Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Most Retroviral Recombinations Occur during Minus-Strand
DNA Synthesis
JIAYOU ZHANG,* LING-YUN TANG, TING LI, YAN MA,ANDCHRISTY M. SAPP Department of Microbiology and Immunology and Markey Cancer Center, University of Kentucky,
Lexington, Kentucky 40536-0096 Received 21 January 1999/Accepted 3 December 1999
Retroviral RNA molecules are plus, or sense in polarity, equivalent to mRNA. During reverse transcription, the first strand of the DNA molecule synthesized is minus-strand DNA. After the minus strand is polymerized, the plus-strand DNA is synthesized using the minus-strand DNA as the template. In this study, a helper cell line that contains two proviruses with two different mutatedgfpgenes was constructed. Recombination between the two frameshift mutant genes resulted in a functionalgfp. If recombination occurs during minus-strand DNA synthesis, the plus-strand DNA will also contain the functional sequence. After the cell divides, all of its offspring will be green. However, if recombination occurs during plus-strand DNA synthesis, then only the plus-strand DNA will contain the wild-type gfp sequence and the minus-strand DNA will still carry the frameshift mutation. The double-stranded DNA containing this mismatch was subsequently integrated into the host chromosomal DNA of D17 cells, which were unable to repair the majority of mismatches within the retroviral double-strand DNA. After the cell divided, one daughter cell contained the wild-typegfpsequence and the other daughter cell contained the frameshift mutation in the gfp sequence. Under fluorescence microscopy, half the cells in the offspring were green and the other half of the cells were colorless or clear. Thus, we demonstrated that more than 98%, if not all, retroviral recombinations occurred during minus-strand DNA synthesis.
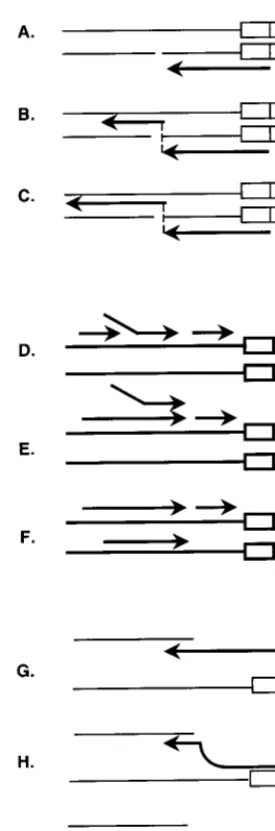
Recombinations in retroviral genes play important roles in retroviral carcinogenesis and in the AIDS epidemic. Two plau-sible models for retrovirus recombination were first proposed 16 years ago. A third model has recently been proposed. How-ever, none of these models have been critically tested. The first model (Fig. 1A to C) proposed that recombination occurred during minus-strand synthesis using RNA as the template (7). This model proposed that retroviral genomes contain consid-erable numbers of breaks within the RNA molecule and that when reverse transcriptase encounters such breaks, it switches to the homologous sequence on the other RNA molecule and continues synthesis. This model is called the forced-copy-choice model. The second model (Fig. 1D to F) proposed that recombination occurs during plus-strand DNA synthesis (15, 16). This model proposed that two minus-strand DNAs are made in one virion using both RNA templates (retrovirus packages two RNA molecules in each virion). Since plus-strand DNA synthesis is initially discontinuous, a fragment of product DNA might be displaced by continuous DNA synthe-sis. The displaced DNA fragment may then hybridize to the minus-strand DNA synthesized from the other molecule of viral RNA used as the template. This model is called the plus-strand replacement model. The third model differs from the original copy choice models because it does not require preexisting breaks in the RNA (6) (Fig. 1G to I). Since reverse transcriptase has a relatively low processivity and because the RNA template is degraded approximately 18 nucleotides from the point of DNA synthesis, it is proposed that an extended single-stranded DNA molecule is left tailing behind. This DNA molecule is free to form a hybrid duplex with another RNA
molecule. If the reverse transcriptase molecule detaches from the template, it is likely that the short (18-bp) hybrid with the original template will be displaced by branch migration of the second RNA and that this RNA molecule will then become the template for continued synthesis. This model is called the mi-nus-strand replacement model.
Data have demonstrated that poly(A) sequences exist in some recombinants of one naturally occurring acute oncogenic virus (14) and in three other experimental systems, including the one we have described previously (22, 25, 28). The poly(A) tracts exist only on the RNA molecules and not on the minus-strand DNA molecules during reverse transcription, since the minus-strand strong stop DNA jumps to the R region [up-stream of the poly(A) tract] to continue synthesis of minus-strand DNA. Therefore, some, if not all, recombination events should use an RNA molecule as the template. Since minus-strand DNA synthesis uses RNA as a template, these results suggest that some retroviral recombinations occur during mi-nus-strand DNA synthesis.
The retroviral RNA molecules are positive sense in polarity, equivalent to mRNA. During reverse transcription, the first strand of DNA synthesized is minus in polarity since it is synthesized from the positive-sense RNA molecule, which is used as the template. The minus-strand DNA is complemen-tary to the plus or sense viral genomic RNA. After the minus-strand DNA is polymerized, the plus-minus-strand DNA is synthe-sized using the minus-strand DNA as the template. By this rationale, if a change results from recombination that occurs during minus-strand DNA synthesis, the plus-strand DNA would also contain this change, because during synthesis of the plus-strand DNA, the minus-strand DNA is used as the tem-plate. However, if changes occurs during plus-strand DNA synthesis, the double-stranded DNA would contain a mis-match. A mismatch within retroviral double-stranded DNA can also be introduced by a preexisting mutation within the * Corresponding author. Mailing address: 206 Combs Research
Bldg., University of Kentucky, 800 Rose St., Lexington, KY 40536-0096. Phone: (606) 257-4456. Fax: (606) 257-8940. E-mail: jzhan1 @pop.uky.edu.
2313
on November 9, 2019 by guest
http://jvi.asm.org/
primer binding site (PBS) of the positive-sense viral RNA genome (3, 8). The PBS is the binding site for a host tRNA within the viral RNA molecule. The PBS is 18 bases in length in Moloney leukemia virus (MLV). The 18-base sequence is reverse transcribed into a minus-strand DNA using the viral RNA genome as the template, while the plus-strand DNA is synthesized using the host tRNA 3⬘ end as the template. If there is a preexisting mutation within this PBS on the viral
RNA molecule, the minus-strand DNA contains the mutation while the plus strand remains the wild type (wt). Therefore, the double-stranded DNA should contain a mismatch within the PBS.
Retroviral double-stranded DNA is then integrated into the cellular chromosome DNA to form a provirus. Most cells con-tain a system to repair different types of mismatches. Some cell lines, such as the HCT 116 cell line, contain defects in their DNA repair systems and are incapable of repairing mis-matches (10, 20). To date it is not clear if the mismatch in the provirus can be repaired by the host cellular DNA repair sys-tem. If the target cells does not repair the mismatch before an infected cell divides, one daughter cell will contain this tion while the other daughter cell will not contain this muta-tion. Therefore, the offspring of the infected cells should carry two different proviruses. However, in the host cell, these two different proviruses would be integrated into the same site of the host chromosome if the integration resulted from a single event. This report demonstrates that D17 cells (target cells) are unable to repair the majority of mismatches within the retroviral double-stranded DNA. This property of D17 cells, therefore, is useful for determining at which step(s) retroviral recombinations occur.
Previous studies that were designed to determine the recom-bination rate of a retrovirus have used selectable genes and cell culture systems (12, 28). In this report, drug selection markers have been exchanged for an unselected color reporter gene,gfp (green fluorescent protein) (5). Cells containing the wt gfp reporter gene appear green under fluorescence microscopy, while cells with a mutantgfpappear colorless or clear (24, 27). A helper cell containing two proviruses that encoded two mu-tatedgfpgenes was constructed. We reasoned that recombina-tion between the two mutarecombina-tions would produce a funcrecombina-tionalgfp gene. Because the plus-strand DNA is synthesized from the minus-strand DNA template—the recombination occurs dur-ing minus-strand DNA synthesis—the plus-strand DNA will also carry the functionalgfpsequence. After the cell divides, all the offspring cells will be green under a fluorescence micro-scope. However, if the recombination occurs during plus-strand DNA synthesis, only the plus-plus-strand DNA will contain the functionalgfpsequence and the minus-strand DNA will contain the mutated gfp. The double-stranded DNA, which contains a mismatch, integrates into the host chromosomal DNA to form a provirus. Because D17 cells are unable to repair this mismatch, after the cell divides into two daughter cells, one of the daughter cells will encode wtgfpand the other daughter cell will encode a mutatedgfp. Growth of the off-spring of these two daughter cells will produce a colony. Under the fluorescence microscope, this colony should be a mixture of green and clear cells.
[image:2.612.100.238.71.487.2]However, a colony of a mixture of green and clear cells is not necessarily a result from a recombination (one event) occur-ring duoccur-ring plus-strand DNA synthesis. Another possibility for the formation of a mixed colony is an infection with two viruses of two daughter cells that have just undergone division from one cell (or two cells that were plated together on a dish) (two separate events). One virus is a parent-type virus (clear), and the other virus is a recombinant formed during minus-strand DNA synthesis (green). If a colony of cells results from two infection events, the resulting colony should be a mixture of two different cells, each of which contains a different provirus resulting from an independent integration event. However, in this scenario these two proviruses will have integrated into two different sites. If a colony of mixed cells results from a recom-bination occurring during plus-strand DNA synthesis, however, the provirus in the green cells should be integrated into the
FIG. 1. Models of retroviral recominations. The light lines represent RNA molecules, and the heavy lines represent DNA molecules. (A to C) Model of forced copy choice for retroviral recombination. This model proposes that ret-roviral genomes contain breaks within the RNA molecule (A) and that when reverse transcriptase encounters such breaks, it switches to the homologous sequence on the other genome (B) and continues minus-strand DNA synthesis (C). (D to F) Model of strand displacement-assimilation for retroviral recombi-nation. This model proposes that two minus-strand DNAs are made within one virion (D). Since plus-strand DNA synthesis is initially discontinuous, a fragment of product DNA might be displaced by continuous DNA synthesis (E). The displaced DNA fragment may then hybridize to the other minus-strand DNA (F). (G to I) Model of minus-strand exchange for retroviral recombination. An extended single-stranded DNA molecule is left tailing behind during synthesis of minus-strand DNA (G). This DNA molecule forms a hybrid duplex with another RNA molecule (H). The reverse transcriptase molecule detaches from the tem-plate and is displaced by the second RNA (I).
on November 9, 2019 by guest
http://jvi.asm.org/
same site as the clear cells in their mixed colony. Using this rationale, we demonstrate that more than 98%, if not all, of retroviral recombinations occur during minus-strand DNA synthesis. In other words, the model of copy choice or minus-strand displacement is the mechanism of most, if not all, ret-roviral recombination events.
MATERIALS AND METHODS
Nomenclature.Plasmids are designated, for example pJZ425 and pJZ468; viruses made from these plasmids are designated, for example, JZ425 and JZ468. Vector constructions.All recombinant techniques were carried out according to conventional procedures (23). All vector sequences are available upon re-quest.
(i) Construction of pLT2.To construct a retroviral vector that contains one base mutation in its PBS region, three fragments were ligated. The first fragment was derived from pLN (19) and contained two MLV long terminal repeats (LTRs) and a neomycin resistance gene (neo). pLN was digested withPshAI and
BstEII.PshAI digested 17 bp upstream of the PBS, whileBstEII digested within the package signal, 537 bp downstream of the PBS. The second fragment was a double-stranded DNA created by annealing two artificially synthesized oligonu-cleotides. The first oligonucleotide was GGGTCTTTCATTTGGGGGCTCGc, while the second oligonucleotide was CCGGgCGAGCCCCCAAATGAAAGA CCC (the lowercase letters represent the nucleotides that are different from those of the wt PBS sequence). The 5⬘end of this DNA was blunt and compatible with thePshAI-digested DNA fragment. The 3⬘double-stranded DNA contained a sticky end, which was identical to the sticky end formed by digestion withXmaI. This double-stranded DNA was designated thePshAI-XmaI fragment. The third
fragment was also derived from pLN. Sequences of pLN were amplified by PCR using two primers. The first primer (GGGGGCTCGcCCGGGATTT) was lo-cated at the PBS; however, the thymidine (T) within the PBS at position 744 was changed to cytosine (C) so that the mutated PBS would be digested withXmaI (cCCGGG) (underlined). The amplified fragment was digested withXmaI and
BstEII. TheBstEII-PshAI fragment isolated from pLN, the double-stranded DNA artificially synthesized withPshAI andXmaI and theXmaI- andBst EII-digested PCR-amplified fragment were ligated. The resulting plasmid was des-ignated pLT2, which was identical to pLN except that the T at position 744 was changed to C so that this plasmid would contain an additionalSmaI site at the PBS. (SmaI recognizes the same CCCGGG asXmaI does, except that the
SmaI-cut fragment is blunt ended while theXmaI-cut fragment forms a sticky end.)
(ii) Construction of a retroviral vector with deletion of the downstream PBS. TheEcoRI fragment which contained the MLV 5⬘LTR was cloned into pSP73 (Promega, Madison, Wis.). To delete the downstream PBS, the MLV vector was amplified by PCR using two primers. The first primer (TAGAACCAGATCTG ATATCATC) was located on pSP73, and the second primer (GACGAGgtCga cAATGAAAGACCCCCGTCGTGGGT) was located at the U5 region of the 3⬘
LTR. The first six nucleotides (CCCCCA) of the PBS region were changed to (gtCgac), so that the amplified fragment could be digested withSalI. The am-plified fragment was digested withBglII andSalI, and thisBglII-SalI fragment did not contain the downstream PBS.
(iii) Construction of a retroviral vector which contains an additionalSmaI site at its PBS and deletes the downstream PBS.The pLN plasmid contains two MLV LTRs and aneogene. Plasmid pLT5 is similar to pLN except that pLT5 contains a T3C transition at the PBS and the PBS downstream of the 3⬘LTR is deleted (Fig. 2A). Plasmid pLT5 was constructed by ligations of five fragments from three vectors. The five fragments are as follows: theSalI-EcoRI fragment FIG. 2. (A) Structure of a retrovirus vector which contains a transition mu-tation on its PBS. The retroviral vector LT5 contains theneogene between two MLV LTRs. Retroviral replication uses host tRNA as its primer. This 18-base PBS (GGGCTCGTCCGGGAT) is complementary to the tRNAPro3⬘end on the
acceptor arm. The T at the 11th nucleotide (underlined) was changed to C so that the 11th to 16th nucleotides within the PBS region could be digested with
SmaI (cCCGGG) (the lowercase letter represents the nucleotide that is different from the nucleotide in the wt PBS sequence.) The PBS downstream from the 3⬘
LTR was deleted. The 18-base sequence is reverse transcribed into a minus-strand DNA using the viral RNA genome as the template; the plus-minus-strand DNA is synthesized using the host tRNA 3⬘end as the template. Because of this preexisting mutation within this PBS region on the viral RNA molecule, the minus-strand DNA contains the mutation (i.e., C) while the plus-strand sequence remains wt (i.e., T). Therefore, the double-stranded DNA should contain a mismatch (C or T) within the PBS region. (B) Electrophoresis of the PCR-amplified fragment of the PBS region of DNA isolated from D17 cells infected with LT5. The cellular DNA of each Neorcolony infected with LT5 was
ampli-fied by PCR, followed by digestion withSmaI. After digestion, the amplified parental fragment was cut into two fragments, 686 and 207 bp in length. If the amplified mutant PBS fragments contained an additionalSmaI site within the PBS region and this amplified fragment was digested withSmaI, there would be three fragments, which would be 558, 207, and 128 bp in length. Lane 1,Sma I-digested amplified DNA fragments from plasmid pLT5; lane 2,SmaI-digested amplified DNA fragments from plasmid pLN; lanes 3 to 26, SmaI-digested amplified DNA fragments from DNA isolated from individual Neorcolonies
infected with pLT5.
on November 9, 2019 by guest
http://jvi.asm.org/
(positions 5324 to 2232), which contains the pUC18 (26) backbone; theEco
RI-AscI fragment of pLN (positions 2233 to 2816), which contains half of the 5⬘
LTR; theAscI-BstEII fragment (positions 2817 to 3519) of pLT2, which contains the other half of the 5⬘LTR and a mutation within the PBS region; theBst
EII-XbaI fragment (positions 3520 to 5027) from pLN, which contains theneogene and half of the 3⬘LTR; and theXbaI-SalI fragment of pLN (positions 5028 to 5323), which contains the second part of the 3⬘LTR and the deleted downstream PBS region.
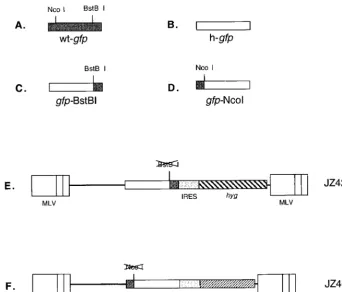
(iv) Introduction of restriction enzyme sites within thegfpgene.Thegfpgene was isolated from the jellyfishAequorea victoria(wt GFP; Clontech, Palo Alto, Calif.). The wtgfpopen reading frame contains anNcoI site at the 5⬘end and a
BstBI site at the 3⬘end (Fig. 3A). Three amino acids between the two sites have been changed so that thegfpgene emits brighter light. In addition, wtgfphas also been engineered to contain silent mutations in all of its amino acid codons to fit the human codon usage preference in order to increase its translational efficiency (Clontech). In this study, thegfpgene containing these silent mutations is des-ignated h-gfp(Fig. 3B).
h-gfpwas mutated by PCR to create aBstBI site within the wtgfp, and the sequence downstream of theBstBI site was replaced with the wtgfpgene. The 5⬘
end upstream of theBstBI site was maintained as the h-gfpsequence. This resulted in the construction of the first chimericgfpgene, which we have desig-natedgfp-BstBI (Fig. 3C). h-gfpwas also mutated so that it would contain anNcoI site at the same 5⬘end site as the wtgfpgene. The sequence upstream of this
NcoI site was also replaced by the 5⬘end of wtgfp. This resulted in the con-struction of the second chimericgfpgene, which we have designatedgfp-NcoI (Fig. 3D). Chimericgfp-BstBI andgfp-NcoI were inserted into retroviral vectors, and the vector DNAs were each introduced into helper cell lines. Cells that contained the two chimericgfpgenes emitted bright green light under a fluores-cence microscope (data not shown). These results indicated that the two chimeric
gfpgenes were functional.
(v) Introduction of a frameshift mutation into thegfpgene.To introduce a frameshift mutation into thegfpgene, thegfp-BstBI gene sequence was digested withBstBI, followed by repair with Klenow fragment.BstBI digestion creates two DNA ends that contain a 2-base overhang. When the overhang was repaired by the Klenow fragment, it created two blunt ends. Ligation of these two blunt ends with T4 ligase created a 2-bp insertion, which shifted thegfpopen reading frame by two (⫹2). Thegfp-NcoI was digested withNcoI, followed by repair with the Klenow fragment and ligation.NcoI digestion created two DNA ends that con-tained a 4-base overhang. As a result, this shifted thegfpopen reading frame by one (⫹1). The firstgfpgene contained a frameshift mutation atBstBI within the 3⬘end of thegfpgene, and the secondgfpgene contained another frameshift mutation at theNcoI site within the 5⬘end of thegfpgene. The two mutatedgfp
genes contained identical sequences between their individual frameshift muta-tion sites, which were separated by about 0.55 kb. Thegfpgene is approximately 0.7 kb in length.
(vi) Construction of pJZ425 and pJZ468. JZ425 (Fig. 3E) contained the frameshift mutationgfp-BstBI gene, the hygromycin resistance gene (hyg), and an internal ribosome entry segment (IRES) sequence located between the two genes. Thegfpgene was expressed by the conventional ribosome scanning model from the viral 5⬘R region, while thehyggene in JZ425 was expressed by the IRES (1, 4). In pJZ425, the 4.6-kbClaI-BamHI fragment (from positions 4055 to 1630) was isolated from pLNCX (19) and contained the two MLV LTRs. The 0.7-kbBamHI-SalI fragment (from positions 1631 to 2389) was derived from
gfp-BstBI (Fig. 3C) and contained a frameshift mutation at theBstBI site. The 0.6-kbSalI-NdeI fragment (from positions 2390 to 2991), which contained the IRES sequence, was isolated from pCITE-1 (Novagen, Madison, Wis.). The 1.1-kbNdeI-ClaI fragment (from positions 2992 to 4054) was isolated from pJD214 Hyg (9) and contained thehygsequence.
[image:4.612.134.476.77.369.2]The retroviral vector JZ468 (Fig. 3F) was similar to JZ425, except that JZ468 contained thegfpgene with the frameshift mutation at theNcoI site (Fig. 3D)
FIG. 3. Structures of retroviral vectors used for determining in which step(s) the recombinations in retroviral replication occur. (A) wtgfpgene. Thegfpgene was isolated from the jellyfishA. victoria, which contains anNcoI site at the 5⬘end and aBstBI site at the 3⬘end. (B) h-gfpgene. wtgfphas also been engineered to contain silent mutations in all of its amino acid codons to fit the human codon usage preference in order to increase the translational efficiency and is designated h-gfp. (C) Chimericgfp-BstBI gene. Thegfp-BstBI gene contains aBstBI site, which was constructed by PCR with the h-gfpsequence to allow the construction of an h-gfp–wt-gfp
chimeric gene. The 5⬘end upstream of theBstBI site remains the h-gfpsequence. (D) Chimericgfp-NcoI gene. Thegfp-NcoI gene contains anNcoI site in wtgfp, and the construction in the h-gfpsequence upstream of theNcoI site was replaced with the wtgfpgene to produce this chimeric gene. (E) Structure of the retroviral vector pJZ425. pJZ425 contains a selectable marker (hyg) and an unselectable color reporter gene,gfp-BstBI. Thegfpgene is transcriptionally expressed from the MLV 5⬘
LTR (containing the promoter and enhancer), and the selectable gene (hyg) is expressed from an IRES. A frameshift mutation is at the 3⬘end of thegfpgene. (F) Structure of the retroviral vector pJZ468. pJZ468 is similar to pJZ425; however, theneogene is replaced with thehyggene and a frameshift mutation is at the 5⬘end of thegfp-NcoI gene.
on November 9, 2019 by guest
http://jvi.asm.org/
and theneogene. In pJZ468, the 5.4-kbNdeI-BamHI fragment (from positions 2993 to 1630) was isolated from pLN and contained theneogene and the two MLV LTRs. The 0.7-kbBamHI-SalI fragment (from positions 1631 to 2391) was derived fromgfp-NcoI (Fig. 3D) and contained a frameshift mutation at theNcoI site. The 0.6-kbSalI-NdeI fragment (from positions 2392 to 2992) was isolated from pCITE-1 (Novagen) and contained the IRES sequence.
Preparation of virus LT5.DNA of pLT5 was used to transfect the xenotropic helper cell line PG13 (18) using Lipofectamine Plus (Life Technologies, Grand Island, N.Y.) according to the manufacturer’s protocol. Three hours after trans-fection, cells were washed four times with Dulbecco modified Eagle medium to remove the residual DNA from the plates. Viruses were detected as early as 6 h by infection of D17 cells and selection for neomycin resistance (Neor). The
viruses were collected 10 to 12 h after transfection.
Isolation of Neorcellular DNA and amplification of the PBS.D17 cells in-fected with LT5 were selected for Neorwith visible colonies formed about 12
days after infection. Well-separated colonies were cloned into 24-well plates. The cells were harvested once they covered the bottom of the well. Cellular chromo-somal DNAs were isolated with a Wizard genomic DNA purification kit (Pro-mega) according to the manufacturer’s protocol.
The PBS region was amplified using primer U3 601 (GGCAAGCTAGCTTA AGTAACGC), located at the U3 region, and primer Neo-rev (CCACCCAAG CGGCCGGAGAA), located at theneogene, for 25 cycles. Primers were re-moved with a Wizard PCR Preps DNA purification kit (Promega) according to the manufacturer’s protocol. The PCR product was further amplified using primer U3-2770 (CCAGATGCGGTCCAGCCC), located between the U3 re-gion (downstream of U3 601) and primer MLVBstEII (AGCAGAAGGTAAC CCAACGTCT), located at the package signal.
Cells, transfection, and infection.The processing of D17 cells (ATCC CRL-8468), PA317 helper cells (ATCC CRL-9078), and PG13 helper cells (ATCC CRL-10686); DNA transfections; virus harvesting; and virus infections were performed as previously described (28).
Fluorescence microscopy.An inverted fluorescence microscope (Zeiss Axio-vert 25) with a mercury arc lamp (100 W) and a fluorescence filter set (CZ909) consisting of a 470- by 40-nm exciter, a 515-nm emitter, and a 500-nm beam splitter was used to detect green fluorescent protein in living cells.
Relations betweenhygandneoRNA frequencies in the step 3 virion.The relations betweenhyg andneo RNA frequencies in the step 3 virion were described previously (28). The relation betweenhygandneoRNA frequencies in step 3 virions can be described in algebraic terms by means of the Hardy-Weinberg equation as follows.
Lethbe the frequency ofhygRNA in STEP 3 virions andnbe the frequency ofneoRNA in step 3 virions, so thath⫹n⫽1.
Assuming random packaging, the frequencies of differenthygandneodimers are determined with the formulah2⫹2hn⫹n2⫽1, whereh2is the frequency
of virions containing twohygRNAs, 2hnis the frequency of virions containing onehygRNA and oneneoRNA, andn2is the frequency of virions containing
twoneoRNAs. Neorcolonies result from infection by all virions containing two
neoRNAs and half virions containing oneneoand onehygRNA. Therefore, the frequency of virions capable of forming Neorcolonies is determined as follows:
Tn⫽n2⫹hn, whereTnis the titer of NeorCFU. The frequency of virions
capable of forming hygromycin-resistant (Hygr) colonies is determined with the
formulaTh ⫽h2⫹hn. These last two equations can be resolved with the
equationh⫽Th/(Tn⫹Th) and alson⫽Tn/(Tn⫹Th).
Therefore, the number of virions containing onehygRNA and oneneoRNA (2hn) is (2Th⫻Tn)/(Tn⫹Th)2.
RESULTS
Construction of a retroviral vector that contained a muta-tion within the PBS.Retroviral replication uses host tRNA as its primer. A host tRNAProbinds to an 18-base sequence of
the MLV genome. This 18-base sequence is called the PBS (8). The 18-base sequence, TGGGGGCTCGTCCGGGAT, is complementary to the tRNAPro3⬘end on the acceptor arm.
The T at the 11th nucleotide (underlined) was changed to C using a PCR technique so that the 11th to 16th nucleotides within the PBS could be digested withSmaI (CCCGGG). This MLV vector was derived from pLN (19). The pLN plasmid contains a neomycin resistance gene between two MLV LTRs. The 3⬘LTR of pLN was cloned from a 5⬘LTR that contained a PBS positioned downstream of the 3⬘LTR. Fifteen percent of the progeny virus utilized the downstream 3⬘LTR PBS to initiate strand DNA synthesis, and a complete minus-strand DNA can be transcribed without a strong stop DNA jump (J. Zhang, unpublished data). To force all progeny vi-ruses to use their natural PBS, the PBS downstream from the 3⬘LTR was deleted using standard PCR techniques. The
vec-tor designated LT5 (Fig. 2A), which contained a deletion of this 3⬘PBS, was constructed so that it contained aSmaI site within its normally positioned PBS.
Majority of mismatches within MLV double-stranded DNA was not corrected by D17 cells. DNA containing the vector LT5 was transfected into a murine retroviral helper cell line PG13 (18). The virus-containing cell supernatants were col-lected 10 h after transfection. The viruses were used to infect D17 cells. Since the target cells did not contain the retrovirus structural genes (gag-polandenv), virus was not released from infected target cells after infection with the retrovirus vector (28). Therefore, this infection assay represented only a single round of replication. Since pLT5 (DNA) contained a mutation in its PBS region (Fig. 2A), the virus RNA transcribed from the viral DNA also contained this mutation. After this viral vector entered D17 cells, the single-stranded RNA was reverse tran-scribed into double-stranded DNA. The PBS within the minus-strand DNA was copied with the viral RNA molecule as its template, while the plus-strand PBS DNA was synthesized with the host tRNAProas its template (8). As a consequence, the
viral double-stranded DNA contained a mismatch within this PBS. The viral double-stranded DNA was then integrated into the host chromosome as a provirus. Infected D17 cells were selected for Neor, and resistant colonies appeared in about 12
days. Well-separated individual colonies were cloned, and the DNAs from cells of these single colonies were isolated. The cellular DNA of each colony was amplified using two primers. The upstream primer (U32770) was located within the U3 region of the LTR, while the downstream primer was located within the packaging signal (MLVBstEII). The amplified frag-ment was 893 bp in length.
If the target cells (D17 cells) were capable of mismatch repair before the infected cells divided, all cells in the Neor
colony would contain either all mutant PBSs or the wt PBS. If the target cells did not repair the mismatch, half of the cells in the colony would contain the mutant PBS and the other half of the cell population would contain the wt PBS. The amplified fragments were digested withSmaI. Amplified fragments with the wild-type PBS contained aSmaI site in the R region. After digestion, the amplified wild-type product was cut into two fragments of 686 and 207 bp in length (Fig. 2B, lane 2). The amplified mutant PBS fragments contained an additionalSmaI site within the PBS. After being digested with SmaI, three fragments of 558, 207, and 128 bp in length (Fig. 2B, lane 1) were observed for the mutant product. Twenty-two of 24 Neor
colonies examined (Fig. 2B, lanes 3 to 26) contained both wt and mutant PBS sequences in the DNA isolated from a single colony. Only one of the 24 clones contained only the wt PBS (Fig. 2B, lane 14) and one of the 24 clones contained only the mutant PBS (Fig. 2B, lane 5). Therefore, the target cells (D17) were unable to repair the majority of mismatches within MLV double-stranded DNA. The frequency of the mismatch repair in the D17 cells was less than 10% before division.
Construction of vectors for the determination of the mech-anisms of retroviral recombination. Two additional MLV vectors were constructed. The first vector contained a drug re-sistance gene (hyg) and an unselected color reporter gene ( gfp-BstBI⫺) between the two MLV LTRs and is designated JZ425 (Fig. 3E). Thegfp-BstBI⫺gene was expressed downstream of the viral 5⬘R region, while thehyggene in JZ425 was expressed downstream of an IRES (1, 4). This IRES allows internal ribosome binding to the transcript expressed from the 5⬘LTR retroviral promoter.gfp-BstBI⫺contained a frameshift muta-tion near its 3⬘end, inside the gene’s open reading frame. As a result of this frameshift, the gfpgene was no longer func-tional. The second vector, JZ468 (Fig. 3F), contained another
on November 9, 2019 by guest
http://jvi.asm.org/
selectable marker,neo, and an unselected color reporter gene, gfp-NcoI⫺, between the two MLV LTRs.gfp-NcoI⫺contained a frameshift mutation at theNcoI site. Cells containing either JZ425 or JZ468 did not emit green light (data not shown). These two frameshift mutations are separated by 0.55 kb, while thegfpgene is about 0.7 kb in length.
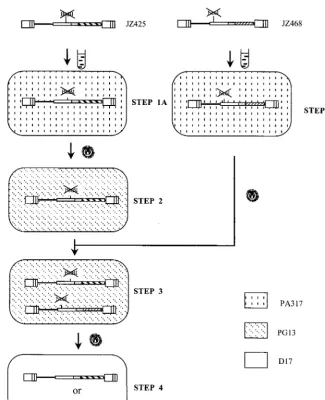
The rates of backward frameshift mutation of JZ425 and JZ468 were very low.pJZ425 was used for transfection into the MLV amphotropic helper cell line PA317 (17). PA317 cells containing pJZ425 were designated step 1A cells (Fig. 4). Vi-ruses from step 1A cells were used to infect the xenotropic helper cell line PG13 (18). Infected cells were selected for Hygr. Four individual Hygrcell clones were isolated and
des-ignated step 2 cells. The pJZ468 vector was used for transfec-tion into fresh PA317 cells. PA317 cells containing JZ468 were designated step 1B cells (Fig. 4). Viruses from step 1B cells were used to infect fresh PG13 cells. Infected cells were se-lected for Neor. Four individual Neorcell clones were isolated.
To confirm that the backward frameshift mutation (or the
secondary frameshift mutation) rate is low, the viruses har-vested from the step 2 Hygrcells were used to infect D17 target
cells. The infected cells were selected for Hygr. The Hygr
colonies were examined under a fluorescence microscope. Likewise, the supernatant containing viruses harvested from PG13 cells which contained JZ468 were used to infect D17 cells. The infected cells were selected for Neor. The Neor
colonies were examined under a fluorescence microscope. A total of 105Hygrcolonies of D17 cells infected with JZ425 and
105 Neorcolonies of D17 cells infected with JZ468 were
ex-amined, and no green colonies were detected. This result in-dicates that the backward frameshift mutation of JZ425 and JZ468 was undetectable.
Determination of the rate of retroviral recombination. Fig-ure 4 shows the outline of an experimental approach for the determination of the rate of retroviral recombination. The virus from step 1B cells was used to infect step 2 Hygrcells. The
[image:6.612.145.470.69.469.2]two vector proviruses were introduced into a single helper cell line by selection for Neor. Individual well-separated Neor
FIG. 4. Outline of an experimental approach for the determination of the rate of recombination during a single cycle of retroviral replication. No plasmid backbone sequences are shown. Transfections are indicated by test tube shapes. Infections are indicated by virion shapes. The different backgrounds represent the indicated cell lines. The lines in the LTR separate the U3, R, and U5 regions. The provirus in step 4 cells is the construct of Neoror Hygrrecombinants, which result from
recombination between the two frameshift mutations located on pJZ425 and pJZ468 (step 3).
on November 9, 2019 by guest
http://jvi.asm.org/
PG13 cells were cloned. PG13 cells containing JZ425 and JZ428 were designated step 3 cells (Fig. 4). Viruses from this step 3 helper cell line can contain two nonidentical RNA mol-ecules, one transcribed from provirus JZ425 and the other transcribed from provirus JZ468. Viruses from this helper cell line were used to infect target D17 cells. Infected cells were selected for Hygrand Neor, respectively. Unlike the helper cell
lines, these target cells do not contain the retroviral structural genes (gag-polandenv), so that after infection with the retro-virus vector, no retro-virus was released from the infected target cells (28). Thus, this infection represented only a single round of replication. Since both proviruses in PG13 cells contain a frameshift mutation within theirgfpgenes, if not mutation or recombination events occur, no green Neor(or Hygr) colonies
should be observed. Green Neor(and Hygr) colonies can occur
by two plausible mechanisms. The first possibility is that a reverse mutation occurs to overcome the frameshift mutation, but this is less likely since the reverse frameshift mutation rate is very low (⬍10⫺5) (see above). The second possibility that
may cause a green colony to form is that a recombination event occurs between the two frameshift mutation sites of these two vectors. The two frameshift mutations are located at a distance of 0.55 kb from one another. The ratio of the number of green colonies to the number of virions which contain both hyg (JZ425) RNA andneo(JZ468) RNAs is the rate of recombi-nation during a single round of retroviral replication. To limit double infections, the multiplicity of infection was lower than 1:1,000. To determine if green colonies resulted from a single infection, 11 green colonies were randomly isolated. The DNA of each colony was digested withBamHI and hybridized with a gfpprobe.BamHI digested JZ425 and JZ468 proviral DNAs 5⬘
of thegfpgene and in cellular flanking sequences 3⬘of thegfp gene. All of 11 colonies analyzed contained only one copy of the gfpgene, indicating that most green cells did not result from double infections (data not shown).
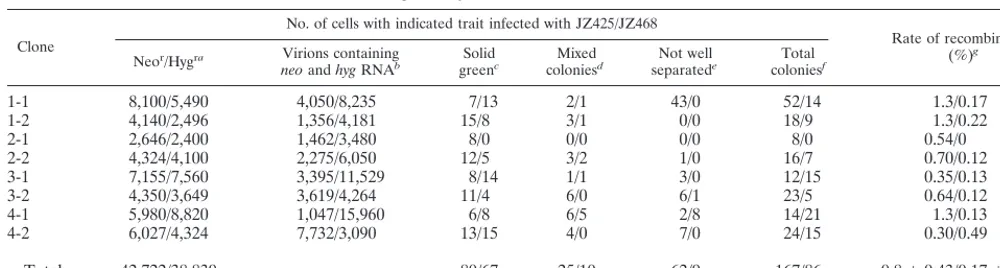
The ratio of the number of green colonies to the number of virions that contained bothhyg(JZ425) RNA andneo(JZ468) RNA was about 0.17%⫾0.14% or 0.80%⫾0.43% per rep-lication cycle, which represent the rate of recombination (Ta-ble 1).
Most retroviral recombinations occur during minus-strand DNA synthesis. If recombination occurred during minus-strand DNA synthesis, the minus-minus-strand DNA would contain a functionalgfpsequence, and since the plus-strand DNA uses the minus-strand DNA as a template, the plus-strand DNA would contain the functionalgfpsequence. After the cell di-vides, all the cells in a Neor(or Hygr) colony would be green.
However, if the recombination occurs during plus-strand DNA synthesis, then only the plus-strand DNA would contain a functionalgfpsequence and the minus-strand DNA would still carry a frameshift mutation. The double-stranded DNA con-taining this mismatch subsequently integrates into the host chromosomal DNA of the D17 cells, which are unable to repair the majority of mismatches within retroviral DNA. After the cell divides, one daughter cell would carry a functionalgfp sequence while the other daughter cell would contain a frame-shift mutation in thegfpsequence. Under fluorescence micros-copy, one-half of the cells in this colony should be green and one-half of the cells in this colony should be clear.
To determine the mechanism of recombination, each Hygr
and Neorcolony was carefully examined under a fluorescence
microscope for green cells. More than 80,000 colonies (42,000 Neor colonies and 38,000 colonies Hygr) were examined.
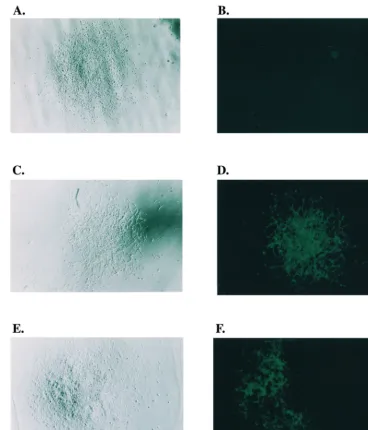
[image:7.612.52.552.83.217.2]Among them there were 105 (77%) well-separated solid green colonies (Fig. 5C and D) and 32 (23%) well-separated mixed colonies (Fig. 5E and F). (In addition, 71 colonies contained green cells that were in such close proximity with other colo-nies that it was difficult to determine the exact nature of the colonies.) A colony of solid green indicates that the recombi-nation had occurred during minus-strand DNA synthesis, while a colony of cells containing both clear and green cells indicates that either the mixture was a result of a recombination that was formed during plus-strand DNA synthesis or that the mixture was the result of the close contact between two cells infected with different viruses. One (green cell) of the two infected cells resulted from a recombination between JZ425 and JZ468 dur-ing minus-strand DNA synthesis, and the other resulted from a parental viral infection (clear cells). The former resulted from one integration event, while the latter resulted from two integration events.
TABLE 1. Microscopic analysis of D17 cells infected with JZ468 and JZ425
Clone
No. of cells with indicated trait infected with JZ425/JZ468
Rate of recombination (%)g Neor/Hygra Virions containing
neoandhygRNAb greenSolidc coloniesMixedd separatedNot welle coloniesTotal f
1-1 8,100/5,490 4,050/8,235 7/13 2/1 43/0 52/14 1.3/0.17
1-2 4,140/2,496 1,356/4,181 15/8 3/1 0/0 18/9 1.3/0.22
2-1 2,646/2,400 1,462/3,480 8/0 0/0 0/0 8/0 0.54/0
2-2 4,324/4,100 2,275/6,050 12/5 3/2 1/0 16/7 0.70/0.12
3-1 7,155/7,560 3,395/11,529 8/14 1/1 3/0 12/15 0.35/0.13
3-2 4,350/3,649 3,619/4,264 11/4 6/0 6/1 23/5 0.64/0.12
4-1 5,980/8,820 1,047/15,960 6/8 6/5 2/8 14/21 1.3/0.13
4-2 6,027/4,324 7,732/3,090 13/15 4/0 7/0 24/15 0.30/0.49
Total 42,722/38,839 80/67 25/10 62/9 167/86 0.8⫾0.43/0.17⫾0.14h
aTotal numbers of colonies observed.
bNumbers of virions which contained bothhygandneoRNAs (or JZ425 and JZ468) as described in Materials and Methods. cNumbers of colonies observed that contained only green cells.
dNumbers of colonies observed that contained both green cells and clear cells.
eNumbers of colonies observed that contained green cells and were in such close proximity with the other colonies that it was difficult to determine the nature of the colonies.
fNumbers of total colonies that contained green cells, i.e., the sum of the numbers of colonies defined in footnotesc,d, ande.
gThe ratio of the total number of colonies as defined in footnotefto the number of virions which contained bothhygandneoRNAs, as defined in footnoteb. hValues are means⫾standard deviations.
on November 9, 2019 by guest
http://jvi.asm.org/
The colonies containing both clear and green cells were subcloned. The cellular DNAs from clear cells and green cells were isolated from these subclones. Cellular DNA was di-gested withBamHI and hybridized with agfpprobe. BamHI digested JZ425 and JZ468 proviral DNA 5⬘of thegfpgene and in cellular flanking sequences 3⬘of thegfpgene. If a recombi-nant colony containing both clear and green cells resulted from a recombination, the provirus in the clear and green cells should integrate at the same site; therefore, the Bam HI-di-gestedgfpfragments from the clear and green cells should be the same length. Southern blot analysis indicated that the pro-viruses in the clear and green cells were integrated at different sites in 11 of 12 mixed colonies analyzed (Fig. 6). The green cells of one mixed colony (Fig. 6, lane 4-1Ng) contained an
[image:8.612.117.486.74.504.2]additional copy of the gfpfragment, suggesting that either a parental infection of one daughter cell followed a plus-strand recombination or a recombinant infection of one daughter cell followed a parental infection. Therefore, about 92% [1⫺(1/ 12)], if not all, of the mixed colonies resulted from two inde-pendent integration events. If the D17 cells could not repair 100% of the mismatches, we estimate that approximately 98%, if not all, of the retroviral recombinations resulted from a mechanism that is engaged during minus-strand DNA synthe-sis {[1⫺23%⫻(1⫺92%)]⫽98%}. Since the D17 cells were still able to repair only about 10% of the mismatches, it ap-pears that the frequency of recombination during plus-strand synthesis is extremely low.
FIG. 5. Microscopic analyses of D17 cells infected with viruses from step 3 cells. (A) Visible light microscopy of a Hygrcolony, which contains the parental type
of Hygrprovirus. (B) Fluorescence microscopy of a Hygrcolony, which contains the parental type of Hygrprovirus. The same colony as that shown in panel A was
examined under a fluorescence microscope. (C) Visible light microscopy of a Hygrcolony which resulted from a recombination between JZ425 and JZ468. (D)
Fluorescence microscopy of a solid green Hygrcolony, which resulted from a recombination between JZ425 and JZ468. The same colony as that shown in panel C was
examined under fluorescence microscopy. (E) Visible light microscopy of a Hygrmixed cell colony, which contains both green and clear cells. (F) Fluorescence
microscopy of a Hygrmixed cell colony, which contains both green and clear cells. The same colony as that shown in panel C was examined under fluorescence
microscopy.
on November 9, 2019 by guest
http://jvi.asm.org/
DISCUSSION
Eukaryotic cells possess several distinct mismatch repair pathways. A mismatch can be introduced in retroviral double-stranded DNA by a preexisting mutation within the PBS of the viral RNA genome. MLV-based vectors with a mutation in their PBSs were used to infect mismatch-repair-competent cell lines as well as mismatch-repair-deficient cell lines. Almost all mismatch-repair-deficient cells colonies analyzed (cell lines HCT 116 and PMS2⫺/⫺) (2, 10, 20, 21) contained both the wild-type and mutated PBS. Therefore, mismatches within ret-roviral double-strand DNA could not be repaired by the mis-match-repair-deficient cells. Only 2 of 45 colonies analyzed showed a pattern corresponding to the presence of a repaired PBS. The two exceptions probably represent an infected cell that divided into two daughter cells and one daughter cell died so that the resulting colony contained only either the wild-type or the mutant PBS (11). Two of 24 (10%) D17 colonies ana-lyzed showed a pattern corresponding to the presence of a repaired PBS, suggesting that D17 is a mismatch-repair-defi-cient cell line.
In contrast, approximately 25% of the mismatch-repair-com-petent cell clones analyzed (cell lines HeLa and PMS2⫹/⫹) were repaired while 75% were not. Therefore, the normal cellular mismatch repair system is not able to repair the ma-jority of mismatches within the viral double-stranded DNA. One possible explanation is that the cells do not have enough time to repair the mismatch after integration of the double-strand DNA before the cell divides. After a retrovirus enters host cells, the single-strand RNA genome is reverse tran-scribed into double-strand DNA within the retroviral capsid in the host cell cytoplasm. Before retroviral integration, the ret-roviral double-stranded DNA exists as a structure called the preintegration complex (PIC) (8). This PIC contains retroviral capsid protein and other proteins. The cellular mismatch re-pair system is probably unable to recognize a DNA mismatch within this PIC. MLV, like other simple retroviruses, can infect only dividing cells, and it has been hypothesized that the PIC enters the nucleus only during mitosis (8, 11). Therefore, the window for the cellular mismatch repair system to repair a mismatch within the retroviral double-stranded DNA would be constrained to mitosis. Only a very low percentage of mis-matches within retroviral double-stranded DNA were repaired in these experiments.
Previous reports suggested that a recombinant may be a result of one or more crossover events (13). In our system, one crossover is sufficient to form a Neorrecombinant. The reverse
transcription growing points synthesize theneogene and the 3⬘
end of thegfpgene using JZ468 as a template and then shift to the JZ425 RNA to complete the functionalgfpgene (Fig. 3E and F). However, the formation of a Hygr recombinant
re-quires at least two crossover events. The reverse transcription growing points at first synthesize thehyggene using JZ425 as the template and then shift the template to copy the 3⬘end of thegfpgene from JZ468. The template is shifted once again to copy the 5⬘end of thegfpgene of JZ425. Results in this report indicate that the Hygr recombinants formed with
approxi-mately equal frequencies as Neor recombinants, which
indi-cates that the two crossover events occurred at the same rate as one crossover event during the minus-strand DNA synthesis. Furthermore, these results suggest that after one recombina-tion occurs, the frequency of a second recombinarecombina-tion rate must be much higher than the average recombination rate.
In this report, we have estimated that most, if not all, re-combinations occur during minus-strand DNA synthesis. Therefore, the mechanism of retroviral recombination is either the copy choice model or the minus-strand replacement model. Further work needs to be done to distinguish between these two minus-strand DNA synthesis models.
ACKNOWLEDGMENTS
We thank W. Bargmann, K. Boris-Lawrie, G. Li, and A. Kaplan for helpful comments on the manuscript.
This research was supported by Public Health Service research grant CA70407 from the National Institutes of Health.
REFERENCES
1.Adam, M. A., N. Ramesh, A. D. Miller, and W. R. Osborne.1991. Internal initiation of translation in retroviral vectors carrying picornavirus 5⬘ non-translated regions. J. Virol.65:4985–4990.
2.Baker, S. M., C. E. Bronner, L. Zhang, A. W. Plug, M. Robatzek, G. Warren, E. A. Elliott, J. Yu, T. Ashley, N. Arnheim, et al.1995. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell82:309–319.
3.Berwin, B., and E. Barklis.1993. Retrovirus-mediated insertion of expressed and non-expressed genes at identical chromosomal locations. Nucleic Acids Res.21:2399–2407.
4.Boris-Lawrie, K. A., and H. M. Temin.1993. Recent advances in retrovirus vector technology. Curr. Opin. Genet. Dev.3:102–109.
5.Chalfie, M., Y. Tu, G. Euskirchen, W. W. Ward, and D. C. Prasher.1994. Green fluorescent protein as a marker for gene expression. Science263:802– 805.
6.Coffin, J. M.1996. Retroviridae: the viruses and their replication, p. 1767– 1847.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Lippincott-Raven Publishers, Philadelphia, Pa.
7.Coffin, J. M.1979. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. J. Gen. Virol.42:1–26.
8.Coffin, J. M., S. H. Hughes, and H. Varmus.1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
9.Dougherty, J. P., and H. M. Temin.1986. High mutation rate of a spleen necrosis virus-based retrovirus vector. Mol. Cell. Biol.6:4387–4395. 10. Drummond, J. T., G. M. Li, M. J. Longley, and P. Modrich.1995. Isolation
of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science268:1909–1912.
11. Hajihosseini, M., L. Iavachev, and J. Price.1993. Evidence that retroviruses integrate into post-replication host DNA. EMBO J.12:4969–4974. 12. Hu, W. S., and H. M. Temin.1990. Genetic consequences of packaging two
RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA87:1556–1560. 13. Hu, W. S., and H. M. Temin.1990. Retroviral recombination and reverse
transcription. Science250:1227–1233.
14. Huang, C. C., C. Hammond, and J. M. Bishop.1985. Nucleotide sequence and topography of chicken c-fps. Genesis of a retroviral oncogene encoding a tyrosine-specific protein kinase. J. Mol. Biol.181:175–186.
15. Junghans, R. P., L. R. Boone, and A. M. Skalka.1982. Products of reverse transcription in avian retrovirus analyzed by electron microscopy. J. Virol. 43:544–554.
16. Junghans, R. P., L. R. Boone, and A. M. Skalka.1982. Retroviral DNA H structures: displacement-assimilation model of recombination. Cell30:53– 62.
17. Miller, A. D., and C. Buttimore.1986. Redesign of retrovirus packaging cell lines to avoid recombination leading to helper virus production. Mol. Cell. Biol.6:2895–2902.
FIG. 6. Southern blot analysis of recombinants between JZ425 and JZ468. Chromosomal DNAs of Hygr(H) or Neor(N) step 4 clones were used. Cellular
DNAs isolated from green (g) cells and clear (c) cells were digested withBamHI and hybridized to agfpgene probe.
on November 9, 2019 by guest
http://jvi.asm.org/
18.Miller, A. D., J. V. Garcia, N. von Suhr, C. M. Lynch, C. Wilson, and M. V. Eiden.1991. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J. Virol.65:2220–2224.
19. Miller, A. D., and G. J. Rosman.1989. Improved retroviral vectors for gene transfer and expression. BioTechniques7:980–990.
20. Parsons, R., G. M. Li, M. J. Longley, W. H. Fang, N. Papadopoulos, J. Jen, A. de la Chapelle, K. W. Kinzler, B. Vogelstein, and P. Modrich.1993. Hypermutability and mismatch repair deficiency in RER⫹tumor cells. Cell 75:1227–1236.
21. Prolla, T. A., S. M. Baker, A. C. Harris, J. L. Tsao, X. Yao, C. E. Bronner, B. Zheng, M. Gordon, J. Reneker, N. Arnheim, D. Shibata, A. Bradley, and R. M. Liskay.1998. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat. Genet. 18:276–279.
22. Raines, M. A., N. J. Maihle, C. Moscovici, L. Crittenden, and H.-J. Kung. 1988. Mechanism of c-erbBtransduction: newly released transducing viruses retain poly(A) tracts oferbBtranscripts and encode C-terminally intacterbB
proteins. J. Virol.62:2437–2443.
23. Sambrook, J., E. F. Fritsch, and T. Maniatis.1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
24. Sapp, C. M., T. Li, and J. Zhang.1999. Systematic comparison of a color reporter gene and drug resistance genes for the determination of retroviral titers. J. Biomed. Sci.6:342–348.
25. Swain, A., and J. M. Coffin.1992. Mechanism of transduction by retroviruses. Science255:841–845.
26. Vieira, J., and J. Messing.1982. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene19:259–268.
27. Zhang, J., and C. M. Sapp.1999. Recombination between two identical sequences within the same retroviral RNA molecule. J. Virol.73:5912–5917. 28. Zhang, J., and H. M. Temin.1993. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science259: 234–238.