site of latency. In epithelial tissues, cells are joined by extensive cell contacts or junctions, and HSV and other herpesviruses spread across these cell junctions. By spreading rapidly from cell to cell through a space that is isolated by tight junctions, HSV races against the mounting immune response and also avoids neutralization by anti-HSV antibodies. It is well known that HSV can cause secondary lesions in the mucosae of individuals who produce high titers of anti-HSV antibodies, and the se-verity of disease does not correlate with antibody titers (12). Thus, anti-HSV antibodies do not contain HSV spread in vivo, supporting the hypothesis that in solid tissues, e.g., epithelium, this form of direct cell-to-cell spread is a primary mode of virus transmission and an important parameter of HSV pathogene-sis. Release of HSV particles from cells also occurs, although in most cultured cells the majority of virions remain cell asso-ciated. Extracellular HSV likely plays an important role in dissemination to other hosts, but in immune individuals expe-riencing recurrent disease a large fraction of the exogenous virus may be neutralized.
Spread of HSV by both the extracellular and cell-to-cell routes requires viral membrane glycoproteins gB, gD, and gH-gL. Mutant HSVs with deletions affecting gB, gD, gH, or gL cannot enter cells, and if the mutants are grown on comple-menting cells (to provide the missing glycoprotein) the viruses can enter cells but do not subsequently spread beyond the initially infected cell (8, 16, 27, 43). Therefore, cell-to-cell
spread and entry of extracellular viruses share essential prop-erties. However, these processes also differ in some significant aspects. Glycoproteins gE and gI, which form a functional complex (gE-gI) (24, 25), play an important role in cell-to-cell spread but do not affect production of infectious virus or the rate at which extracellular virus particles bind to or enter cells, whether the virus is applied to the apical or basolateral sur-faces of the cells (2, 14, 17a). Similar results were reported for the gE and gI homologues of the related alphaherpesvirus pseudorabies virus (PrV) (57). The reduced cell-to-cell spread of gE or gI mutant viruses was most evident in plaque assays involving cells that form extensive cell junctions, i.e., normal human fibroblasts and epithelial cells, rather than transformed cells. For example, as few as 3 to 7 normal human epithelial cells may become infected by a gE mutant, F-gEb, compared with the 150 to 500 cells that become infected by wild-type HSV serotype 1 (HSV-1) strain F in plaque assays involving media overlays containing HSV-neutralizing antibodies (13). Similarly, wild-type HSV-1 could infect and spread to approx-imately four times the number of cultured rat neurons than were infected by F-gEb(15).
Apparently related to this inability to spread from cell-to-cell is the fact that gE-gI mutants are also severely attenuated in vivo. In the corneal epithelium and skin of mice, gE-negative (gE2) and gI2mutants form small lesions, spreading poorly
beyond the initial site of infection, and over time produce much less infectious virus than wild-type HSV (2, 14). In ad-dition, gE2and gI2mutants fail to spread efficiently into and
within the nervous system and cause less neurological disease than wild-type HSV (2, 14, 15, 26, 35, 40, 41). We have pro-posed that gE-gI facilitates the movement of HSV across the extensive junctions formed between epithelial cells, fibroblasts, and neurons in vivo (15). However, there is also evidence that PrV gE-gI may function in a more complex fashion in the * Corresponding author. Mailing address: L-220 Dept. of Molecular
Microbiology and Immunology, Oregon Health Sciences University, 3181 S.W. Sam Jackson Park Rd., L220, Portland, OR 97201. Phone: (503) 494-0834. Fax: (503) 494-6862. E-mail: [email protected].
8933
on November 9, 2019 by guest
http://jvi.asm.org/
nervous system, mediating entry into some neuronal circuits but not others and affecting neurovirulence (11, 48).
The process by which enveloped viruses spread from cell to cell in solid tissues such as skin or mucosa is poorly under-stood, yet several virus families, herpesviruses and poxviruses being examples (44, 52), rely heavily on direct cell-to-cell spread. Since HSV gE-gI appears to function in cell-to-cell spread but not in entry of extracellular virus, this glycoprotein complex provides an important molecular tool with which to study cell-to-cell spread. We expressed gE and gI by using adenovirus (Ad) vectors in human epithelial cells, cells that are particularly important in terms of the biology of HSV, in order to study the subcellular localization of gE-gI and effects of these glycoproteins on cells without the pleiotropic effects of other HSV polypeptides. The gE-gI complex accumulated spe-cifically on those lateral surfaces of epithelial cells that were forming cell junctions but was not found on those lateral sur-faces that were not in contact with another cell. By contrast, another HSV glycoprotein, gD, was found predominantly on the apical surfaces of the cells. These results support the hy-pothesis that gE-gI binds to components of epithelial cell junc-tions.
MATERIALS AND METHODS
Cells and viruses. HEC-1A endometrial epithelial cells (4) (a gift of Jay Nelson, Oregon Health Sciences University) were grown in RPMI medium (BioWhittaker Inc., Walkersville, Md.) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; BioWhittaker). 293 cells (17) and Vero cells were grown in Dulbecco’s modified minimal essential medium (DMEM; Bio-Whittaker) supplemented with 10 and 5% FBS, respectively. HSV-1 strains F (obtained from P. G. Spear, Northwestern University Medical School, Chicago, Ill.), F-US7kan (25), and F-gEb(14) were propagated on, and their titers were determined on, Vero cells. Two replication-competent Ad vectors, AdgE and AdgI, that were described previously (19) will be denoted Ad(E11)gE and
Ad(E11)gI here. Ad(E12)gE, Ad(E12)gI, AdgD1(E12) (7), and AddlE1, which
contains no HSV sequences, are all replication-defective (E12) Ad vectors, and
they were propagated on, and their titers were determined on, 293 cells.
Antibodies.Monoclonal antibody (MAb) 3104, specific for gI, and MAb 3114, specific for gE (25), were gifts of Anne Cross and Nigel Stow (Institute of Virology, Glasgow, United Kingdom). MAb II-481, specific for gE, was a gift of Patricia Spear (Northwestern University Medical School). MAb DL-6, specific for gD (22), was a gift of Gary Cohen and Roselyn Eisenberg (University of Pennsylvania, Philadelphia). A mouse MAb directed againstb-catenin and a mouse MAb specific for E-cadherin were obtained from Transduction Labora-tories (Lexington, Ky.), and rabbit polyclonal antibodies directed againstb -cate-nin were from Sigma. A rabbit anti-ZO-1 serum was obtained from Zymed Laboratories (South San Francisco, Ca.). Texas red-coupled goat anti-rabbit immunoglobulin G (IgG) and fluorescein isothiocyanate (FITC)- and Cy3-cou-pled goat anti-mouse IgG were obtained from Jackson ImmunoResearch Labs Inc. (West Grove, Pa.). BODIPY-coupled goat anti-FITC IgG was obtained from Molecular Probes (Eugene, Oreg.). The FITC analogue Oregon green was directly coupled to MAb 3114 by using a FluoReporter Oregon green-488 pro-tein labelling kit (Molecular Probes) as described by the manufacturer. Horse-radish peroxidase-conjugated goat anti-mouse IgG was obtained from Amer-sham Life Sciences, Inc. (Arlington Heights, Ill.).
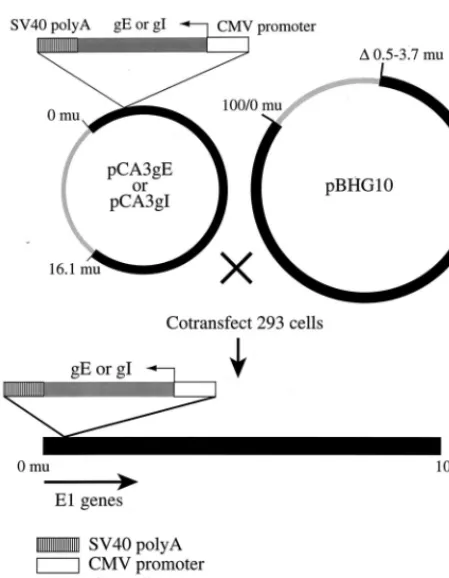
Construction of replication-defective recombinant Ad vectors expressing ei-ther gE or gI.All restriction and DNA modification enzymes were obtained from New England Biolabs (Beverly, Mass.). Plasmid DNA was prepared by using Qiagen 500 columns (Qiagen Inc., Chatsworth, Calif.). The full-length gE and gI genes were excised from plasmids pSV2X3gE and pSV2X3gI (19), respectively, and subcloned into plasmid pCA3 (20) by using the restriction enzymes EcoRI and XbaI. The resulting plasmids, pCA3gE and pCA3gI, respectively, contained either the gE or gI gene coupled to the human cytomegalovirus (HCMV) im-mediate-early promoter and followed by a simian virus 40 (SV40) poly(A) se-quence. Recombinant Ads expressing either gE [Ad(E12)gE] or gI [Ad(E12)gI]
were obtained following cotransfection of either pCA3gE or pCA3gI with pBHG10 (6) into 293 cells by the calcium phosphate technique as previously described (20). Recombinant viruses were plaque purified three times.
Labelling of cells with [35S]methionine-[35S]cysteine, immunoprecipitation of
proteins, gel electrophoresis, and Western blotting.Monolayers of HEC-1A cells grown in 35-mm-diameter dishes were infected either with 20 PFU of HSV-1 (strain F) per cell or with 400 PFU of either Ad(E12)gE or Ad(E12)gI per cell,
were coinfected with Ad(E12)gE and Ad(E12)gE (400 PFU of each per cell), or
left uninfected. Six hours after infection with HSV or 48 h after infection with Ad vector, the cells were washed twice with DMEM lacking methionine and cysteine and containing 1% dialyzed FBS and then labelled with [35S]methionine and
[35S]cysteine (150mCi/ml; NEN) in DMEM lacking methionine and cysteine for
3 h. Cells were lysed in Nonidet P-40 (NP-40)–deoxycholate (DOC) extraction buffer (1% NP-40, 0.5% DOC, 50 mM Tris-Cl [pH 7.5], 100 mM NaCl) contain-ing 2 mg of bovine serum albumin per ml and 1 mM phenylmethylsulfonyl fluoride and stored at270°C. Lysates were thawed, centrifuged at 50,000 to 100,0003g for 45 to 60 min, and mixed with anti-gE MAb 3114 or anti-gI MAb 3104 for 2 h at 4°C. Protein A-Sepharose (Pharmacia, Dorval, Quebec, Canada) was added to the lysates, which were then incubated for an additional 2 h; subsequently, the protein A-Sepharose was washed with NP-40–DOC extraction buffer, and immunoprecipitated proteins were subjected to electrophoresis on 12% polyacrylamide gels (14). The gels were fixed, enhanced with Enlightning (Dupont), dried, and then exposed to X-ray film. For Western blotting, proteins were transferred to Imobilon membranes (Millipore, Bedford, Mass.), the mem-branes were air dried and then incubated with 5% nonfat skim milk in phos-phate-buffered saline (PBS) containing 0.1% Tween 20 (W buffer). The blots were then incubated with MAb II-481 or an MAb specific for E-cadherin for 1 h; then they were washed, incubated with horseradish peroxidase-conjugated goat anti-mouse IgG, and washed again, and proteins were detected by enhanced chemiluminescence (ECL kit; Amersham).
Immunofluorescence of HSV- or Ad-infected HEC-1A cells.HEC-1A cells grown on glass Transwell slides (Lab Tek, Naperville, Ill.) either were left uninfected or were infected apically with either 20 PFU of HSV-1 strain F per cell, 400 PFU of Ad(E12)gE or Ad(E12)gI per cell, or 400 PFU each of both
Ad(E12)gE and Ad(E12)gI per cell. After either 8 or 11 h of infection with
HSV-1 or after 48 h of infection with Ad vector, cells were fixed in 4% (wt/vol) paraformaldehyde in PBS and then, in some cases, permeabilized with 0.2% Triton X-100 (TX100) for 5 min. Samples were incubated with blocking buffer (PBS with 2% normal goat serum, 2% bovine serum albumin, and 0.02% Tween 20) for 30 min. Staining for gE and b-catenin involved (i) incubation with anti-b-catenin mouse MAb for 1 h, (ii) a wash step, (iii) incubation with Cy3-coupled anti-mouse IgG for 1 h, (iv) another wash step, (v) incubation with Oregon Green-coupled MAb 3114 (specific for gE) for 1 h, (vi) a third wash step, and (vii) incubation with BODIPY-coupled anti-FITC (which reacts with Oregon Green) antibodies for 1 h. Staining for gD andb-catenin involved incubating cells simultaneously with anti-gD MAb DL6 and rabbit polyclonal antibodies specific forb-catenin, washing the cells, and then incubating the cells with FITC-coupled goat anti-mouse IgG antibodies and Texas Red-conjugated goat anti-rabbit IgG antibodies. Cells were stained for ZO-1 and either gE, gI, or gD by simultaneously incubating them with rabbit anti-ZO-1 antibodies and either anti-gE MAb 3114, anti-gI MAb 3104, or anti-gD MAb DL6 for 1 h; the cells were subsequently washed and then incubated with FITC-coupled goat anti-mouse IgG antibodies and Texas Red-coupled goat anti-rabbit antibodies for 1 h. Samples were mounted on microscope slides by using Vectashield (Vector Lab-oratories, Burlingame, Calif.) and viewed with a Leica confocal microscope.
TER and paracellular permeability.HEC-1A cells (53105per insert) were
grown for approximately 4 to 5 days on membrane filter inserts (12-mm diameter, 0.4-mm pore size; Millipore) precoated with rat tail collagen (Boehringer Mann-heim) until the transepithelial resistance (TER; net resistance5total resis-tance3resistance of membrane alone) reached at least 400Vcm2. TER was
measured with a Millipore Voltohmmeter. Monolayers were infected with Ad(E12)gE and Ad(E12)gI (400 PFU of each/cell) or AddlE1 (800 PFU/cell) or
were left uninfected; then, 48 h later, the TER was measured. Similarly, cells were either infected with Ad vector or left uninfected, and after 48 h, paracellular permeability was measured by adding 1mCi of [14C]inulin/ml in 200ml of RPMI
medium to the upper chamber. At 20-min intervals, the inserts were transferred to fresh wells and the quantity of radioactivity that had accumulated in the lower compartment was measured with ab-counter.
Determination of TX100 insolubility of gE.HEC-1A cells in 12-well dishes were coinfected with Ad(E12)gE and Ad(E12)gI (400 PFU of each/cell) for 48 h
or were left uninfected. The cells were washed twice with Tris-saline (15 mM Tris-Cl [pH 7.5], 150 mM NaCl) and then extracted with 300ml of ice-cold extraction buffer (1 mM CaCl2, 1 mM MgCl2, 15 mM Tris-Cl [pH 7.5], and 150
mM NaCl) containing a TX100 concentration of 0, 0.05, 0.1, 0.2, 0.4, 0.5, 0.75, or 1% (wt/vol) for 10 min. The cells were then scraped into the extraction buffer, transferred to 1.5-ml centrifuge tubes, and centrifuged at 20,0003g for 30 min at 4°C. Supernatants were transferred to fresh tubes, and sodium dodecyl sulfate (SDS) andb-mercaptoethanol were added to 2% each. The pellets were resus-pended in 400ml of 50 mM Tris-HCl, pH 6.8, containing 2% SDS and 2% b-mercaptoethanol. The samples were boiled and subjected to electrophoresis on 7.5% polyacrylamide gels, and then proteins were detected by Western blot-ting.
RESULTS
Construction of replication-defective Ad vectors expressing gE or gI.In order to study the subcellular localization of gE-gI and effects of this protein in epithelial cells, it was useful to express the glycoproteins without other HSV polypeptides. HSV infection inhibits host protein synthesis and leads to al-terations in the cytoskeleton, cell rounding, and changes in
8934 DINGWELL AND JOHNSON J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
host membranes, including disruption of the Golgi apparatus (9). We chose to construct replication-defective (E12) Ad
vec-tors expressing gE and gI. Such vecvec-tors have advantages in that Ad proteins are not expressed at significant levels and foreign proteins can be expressed at high levels and in combination in different cells. The complete coding sequences of gE and gI, derived from HSV-1 strain KOS, were excised from plasmids pSV2X3gE and PSV2X3gI (19), respectively, using the restric-tion enzymes EcoRI and XbaI and inserted into the shuttle vector pCA3 (20), so that the gE and gI genes were inserted between the HCMV immediate-early promoter and the SV40 polyadenylation sequence in a right-to-left orientation with respect to the flanking Ad E1 sequences (Fig. 1). 293 cells (which supply E1) were cotransfected with either pCA3gE or pCA3gI and a second plasmid, pBHG10, which supplies the right end of the Ad genome, and viruses Ad(E12)gE and
Ad(E12)gI were produced, respectively.
Expression of gE and gI was examined after infection of HEC-1A human epithelial cells with either Ad(E12)gE or
Ad(E12)gI, with both Ad(E12)gE and Ad(E12)gI, or with
HSV-1. Under these conditions, the Ad vectors did not cause obvious cell toxicity or alter cellular metabolism, protein syn-thesis, or morphology. The cells were labelled with [35 S]methi-onine-[35S]cysteine, beginning 48 h after infection with Ad vector or 8 h after infection with HSV-1. gE and gI were immunoprecipitated from extracts of cells by using MAbs 3114 and 3104, respectively. The gE-specific MAb 3114 and the
gI-specific MAb 3104 each precipitated two protein species from either Ad(E12)gE- or Ad(E12)gI-infected cell extract
which corresponded to the immature and mature form of ei-ther gE or gI, respectively (Fig. 2A). The gE-gI complex could be precipitated from extracts of cells coinfected with both Ad(E12)gE and Ad(E12)gI by using MAb 3104, and the
quan-tities of labelled gE-gI complex produced in the cells were similar to those produced in cells infected with HSV-1 (Fig. 2A). Western blot analysis indicated that the quantity of gE that accumulated by 36 or 48 h of infection with Ad(E12)gE
and Ad(E12)gI was greater than that observed in
HSV-in-fected cells, 8 h after infection (Fig. 2B).
Cell surface transport of gE requires gI in epithelial cells. Nonpolarized cell lines (e.g., Vero, Hep-2, and R970 cells) have been used extensively to examine the functions of HSV glycoproteins during viral replication. However, the effects of gE-gI on cell-to-cell spread are less evident in these nonpolar-ized cells and much more evident in primary fibroblasts or epithelial cells. Therefore, we studied the subcellular localiza-tion of and effects of expressing gE-gI in HEC-1A cells, human mucosal epithelial cells that display all of the functional char-acteristics of fully polarized epithelial cells: (i) high TER; (ii) asymmetrical budding of influenza virus and vesicular stoma-titis virus from the apical and basolateral domains, respec-tively; and (iii) polarized distribution of endogenous cellular proteins (e.g., polyimmunoglobulin receptor) (4).
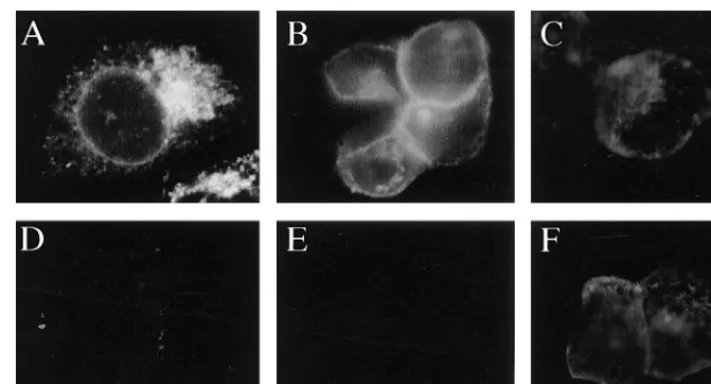
When HEC-1A cells were infected with Ad(E12)gE, gE was
predominantly found in a perinuclear region, often localized to one side of the nucleus, and did not appear to be present on the plasma membrane (Fig. 3A). When cells were coinfected with Ad(E12)gE and Ad(E12)gI, the staining pattern of gE
changed dramatically; gE staining was primarily found at boundaries of cells, in a ring-like pattern separating adjoining cells, and there was little perinuclear staining (Fig. 3B).
How-FIG. 1. Construction of replication-defective (E12) Ad vectors expressing gE
or gI. The full-length gE and gI genes were subcloned into the Ad shuttle plasmid pCA3, which contains the left end of the Ad type 5 genome. In pCA3gE and pCA3gI, the gE and gI genes, respectively, were flanked by the HCMV imme-diate-early promoter and the SV40 poly(A) sequence, so that transcription was in the leftward direction, opposite the direction of E1 transcription. Recombi-nant Ads expressing either gE [Ad(E12)gE] or gI [Ad(E12)gI]) were obtained
[image:3.612.58.283.67.356.2]by cotransfecting 293 cells with pBHG10 and either pCA3gE or pCA3gI by the calcium phosphate technique. mu, map units.
FIG. 2. Expression of gE and gI by recombinant Ad vectors. (A) Human HEC-1A epithelial cells were left uninfected (mock) or were infected with 400 PFU of either Ad(E12)gE or Ad(E12)gI per cell, with both Ad(E12)gE and
Ad(E12)gI, each at 400 PFU/cell, or with HSV-1 at 20 PFU/cell. The cells were
labelled with [35S]methionine and [35S]cysteine for 3 h, beginning 48 h after
infection with Ad vector or 8 h after infection with HSV. Detergent extracts of the cells were mixed with MAbs specific for gE (3114) or gI (3104) to immuno-precipitate these proteins. Positions of molecular size markers in kilodaltons are shown on the left. (B) HEC-1A cells were either infected with HSV-1 or coin-fected with Ad(E12)gE and Ad(E12)gI. After 8 h of infection with HSV-1 or
after 36 or 48 h of infection with the Ad vectors, the cell monolayers were scraped into SDS gel electrophoresis buffer. Samples were boiled, and the pro-teins were separated by polyacrylamide gel electrophoresis and then transferred to nylon membranes. gE was detected by incubating the blots with MAb II-481. The blots were washed and then incubated with horseradish peroxidase-coupled anti-mouse antibodies, and these antibodies were detected by enhanced chemi-luminescence.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.312.548.69.219.2]ever, regardless of whether gI was expressed, gE could not be detected on the apical surfaces of the cells; i.e., the glycopro-tein was not detected if the cell monolayer was not permeabil-ized with detergent to allow access of antibodies to the cyto-plasm and basolateral cyto-plasma membrane (Fig. 3D and E). Similar results were observed when HEC-1A cells were in-fected with replication-competent (E11) Ad vectors expressing
gE or gE plus gI (19). In this case, lower multiplicities of infection (20 PFU/cell) were used and gE-gI expression was observed early (18 h) after infection, but again, when gE was expressed alone (without gI), the glycoprotein was retained in a perinuclear region of the cytoplasm (data not shown). An-other HSV glycoprotein, gD, expressed by using a replication-defective (E12) Ad vector, Ad(E12)gD, was readily detected
by anti-gD antibodies without permeabilization of the cells and was thus on the apical surfaces of the cells (Fig. 3F). Therefore, transport of gE to the cell surface in these polarized epithelial cells requires coexpression of gI, and gE-gI accumulates on the basolateral plasma membrane and not on the apical surface. These results were in contrast to our previous observations with human R970 cells, highly transformed, nonepithelial cells that do not form extensive cell junctions (19). When gE was expressed without gI in R970 cells (using the E11Ad vectors),
gE was largely processed to the mature form, could be iodin-ated on the cell surface by using lactoperoxidase, and bound IgG-coated red blood cells on the cell surface.
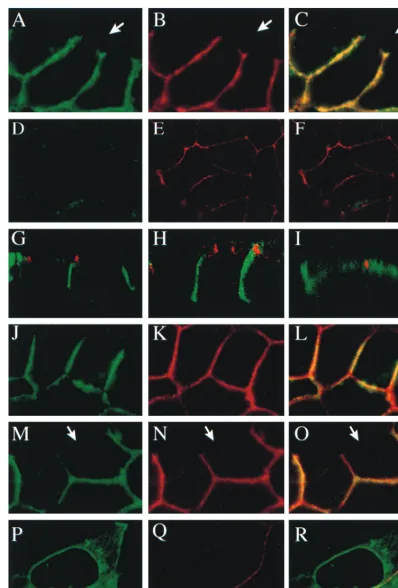
gE and gI expressed by Ad vectors accumulates along lateral but not basal or apical surfaces of epithelial cells. Confocal microscopy was used to examine the distribution of gE in more detail. HEC-1A cells were coinfected with Ad(E12)gE and
Ad(E12)gI and stained simultaneously for gE and either ZO-1
orb-catenin. ZO-1 is a cytoplasmic protein and a component of tight junctions that is important for tight-junction integrity (reviewed in reference 3). Tight junctions are localized just below the apical membranes of epithelial cells and are respon-sible for polarizing the apical and basolateral domains of the plasma membrane and for restricting the movement of solutes from the apical to the basolateral compartments.b-Catenin is a cytoplasmic protein and a component of adherens junctions, cell junctions that are located more uniformly along the lateral plasma membrane beginning just below tight junctions and
extending to the basal surface. Adherens junctions are formed through homophilic binding of cadherins proteins, Ca-depen-dent cell adhesion molecules (CAMs) which are linked to the cytoskeleton byb- anda-catenins (reviewed in reference 54). Extensive colocalization of gE andb-catenin was observed along the entire lateral plasma membrane of the HEC-1A cell, beginning just below the level of the tight junctions. Represen-tative images of a section near the middle of the lateral surface of a HEC-1A cell, with gE in green andb-catenin in red, are shown in Fig. 4A to C, but a similar pattern was observed along the entire lateral surface, except near the apical surface. At these subapical domains, there was intense staining by anti-ZO-1 antibodies but not by anti-gE antibodies (Fig. 4D to F). There was also little or no gE in the cytoplasm or on the apical or basal surfaces.
Confocal sections were also taken through the z axis of cells expressing gE-gI, i.e., on a plane perpendicular to the images in Fig. 4A to F, so that the view is from the side of the monolayer and the apical membrane extends horizontally along the top of the image. z-axis images showed clearly that gE (green) accumulated along most of the lateral borders between cells, extending from the bottom of the image (basal membrane) to near the apical membrane (Fig. 4G). There was little or no gE found on either the apical or basal surfaces, and gE was excluded from tight junctions, which were stained by anti-ZO-1 antibodies (red). Moreover, gI was distributed in Ad(E12)gE- and Ad(E12)gI-infected HEC-1A cells in a
man-ner similar to gE, predominantly along the lateral plasma membrane, below the level of the tight junction (Fig. 4H), and extensively colocalized with b-catenin (data not shown). gE and gI also accumulated along the lateral plasma membranes after expression with replication-competent (E11) Ad vectors
(data not shown). By contrast, HSV glycoprotein gD, ex-pressed by Ad(E12)gD, accumulated primarily along the
api-cal plasma membranes of HEC-1A cells (Fig. 4I). The obser-vations that gE-gI was exclusively along the lateral surface and that gD was primarily on the apical surface extended to MDCK canine epithelial cells and A431 human epithelial cells (data not shown).
[image:4.612.121.477.69.261.2]Subcellular distribution of gE-gI and gD in HSV-infected epithelial cells.As with epithelial cells in which gE and gI were
FIG. 3. gE requires gI to reach the surface of epithelial cells. HEC-1A cells were infected with either Ad(E12)gE (A and D), Ad(E12)gE plus Ad(E12)gI (B and
E), or Ad(E12)gD (C and F) for 48 h. The cells were fixed with paraformaldehyde, either permeabilized with 0.2% TX100 (A to C) or left unpermeabilized (D to F),
and then incubated with MAb 3114, specific for gE (A, B, D, and E), or MAb DL-6, specific for gD (C and F). The cells were washed, incubated with FITC-coupled goat anti-mouse antibodies, washed again, mounted on coverslips, and viewed under a Nikon epifluorescence microscope.
8936 DINGWELL AND JOHNSON J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 4. Subcellular (confocal) localization of gE, gI, and gD in polarized epithelial cells. HEC-1A cells were coinfected with Ad(E12)gE and Ad(E12)gI (A to H)
or infected with Ad(E12)gD (I) for 48 h or were infected with HSV-1 for 8 h (J to L) or for 11 h (M to R). The cells were fixed with paraformaldehyde, permeabilized
with 0.2% TX100, washed, and then incubated with various antibodies as follows. (A to C) Anti-b-catenin MAb (red) followed by Cy3-coupled anti-mouse antibodies and then with anti-gE MAb 3114 (green) coupled directly to Oregon Green followed by BODIPY-coupled anti-FITC antibodies (which react with Oregon Green); (D to G) rabbit anti-ZO-1 (red) and, simultaneously, anti-gE MAb 3114 (green) followed by FITC-coupled anti-mouse IgG antibodies and Texas Red-coupled anti-rabbit antibodies; (H) rabbit anti-ZO-1 (red) and, simultaneously, anti-gI MAb 3104 (green) followed by FITC-coupled antimouse IgG antibodies and Texas Red-coupled anti-rabbit antibodies; (I) rabbit anti-ZO-1 (red) and, simultaneously, anti-gD MAb DL6 followed by Texas Red-coupled anti-rabbit antibodies and FITC-coupled anti-mouse IgG antibodies; (J to O) as in A to C, anti-b-catenin MAb (red) followed by anti-gE MAb (green); (P to R) rabbit anti-b-catenin (red) and, simultaneously, anti-gD MAb DL6 followed by FITC-conjugated anti-mouse and Texas Red-coupled anti-rabbit antibodies. The left panels show HSV gE or gD in green, the same image withb-catenin or ZO-1 stained in red in the middle panels, and the two signals are combined in the right panels, except in panels G to I, which show combined signals of ZO-1 and either gE (G), gI (H), or gD (I). The arrows indicate borders of cells not in contact with other cells and without gE-gI andb-catenin.
8937
on November 9, 2019 by guest
expressed by Ad vectors, gE-gI accumulated along the lateral plasma membranes of HSV-1-infected HEC-1A cells. After 8 h of infection with HSV-1, gE largely accumulated in the lateral membranes, often being restricted to patches in the central region of the membrane, but there was also a small fraction of gE present in cytoplasmic membranes (Fig. 4J to L). By 11 h, gE was distributed more uniformly along and across the lateral plasma membranes, and there was little or no gE found in the cytoplasm (Fig. 4M to O). As with cells infected with the Ad vectors, there was no gE on the apical surfaces of HSV-in-fected cells. A pattern similar to this was observed for gI, and the viral inoculum did not contribute to this pattern of glyco-protein expression (data not shown). By contrast, gD was found more widely distributed on all plasma membrane sur-faces, including the apical sursur-faces, and within the cytoplasm of HSV-infected HEC-1A cells, and although there was also some gD colocalizing withb-catenin on the lateral surfaces, gD did not accumulate at sites of cell-cell contact to the extent that was observed for gE-gI (Fig. 4P to R). Therefore, gE-gI, pro-duced in Ad- or HSV-infected cells, accumulates along the lateral surfaces of epithelial cells, below the level of the tight junctions, whereas gD is primarily present on the apical plasma membrane, as well as in the cytoplasm.
gE accumulates at cell junctions but not on lateral surfaces that are not forming junctions.In subconfluent monolayers of HEC-1A cells,b-catenin was concentrated at cell junctions but was not found on those lateral membranes that were not in contact with another cell (Fig. 4B). The white arrows in Fig. 4 indicate cell borders where cells are not in contact with other cells. This was to be expected because cadherins and adherens junctions form only at sites at which cells contact one another. In both AdgE/gI- and HSV-infected cells, gE was found only at sites of cell-cell contact and was absent from those lateral surfaces not in contact with an opposing cell (Fig. 4A and M). This observation has important implications because the spe-cific accumulation of gE-gI at cell junctions was independent of cell polarization; there was no requirement that tight junctions and other cell junctions must encircle the cells. Therefore, targeting of gE-gI to the basolateral membrane and its reten-tion there by virtue of the barrier imposed by tight juncreten-tions cannot account for the accumulation of gE-gI along the lateral surfaces of epithelial cells.
Solubility of gE-gI in nonionic detergent.In polarized epi-thelial cells, the accumulation of cellular proteins at cell junc-tions often involves binding to components of the cytoskeleton (reviewed in reference 31). For example, the Na1,K1-ATPase
and E-cadherin bind to components of the cortical cytoskele-ton, and this correlates with an increase in resistance to ex-traction with nonionic detergents (e.g., TX100) at concentra-tions greater than 0.5% (18, 34, 36). It was of interest to determine whether gE-gI would resist extraction with low con-centrations of TX100, providing evidence that gE-gI was bound to the cytoskeleton. HEC-1A cells were coinfected with Ad(E12)gE and Ad(E12)gI, and cell extracts were made by
using increasing concentrations of TX100, from 0 to 1% (wt/ vol). The extracts were centrifuged to pellet the insoluble cy-toskeletal fraction, and then gE or E-cadherin was detected by Western blotting. As shown in Fig. 5, a substantial fraction of the gE could be solubilized with 0.05% TX100, and the ma-jority of the glycoprotein could be solubilized with 0.1% TX100. By contrast, little of the E-cadherin was solubilized with 0.05% TX100, and only a fraction of the protein was solubilized with 0.1% TX100. There were significant fractions of the total E-cadherin that were not solubilized with 0.2, 0.4, or 0.75% TX100, conditions under which the majority of gE was solubilized. These results suggest either that gE-gI does
not bind tightly to the cytoskeleton or that its interaction with the cytoskeleton is disrupted by relatively low concentrations of TX100.
Expression of gE does not disrupt epithelial cell junctions. Previously, it was reported that there was a redistribution of the cell junction markers ZO-1, B-cadherin, andb-catenin in HSV-infected human retinal epithelial cells (28). It was sug-gested that gE-gI might contribute to this phenomenon, lead-ing to increases in paracellular permeability (28). In our ex-periments, gE-gI expression did not cause an obvious redistribution of either ZO-1 orb-catenin in HEC-1A, MDCK, or A431 epithelial cells, and there were no obvious alterations in the cell junctions as determined by light microscopy. Cell rounding and redistribution of junctions were observed after HSV infection, but only late (16 to 18 h) after infection, and there were no obvious differences when gE or gI was absent (mutants F-gEb and F-US7kan, respectively) (data not shown).
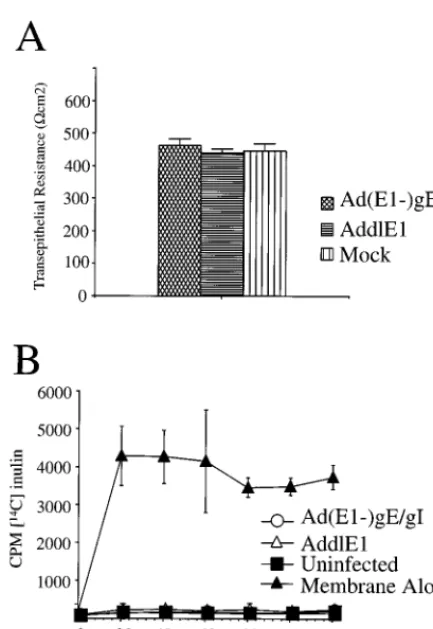
To assess the integrity of tight junctions, we examined the resistance of epithelial monolayers (TER). HEC-1A cells were grown on filter supports until a polarized monolayer was es-tablished, and then the cells were infected with Ad(E12)gE
and Ad(E12)gI or with a replication-defective control vector,
AddlE1, that does not express gE or gI. More than 90% of the cells grown on filters and infected with Ad(E12)gE and
Ad(E12)gI stained positive for gE and gI (data not shown),
and the quantity of gE-gI expressed in the (“mock”) cells was larger than that in HSV-1-infected cells (Fig. 2B). There were no significant differences in TER when uninfected cells or cells infected with AddlE1 were compared to cells infected with Ad(E12)gE and Ad(E12)gI; in all cases, the resistance of the
cell monolayer was greater than 400Vcm2(Fig. 6A). We also examined the paracellular transport of14C-labelled inulin (a small molecule of ;5,000 Da) across epithelial cell monolayers, from the apical to the basolateral compartments. After HEC-1A cells were allowed to become polarized, they were infected either with AddlE1 or with Ad(E12)gE plus
Ad(E12)gI or were left uninfected, and then14C-labelled in-ulin was added to the apical compartment of the monolayer and the accumulation of radiolabel in the basal compartment was measured. With membrane inserts alone, the labelled in-ulin was able to accumulate rapidly in the basal compartment, with maximum levels being achieved within 20 min (Fig. 6B). Movement of [14C] inulin from one compartment to the other was markedly reduced when HEC-1A cells were grown on the
FIG. 5. Solubility of gE in the nonionic detergent TX100. HEC-1A cells were infected with both Ad(E12)gE and Ad(E12)gI for 48 h, washed with Tris-saline,
and then scraped into extraction buffer containing 0.1% TX100. Cell extracts were centrifuged at 20,0003g for 30 min. Proteins in the pellets (P) and supernatants (S) were denatured in buffer containing 2% SDS and 2%b -mer-captoethanol; then the proteins were boiled, subjected to electrophoresis, and transferred to nitrocellulose. gE was detected by incubating the blots with MAb II-481, and E-cadherin was detected with an anti-E-cadherin MAb; in each case, this was followed by incubation with horseradish peroxidase-coupled anti-mouse IgG antibodies and ECL (Amersham).
8938 DINGWELL AND JOHNSON J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
membranes (Fig. 6B). Infection of the cells either with Ad(E12)gE plus Ad(E12)gE or with AddlE1 had no effect.
Therefore, the expression of gE-gI without other HSV proteins does not cause redistribution of junction proteins or affect the function of tight junctions in human HEC-1A cells.
DISCUSSION
Previous work with HSV and the related alphaherpesviruses PrV and varicella-zoster virus has clearly demonstrated that gE-gI mediates or facilitates cell-to-cell spread, especially in solid tissues such as epithelium, in the nervous system, and with certain cultured cells, e.g., normal fibroblasts, epithelial cells, and neurons, cells that form extensive cell junctions (2, 24, 25). Other HSV glycoproteins function similarly in cell-to-cell spread, but unlike these glycoproteins, gE-gI functions in cell-to-cell spread but not in the entry of extracellular virions. Therefore, interactions between gE-gI and cell junctions may provide molecular details of how a herpesvirus moves directly from cell to cell.
When gE-gI was expressed in HEC-1A cells or in other epithelial cells by recombinant Ad vectors, the protein
accu-mulated specifically along the lateral surfaces of cells and was not found on either the apical or basal surfaces or at tight junctions. Sorting of gE-gI to the basolateral surfaces of these cells cannot explain the subcellular localization of gE-gI. This specific accumulation did not include the basal surface and was observed in subconfluent monolayers of cells, before the cells became polarized and before tight junctions encompassed the cell, excluding glycoprotein movement to the apical surface. This observation has important implications for understanding how gE-gI mediates cell-to-cell spread. In those epithelial cells that were not in contact with other cells along one cell border, gE-gI exclusively accumulated at cell junctions. Thus, glyco-proteins inserted into either lateral membrane should be ca-pable of diffusion to apical or other basolateral domains, with-out restriction by tight junctions. We observed a similar distribution of gE-gI in HSV-infected HEC-1A cells, but a second HSV glycoprotein expressed by Ad vectors, gD, pre-dominantly accumulated on the apical surface.
The simplest interpretation of our results is that gE-gI is retained at epithelial cell junctions by binding to components of the cell junctions. This is the case for cellular CAMs and other cell junction components which are found specifically at sites of cell-cell contact and not at lateral surfaces which are not in contact with other cells. For example, cadherins are localized to cell junctions by virtue of homophilic interactions with other cadherins, and without such interactions the cad-herins are rapidly endocytosed (reviewed in reference 54). Certain cellular proteins accumulate at cell junctions through interactions with the cytoskeletal elements (31, 46), but in our experiments involving TX100 extraction, gE-gI did not appear to be tightly bound to the cytoskeleton. However, these results must be viewed with some caution because gE-gI may interact with the cytoskeleton, directly or indirectly, by virtue of inter-actions that are sensitive to TX100. The uniform distribution of gE-gI along the lateral surfaces of cells, underneath the tight junctions, was similar to that of b-catenin, a component of adherens junctions; thus, likely candidates for cellular ligands of gE-gI are the cadherins or other components of adherens junctions. gE and gI mutants also show defects in cell-to-cell spread in normal human fibroblasts and neurons (14, 15), cells that form well-defined cell junctions but over a smaller per-centage of the cell membrane. Therefore, we expect that pu-tative cellular ligands for gE-gI will not be restricted to epi-thelial cells.
In other cell types, primarily highly transformed cell lines— e.g., Vero or R970 cells—that do not form extensive cell junc-tions, gE-gI does not affect cell-cell spread. Previous studies involving nonpolarized cell lines such as L cells, Vero cells, and human R970 and HeLa cells indicated that gE moved to the cell surface without gI (5, 19, 45). Here, we found that gE accumulated in perinuclear, cytoplasmic vesicles in epithelial cells, consistent with reports that HSV gE and alphaherpesvi-rus gE homologues require coexpression of gI for efficient transport to the cell surface in some but not all types of cells (29, 33, 49, 50, 53). Therefore, there appears to be retention in the endoplasmic reticulum and possibly misfolding of gE when the glycoprotein is expressed without gI, at least in some types of cells. These results highlight the importance of considering gE-gI as a functional and structural complex and of character-izing gE-gI trafficking and function in cells that closely approx-imate those infected in vivo.
Previously, there have been reports that certain HSV-2 gly-coproteins were transported to either the basolateral domain of bovine and monkey epithelial cells (47) or the apical do-mains of neuroblastoma cells (37) when the proteins were expressed by HSV-2 infection. It is not clear why our
observa-FIG. 6. Expression of gE-gI does not disrupt the functional integrity of tight junctions. HEC-1A cells were grown on membrane filter supports for 5 days, until the TER exceeded 400Vcm2; subsequently, the cells were coinfected with
Ad(E12)gE and Ad(E12)gI were infected with AddlE1 (a control Ad vector) or
were left uninfected (Mock) for a further 2 days. (A) TER was measured with a Millipore Voltohmmeter. Note that the values shown are resistance values for the cell monolayers, since the resistance of the membrane support was subtracted from the total resistance. (B) Paracellular permeability was determined by in-troducing [14C]inulin into the upper compartment and measuring the labelled
inulin in the lower chamber at various time points. In each case, three separate wells were used for each measurement, and error bars represent the standard deviations.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.59.276.66.381.2]tions differ from these previous results; however, we observed accumulation of gE-gI at junctions with several other epithelial cell types, including canine MDCK cells and human A431 epithelial cells. Our observation that gE-gI is not present on the apical surface was interesting in light of previous reports that gE-gI is present in the virion envelope (25, 39). Therefore, virus particles produced in polarized epithelial cells may not contain gE-gI; alternatively, there may be very few virions on the apical surfaces of these cells. This would represent a strik-ing difference between cells that form extensive cell junctions and those that do not. Moreover, the finding that gE-gI is not present on the apical surface complicates the notion that this complex acts as an Fc receptor (reviewed in reference 55) because, at least in these cells, gE-gI does not have access to IgG.
The accumulation of gE-gI on the lateral surfaces of epithe-lial cells suggests that there are sorting mechanisms that spec-ify transport of this complex to the basolateral rather than apical membranes. Basolateral sorting in polarized cells is me-diated by a variety of signals in the cytoplasmic tails of mem-brane glycoproteins (reviewed in references 30, 32, and 42). Many of these sequence motifs are related to or overlap with signals that cause proteins to be recognized by adaptor pro-teins and directed to the trans-Golgi network (TGN) and en-dosomes or incorporated into cell surface clathrin-coated pits and endocytosed, e.g., tyrosine-based motifs and dileucine mo-tifs (21, 30, 32). The cytoplasmic domain of HSV-1 gE contains a tyrosine motif, YADW, and gI contains a dileucine motif, both of which may affect TGN localization and specific trans-port to basolateral surfaces. Similarly, varicella-zoster virus gE contains a Tyr-based motif and an acidic domain that target
the glycoprotein to the TGN and specify endocytosis (1, 38, 56).
The accumulation of gE-gI at epithelial cell junctions in the absence of other HSV polypeptides provides important clues as to how gE-gI facilitates epithelial cell-to-cell spread. We have considered three models of how gE-gI functions, based on the present observations. The first model is based on work of Maidji et al. (28), who suggested that gE-gI could promote cell-to-cell spread by disrupting cell junctions. In our experi-ments, expression of gE-gI without other HSV proteins did not lead to redistribution ofb-catenin or ZO-1 or to alterations in paracellular movement of small molecules in a number of types of human and animal epithelial cells. In addition, we found no differences in the kinetics with which cell junctions were dis-rupted by wild-type versus gE2HSV-1 when all of the cells in
a monolayer were infected. Therefore, it appears unlikely that gE-gI alone can disrupt epithelial cell junctions.
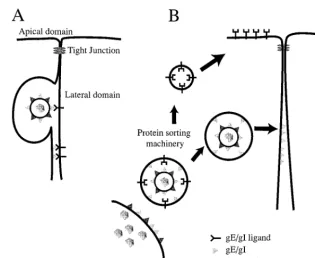
[image:8.612.140.455.67.325.2]Two other models describing how gE-gI could affect cell-to-cell spread are depicted in Fig. 7. Model A suggests that gE-gI binds to cellular ligands concentrated at cell junctions and, as a consequence of these interactions, facilitates transfer of HSV particles into the space between cells, their movement across this space, or their fusion with the uninfected-cell membrane. It is not clear at this point whether gE-gI that is part of the virion envelope (Fig. 7A, top) or part of the plasma membrane (Fig. 7A, bottom) mediates cell-to-cell spread. Related to this point, PrV mutants in which gE was expressed, but not incor-porated into the virion, formed relatively small plaques on monolayers of bovine epithelial cells (48), consistent with the view that gE must be in the virion envelope in order to facil-itate PrV cell-to-cell spread. However, it is also possible that
FIG. 7. Models for how gE-gI facilitates epithelial cell-to-cell spread. (A) gE-gI (triangles) accumulates at junctions formed between an infected cell (left) and an uninfected cell (right) by virtue of interactions with cellular ligands which are expressed in the lateral membranes of the uninfected cell. Presumably, binding of gE-gI to these cellular ligands enhances the transfer of virus across cell junctions so that virus particles can move into the space between the cells and then fuse with the opposing, uninfected cell membrane. It is not clear whether gE-gI functions in this capacity as part of the virion envelope (top) or as a constituent of the plasma membrane of the infected cell (bottom). (B) Alternatively, gE-gI may act as a trafficking signal to direct enveloped particles to the lateral plasma membrane rather than to the apical surface (as with apically sorted proteins). Since there is some evidence that the cytoplasmic domains of gE and gI have motifs that affect intracellular transport, it is likely that these cytosolic domains serve to target virions present within cytoplasmic vesicles to sites of cell-cell contact.
8940 DINGWELL AND JOHNSON J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
these mutant PrV gE molecules were mislocalized in cells. Whether gE-gI acts as part of the virion envelope or the plasma membrane, it is difficult to explain the accumulation of this complex at cell junctions without proposing that it binds some cellular component of junctions. Close apposition be-tween the two plasma membranes at junctions would allow gE-gI to interact with a cellular ligand on an adjoining cell, in a similar fashion to CAMs. In this manner, gE-gI may provide a favorable environment for gB, gD, or gH-gL to promote entry of virus into the apposing cell. This model is also consis-tent with the observation that gI (and perhaps even gE) is structurally or evolutionarily related to a family of S-compo-nent glycoproteins including gD and gG (31a), and gD is known to act as a receptor binding protein (10, 23, 51).
The second model (Fig. 7B) suggests that gE-gI acts by sorting virus particles, so that virions are preferentially trans-ported to lateral surfaces and cell junctions rather than to the apical plasma membrane. Since most cytoplasmic virions are within membrane vesicles, it is likely that this sorting involves the cytoplasmic tails of gE or gI that are part of these vesicles. Thus, the gE-gI cytoplasmic tails may interact with the cellular sorting machinery to direct virus particles to sites of cell-cell contact. To explain the accumulation of gE-gI at cell junctions in this model, we propose that junctions are the final destina-tion, so that gE-gI accumulates there after long-term expres-sion. It is important to note that these two models are not necessarily mutually exclusive. It will be important to study the effects of mutating gE and gI on the subcellular trafficking of gE-gI, its localization to cell junctions, and its cell-to-cell spread.
ACKNOWLEDGMENTS
We thank Kenneth Fish, Jodi Engstrom, and Sally Hanson for their patience and help with the confocal microscopy. We are grateful to the members of the Johnson and Jay Nelson labs for many helpful discus-sions.
Support for this research was provided by grants from the Medical Research Council of Canada (MRCC) and the NIH (CA73996). Dur-ing a portion of this work, K.S.D. held a research studentship from the MRCC and D.C.J. was a senior research scholar of the National Cancer Institute of Canada.
REFERENCES
1. Alconada, A., U. Bauer, and B. Hoflack. 1996. A tyrosine-based motif and a casein kinase II phosphorylation site regulate the intracellular trafficking of the varicella-zoster virus glycoprotein I, a protein localized in the trans-Golgi network. EMBO J. 15:6096–6110.
2. Balan, P., N. Davis-Poynter, S. Bell, H. Atkinson, H. Browne, and T. Minson. 1994. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 75:1245–1258.
3. Balda, M. S., and K. Matter. 1998. Tight junctions. J. Cell Sci. 111:541–547. 4. Ball, J. M., Z. Moldoveaunu, L. R. Melsen, P. A. Kozlowski, S. Jackson, M. J.
Mulligan, J. F. Mestecky, and R. W. Compans.1995. A polarized human endometrial cell line that binds and transports polymeric IgA. In Vitro Cell Dev. Biol. 31:196–206.
5. Bell, S., M. Cranage, L. Borysiewicz, and T. Minson. 1990. Induction of immunoglobulin G Fc receptors by recombinant vaccinia viruses expressing glycoproteins E and I of herpes simplex virus type 1. J. Virol. 64:2181–2186. 6. Bett, A. J., W. Haddara, L. Prevec, and F. L. Graham. 1994. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA. 91:8802–8806. 7. Brunetti, C. R., K. S. Dingwell, C. Wale, F. L. Graham, and D. C. Johnson. 1998. Herpes simplex virus gD and virions accumulate in endosomes by mannose 6-phosphate-dependent and -independent mechanisms. J. Virol.
72:3330–3339.
8. Cai, W. H., B. Gu, and S. Person. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 62:2596–2604. (Erratum, 62:4438.)
9. Campadelli-Fiume, G., R. Brandimarti, C. Di Lazzaro, P. L. Ward, B.
Roiz-man, and M. R. Torrisi.1993. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the
Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 90:2798–2802.
10. Campadelli-Fiume, G., M. Arsenakis, F. Farabegoli, and B. Roizman. 1988. Entry of herpes simplex virus 1 in BJ cells that constitutively express viral glycoprotein D is by endocytosis and results in degradation of the virus. J. Virol. 62:159–167.
11. Card, J. P., and L. W. Enquist. 1995. Neurovirulence of pseudorabies virus. Crit. Rev. Neurobiol. 9:137–162.
12. Corey, L., and P. G. Spear. 1986. Infections with herpes simplex viruses. N. Engl. J. Med. 314:686–691.
13. Dingwell, K. S. Unpublished data.
14. Dingwell, K. S., C. R. Brunetti, R. L. Hendricks, Q. Tang, M. Tang, A. J.
Rainbow, and D. C. Johnson.1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834–845.
15. Dingwell, K. S., L. C. Doering, and D. C. Johnson. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69: 7087–7098.
16. Forrester, A., H. Farrell, G. Wilkinson, J. Kaye, N. Davis-Poynter, and T.
Minson.1992. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341– 348.
17. Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–72.
17a.Griffiths, A., S. Renfrey, and T. Minson. 1998. Glycoprotein C-deficient mutants of two strains of herpes simplex virus type 1 exhibit unaltered adsorption characteristics on polarized and non-polarized cells. J. Gen. Vi-rol. 79:807–812.
18. Hammerton, R. W., K. A. Krzeminski, R. W. Mays, T. A. Ryan, D. A.
Wollner, and W. J. Nelson.1991. Mechanism for regulating cell surface distribution of Na1,K1-ATPase in polarized epithelial cells. Science 254:
847–850.
19. Hanke, T., F. L. Graham, V. Lulitanond, and D. C. Johnson. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vec-tors expressing glycoproteins E and I. Virology 177:437–444.
20. Hitt, M., A. J. Bett, C. L. Addison, L. Prevec, and F. L. Graham. 1995. Techniques for human adenovirus vector construction and characterization. Methods Mol. Genet. 78:13–30.
21. Hunziker, W., and C. Fumey. 1994. A di-leucine motif mediates endocytosis and basolateral sorting of macrophage IgG Fc receptors in MDCK cells. EMBO J. 13:2963–2969.
22. Isola, V. J., R. J. Eisenberg, G. R. Siebert, C. J. Heilman, W. C. Wilcox, and
G. H. Cohen.1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 63:2325–2334.
23. Johnson, D. C., R. L. Burke, and T. Gregory. 1990. Soluble forms of herpes simplex virus glycoprotein D bind to a limited number of cell surface recep-tors and inhibit virus entry into cells. J. Virol. 64:2569–2576.
24. Johnson, D. C., and V. Feenstra. 1987. Identification of a novel herpes simplex virus type 1-induced glycoprotein which complexes with gE and binds immunoglobulin. J. Virol. 61:2208–2216.
25. Johnson, D. C., M. C. Frame, M. W. Ligas, A. M. Cross, and N. D. Stow. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347–1354. 26. Kudelova, M., M. Kostal, L. Cervenakova, J. Rajcani, and H. C. Kaerner. 1991. Pathogenicity and latency competence for rabbits of the herpes simplex virus type 1 ANGpath gC and gE defective mutants. Acta Virol. 35:438–449. 27. Ligas, M. W., and D. C. Johnson. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced byb-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494. 28. Maidji, E., S. Tugizov, T. Jones, Z. Zheng, and L. Pereira. 1996. Accessory
human cytomegalovirus glycoprotein US9 in the unique short component of the viral genome promotes cell-to-cell transmission of virus in polarized epithelial cells. J. Virol. 70:8402–8410.
29. Mallory, S., M. Sommer, and A. M. Arvin. 1997. Mutational analysis of the role of glycoprotein I in varicella-zoster virus replication and its effects on glycoprotein E conformation and trafficking. J. Virol. 71:8279–8288. 30. Matter, K., and I. Mellman. 1994. Mechanisms of cell polarity: sorting and
transport in epithelial cells. Curr. Opin. Cell Biol. 6:545–554.
31. Mays, R. W., K. A. Beck, and W. J. Nelson. 1994. Organization and function of the cytoskeleton in polarized epithelial cells: a component of the protein sorting machinery. Curr. Opin. Cell Biol. 6:16–24.
31a.McGeoch, D. J. 1990. Evolutionary relationships of virion glycoprotein genes in the S regions of alphaherpesvirus genomes. J. Gen. Virol. 71:2361–2367. 32. Mellman, I. 1996. Endocytosis and molecular sorting. Annu. Rev. Cell Dev.
Biol. 12:575–625.
33. Mijnes, J. D. F., L. M. van der Horst, E. van Anken, M. C. Horzinek, P. J. M.
Rottier, and R. J. de Groot.1996. Biosynthesis of glycoproteins E and I of feline herpesvirus: gE-gI interaction is required for intracellular transport. J. Virol. 70:5466–5475.
34. Morrow, J. S., C. D. Cianci, T. Ardito, A. S. Mann, and M. Kashgarian. 1989. Ankyrin links fodrin to the alpha subunit of Na,K-ATPase in Madin-Darby
on November 9, 2019 by guest
http://jvi.asm.org/
canine kidney cells in intact renal tubule cells. J. Cell Biol. 108:455–465. 35. Neidhardt, H., C. H. Schro¨der, and H. C. Kaerner. 1987. Herpes simplex
virus type 1 glycoprotein E is not indispensable for viral infectivity. J. Virol.
61:600–603.
36. Nelson, W. J., and P. J. Veshnock. 1987. Ankyrin binding to (Na11K1)ATPase and implications for the organization of membrane
do-mains in polarized cells. Nature 328:533–536.
37. Nielsen, L. N., R. J. Whitley, and S. Chatterjee. 1991. Apical expression of herpes simplex virus type 2 glycoproteins in human neuroblastoma cells. Virology 185:908–910.
38. Olson, J. K., and C. Grose. 1997. Endocytosis and recycling of varicella-zoster virus Fc receptor glycoprotein gE: internalization mediated by a YXXL motif in the cytoplasmic tail. J. Virol. 71:4042–4054.
39. Para, M. F., R. B. Baucke, and P. G. Spear. 1982. Glycoprotein gE of herpes simplex virus type 1: effects of anti-gE on virion infectivity and on virus-induced Fc-binding receptors. J. Virol. 41:129–136.
40. Rajcani, J., U. Herget, and H. C. Kaerner. 1990. Spread of herpes simplex virus (HSV) strains SC16, ANG, ANGpath and its GlyC minus and GlyE minus mutants in DBA-2 mice. Acta Virol. 34:305–320.
41. Rajcani, J., U. Herget, M. Kostal, and H. C. Kaerner. 1990. Latency com-petence of herpes simplex virus strains ANG, ANGpath and its gC and gE minus mutants. Acta Virol. 34:477–486.
42. Robinson, R. 1997. Coats and vesicle budding. Trends Cell Biol. 7:99–102. 43. Roop, C., L. Hutchinson, and D. C. Johnson. 1993. A mutant herpes simplex
virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 67:2285–2297.
44. Roper, R. L., E. J. Wolffe, A. Weisberg, and B. Moss. 1998. The envelope protein encoded by the A33R gene is required for formation of actin-containing microvilli and efficient cell-to-cell spread of vaccinia virus. J. Vi-rol. 72:4192–4204.
45. Rosenthal, K. L., J. R. Smiley, S. South, and D. C. Johnson. 1987. Cells expressing herpes simplex virus glycoprotein gC but not gB, gD, or gE are recognized by murine virus-specific cytotoxic T lymphocytes. J. Virol. 61: 2438–2447.
46. Scannevin, R. H., H. Murakoshi, K. J. Rhodes, and J. S. Trimmer. 1996. Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K1channel. J. Cell Biol. 135:1619–1632.
47. Srinivas, R. V., N. Balachandran, F. V. Alonso-Caplen, and R. W. Compans.
1986. Expression of herpes simplex virus glycoproteins in polarized epithelial cells. J. Virol. 58:689–693.
48. Tirabassi, R. S., R. A. Townley, M. G. Eldridge, and L. W. Enquist. 1997. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: evidence for envelope incorporation, virulence, and neu-rotropism domains. J. Virol. 71:6455–6464.
49. van Vliet, K. 1993. Ph.D. thesis. Utrecht University, Utrecht, The Nether-lands.
50. Whitbeck, J. C., A. C. Knapp, L. W. Enquist, W. C. Lawrence, and L. J. Bello. 1996. Synthesis, processing, and oligomerization of bovine herpesvirus 1 gE and gI membrane proteins. J. Virol. 70:7878–7884.
51. Whitbeck, J. C., C. Peng, H. Lou, R. Xu, S. H. Willis, M. Ponce De Leon, T.
Peng, A. V. Nicola, R. I. Montgomery, M. S. Warner, A. M. Soulika, L. A. Spruce, W. T. Moore, J. D. Lambris, P. G. Spear, G. H. Cohen, and R. J. Eisenberg.1997. Glycoprotein D of herpes simplex virus (HSV) binds di-rectly to HVEM, a member of the tumor necrosis factor receptor superfam-ily and a mediator of HSV entry. J. Virol. 71:6083–6093.
52. Wolffe, E. J., E. Katz, A. Weisberg, and B. Moss. 1997. The A34R glyco-protein gene is required for induction of specialized actin-containing mi-crovilli and efficient cell-to-cell transmission of vaccinia virus. J. Virol. 71: 3904–3915.
53. Yao, Z., W. Jackson, B. Forghani, and C. Grose. 1993. Varicella-zoster virus glycoprotein gpI/gpIV receptor: expression, complex formation, and antige-nicity within the vaccinia virus-T7 RNA polymerase transfection system. J. Virol. 67:305–314.
54. Yap, A. S., W. M. Brieher, and B. M. Gumbiner. 1997. Molecular and functional analysis of cadherin-based adherens junctions. Annu. Rev. Cell Dev. Biol. 13:119–146.
55. York, I. A., and D. C. Johnson. 1994. Inhibition of humoral and cellular immune recognition by herpes simplex viruses, p. 89–110. In G. McFadden (ed.), Viroceptors, virokines and related immune modulators encoded by DNA viruses. R. G. Landes Company, Austin, Tex.
56. Zhu, Z., Y. Hao, M. D. Gershon, R. T. Ambron, and A. A. Gershon. 1996. Targeting of glycoprotein I (gE) of varicella-zoster virus to the trans-Golgi network by an AYRV sequence and an acidic amino acid-rich patch in the cytosolic domain of the molecule. J. Virol. 70:6563–6575.
57. Zsak, L., F. Zuckermann, N. Sugg, and T. Ben-Porat. 1992. Glycoprotein gI of pseudorabies virus promotes cell fusion and virus spread via direct cell-to-cell transmission. J. Virol. 66:2316–2325.
8942 DINGWELL AND JOHNSON J. VIROL.