0022-538X/07/$08.00⫹0 doi:10.1128/JVI.01495-07
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Entry of Hepatitis Delta Virus Requires the Conserved Cysteine
Residues of the Hepatitis B Virus Envelope Protein Antigenic
Loop and Is Blocked by Inhibitors of
Thiol-Disulfide Exchange
䌤
Georges Abou-Jaoude
´
1and Camille Sureau
1,2*
Laboratoire de Virologie Mole´culaire, INTS, Paris, France,1and Department of Virology and Immunology,
Southwest Foundation for Biomedical Research, San Antonio, Texas2 Received 9 July 2007/Accepted 12 September 2007
Hepatitis delta virus (HDV) particles are coated with the envelope proteins (large, middle, and small) of the hepatitis B virus (HBV). The large protein bears an infectivity determinant in its pre-S1 domain, whereas a second determinant has been proposed to map to the cysteine-rich antigenic loop (AGL) within the S domain of all three envelope proteins (G. Abou Jaoude´ and C. Sureau, J. Virol. 79:10460–10466, 2006). In this study, the AGL cysteines were substituted by serine or alanine, and the mutants were evaluated for their function at viral entry using HDV particles and susceptible HepaRG cells. Mutations of cysteines 121 to 149 were tolerant of the production of HDV virions. The mutations altered the structure and antigenicity of the conserved “a” determinant of the AGL, as measured by conformation-sensitive antibodies, and they created a block to infectivity. Substitution of Cys-90 or Cys-221, located outside of the AGL, had no impact on the “a” determi-nant or viral entry. Furthermore, infectivity was maintained when the AGL CxxC motif at position 121 to 124 was modified by single-amino-acid deletion or insertion, suggesting that cysteines 121 and 124 are not catalyzers of thiol/disulfide exchange. However, membrane-impermeable inhibitors of thiol/disulfide isomera-zation demonstrated a dose-dependent inhibition of infection in an in vitro assay when applied to the virus prior to inoculation or during the virus-cell interaction period. Overall, the results demonstrate the essential role of the AGL cysteines at viral entry, and they establish a correlation between the cysteine disulfide network, the conformation of the “a” determinant, and infectivity.
Hepatitis B virus (HBV) is responsible for acute and chronic liver disease that affects more than 400 million individuals worldwide (12). HBV is characterized by a very narrow host range that is likely to reflect a highly specific interaction be-tween the viral envelope proteins and the receptor(s) at the surface of human hepatocytes. The mechanism of HBV entry is still poorly understood—the receptor remains unknown— and only recently have determinants of infectivity been mapped to discrete domains within the amino acid sequence of the envelope proteins (2, 4, 16, 24). HBV particles bear three sequence-related envelope proteins: the small protein (S-HBsAg) consists of a 226-amino-acid-long transmembrane protein, the middle protein (M-HBsAg) includes the S domain and an additional N-terminal pre-S2 ectodomain that is 55 amino acids in length, and the large protein (L-HBsAg) com-prises a N-terminal pre-S1 domain (109 amino acids) in addi-tion to pre-S2 and S domains (20). Synthesis occurs at the endoplasmic reticulum (ER) membrane, but it leads almost exclusively to the secretion of empty subviral particles (SVPs). The assembly of mature HBV virions is a rare event that results from an interaction between the matrix domain of L-HBsAg and the HBV nucleocapsid (6).
The HBV envelope proteins also have the capacity to
inter-act with the hepatitis delta virus (HDV) ribonucleoprotein (RNP) in cases of HBV/HDV coinfection (5, 41). This inter-action leads to the formation of HDV virions (35, 41). HDV is thus considered an occasional satellite of HBV, because its capacity to propagate depends on the envelope proteins of the latter (13). Because the coats of HBV and HDV particles are identical, a study of the HBV envelope proteins functions at viral entry can be conducted using the HDV model (2, 3, 36). It is well established that infectivity of HBV or HDV particles is directly dependent on L-HBsAg, which bears a receptor binding domain (RBD) within its N-terminal pre-S1 moiety (2, 4, 16, 24). The latter is myristoylated at glycine 2, and this modification is indispensable for infectivity (8, 18). The RBD is responsible for tissue and species specificity as demonstrated by the activity of anti-pre-S1 antibodies in neutralizing infec-tion and in preventing interacinfec-tion between hepatocyte mem-brane preparations and virions (14, 29, 37). Furthermore, myr-istoylated synthetic peptides specific for the N-terminal 47 amino acids of the pre-S1 domain are potent inhibitors of viral entry (2, 16, 17).
In a recent study, we have presented evidence for the pres-ence of a second infectivity determinant located in the anti-genic loop (AGL) of the envelope protein S domain, but the mechanism by which this motif participates in entry is as yet unclear (21). The AGL is known to bear the major HBV-neutralizing epitopes (30) and a conserved immunodominant determinant, referred to as “a.” It also contains eight cysteine residues described as engaged in disulfide bonds that are
in-* Corresponding author. Mailing address: Laboratoire de Virologie Mole´culaire, Institut National de la Transfusion Sanguine, 6 Rue Alexandre-Cabanel, 75739 Paris, France. Phone: (33) 1 44 49 30 56. Fax: (33) 1 44 49 30 59. E-mail: csureau@ints.fr.
䌤Published ahead of print on 26 September 2007.
13057
on November 8, 2019 by guest
http://jvi.asm.org/
strumental in defining the structure of the “a” determinant (25, 27). Furthermore, cysteines at positions 121 and 124 constitute a CxxC motif that is generally found on protein-disulfide isomerase (PDI)-related proteins (38). Their substitution by serine was shown to be detrimental to infectivity (21). In view of these results, it is tempting to speculate that after the initial binding of the virus to its receptor, the completion of the entry process requires a mechanism for disassembly of the virion envelope through isomerization of disulfide bonds (42). Whether a PDI activity is borne by the HBV envelope proteins is uncertain, and there exists the possibility that the negative impact caused by cysteine mutations in the CxxC motif is due only to a modification of the AGL structure. The AGL, includ-ing the “a” determinant, could, for instance, cooperate with pre-S1 RDB in binding to a primary receptor; it could also mediate an interaction with a secondary receptor or be instru-mental in a disassembly mechanism engaged after internaliza-tion.
Our findings demonstrate that cysteine residues of the HBV envelope protein AGL are essential to HDV infectivity and that viral entry is blocked by inhibitors of thiol/disulfide ex-change reactions.
MATERIALS AND METHODS
Reagents.[2-(Trimethylammonium) ethyl] methanethiosulfonate bromide (MTSET),
4-(N-maleimido) benzyl-alpha trimethylammonium iodide (M135), and
Tris-(2-carboxyethyl) phosphine (TCEP) were from Toronto Research Chemicals, Inc.
Dithiothreitol (DTT) and 5,5⬘dithiobis (2-nitrobenzoic acid) (DTNB) were from
Sigma. 4-Acetamido-4⬘-maleimidylstilbene 2,2⬘-disulfonic acid (AMS) was from
Invitrogen Molecular Probes. The Monolisa HBsAg Ultra enzyme-linked immu-nosorbent assay (ELISA) kit was from Bio-Rad, and ETI-MAK-4 HBsAg was from Dia-Sorin.
Site-directed mutagenesis. Amino acid substitutions in the HBV envelope proteins were carried out by mutagenesis of pT7HB2.7 plasmid DNA using the PCR overlap extension method (22). All PCR-generated fragments that were cloned in pT7HB2.7 were sequenced using a Big Dye Terminator sequencing protocol (Applied Biosystems). The mutations were designated by the one-letter code for cysteine followed by its position in the S domain of the envelope protein and the one-letter code for the substituted amino acid.
Production of HDV particles in HuH-7 cells.For production of HDV particles, HuH-7 cells were transfected with a mixture of the pSVLD3 plasmid for pro-duction of HDV RNPs and pT7HB2.7 or its derivatives for the supply of the wild-type (wt) or mutant HBV envelope proteins, respectively (3). Transfections were carried out by the use of FuGENE 6 reagent (Roche) as described previ-ously (21). Culture medium was harvested on days 5, 7, and 9 posttransfection and analyzed for the presence of viral particles, by immunoblotting for the detection of HBV envelope proteins and by Northern blotting for the detection of HDV RNA (4).
Characterization of HDV particles produced in HuH-7 cells.Culture fluids harvested on days 5, 7, and 9 after transfection were pooled and clarified by
centrifugation at 5,000⫻g at 4°C for 30 min. Viral particles from the clarified
medium were subjected to sedimentation by centrifugation for 2 h at 50,000 rpm in an SW55 rotor (Beckman) on 1 ml of a 30% sucrose cushion in 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 1 mM EDTA. After centrifugation, the particle-containing pellet was resuspended in sodium dodecyl sulfate (SDS) protein disruption buffer. Solubilized proteins were subjected to SDS-polyacryl-amide gel electrophoresis (SDS-PAGE), transfer to polyvinylidene difluoride (PVDF) membranes, and incubation with a mixture of anti-S and anti-pre-S2 antibodies (4). Membranes were then incubated either in the presence of anti-rabbit antibodies coupled to horseradish peroxidase (HRP) at a 1:5,000 dilution
or 125I-labeled (1 Ci/ml). Immunoblots were developed using enhanced
chemiluminescence (ECL) reagents (Amersham Pharmacia Biotech). For
im-munoblot assays performed with125I-labeled secondary antibodies (1Ci/ml;
PerkinElmer), signal quantification was achieved using a phosphorimager (FUJIFILM BAS-1800 imaging plate reader) and FUJIFILM image reader V1.8
software. HDV RNA was extracted from 140l of cell culture supernatant and
analyzed by electrophoresis through a 1.2% agarose–2.2 M formaldehyde gel,
transfer to a nylon membrane (Roche), and hybridization to a32P-labeled RNA
probe specific for genomic HDV RNA as described previously (4).
To test the sensitivity of HDV virions to AMS, TCEP, or NP-40, aliquots of HDV particles were mock treated or treated with 1:2 dilutions of 2 mM AMS, 2 mM TCEP, or 0.1% NP-40 for 1 h at 37°C. Viral RNA was then extracted and analyzed as described above. To analyze the antigenicity of viral particles treated with AMS, TCEP, or NP-40, aliquots were spotted on a PVDF membrane using a Bio-Dot microfiltration apparatus (Bio-Rad). Membrane was then blocked in 20 mM Tris-HCl (pH 7.4)–0.5 M NaCl (Tris-buffered saline [TBS]) and 1% casein (TBS-casein) for 1 h. Immunodetection was achieved by incubating the membrane for 2 h with rabbit polyclonal anti-pre-S2 (R257) or mouse monoclo-nal anti-S (A1.2) antibodies at 1:1,000 dilutions in TBS-casein. After washes in TBS-0.3% Tween 20, the membrane was incubated for 1 h in TBS-casein con-taining anti-rabbit or anti-mouse antibodies coupled to HRP at a 1:5,000 dilu-tion. The immunoblots were developed using ECL reagents (Amersham Phar-macia Biotech).
In vitro infection assays.For infection assays, HepaRG cell cultures were treated with 2% dimethyl sulfoxide for 2 weeks prior to inoculation with HDV particles (19). Inocula consisted of culture fluids collected from HuH-7 cells at days 5, 7, and 9 posttransfection, which were pooled and clarified by
centrifuga-tion at 5,000⫻gfor 30 min at 4°C. HepaRG cells (3.3⫻105
cells/20-mm-diameter well) were exposed to 108genome equivalents (GE) of HDV virions for
16 h, in the presence of 5% polyethylene glycol 8000. When cells were inoculated in the presence of TCEP, polyethylene glycol was omitted to prevent the forma-tion of precipitates. Cells were harvested at day 7 postexposure for measurement of intracellular HDV RNA that served as a marker of infection. HDV RNA
signals were detected by Northern blot analysis using a32P-labeled RNA probe
and quantified using a phosphorimager (3).
RESULTS
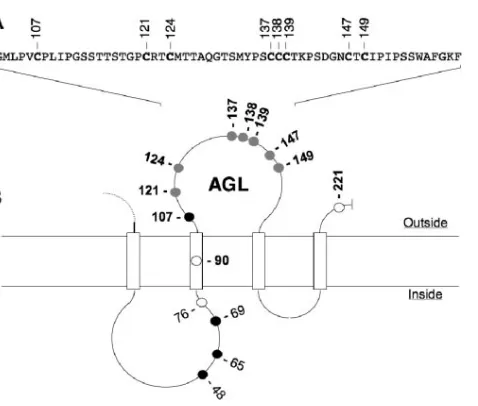
At the surface of viral particles, the HBV envelope proteins are assumed to establish an intermolecular disulfide network through cysteine residues of the AGL (Fig. 1) (25–27, 42). To study the importance of AGL cysteines at viral entry, we used the HDV/HepaRG model (19, 21), in which envelope proteins carrying cysteine substitutions were assayed for function at viral entry at the surface of HDV virions. Infectivity was
as-FIG. 1. Schematic representation of the HBV envelope protein AGL. (A) AGL amino acid sequence. The positions of cysteine resi-dues are indicated. (B) A secondary-structure model for the S domain of HBV envelope proteins is represented. Open boxes represent hy-drophobic transmembrane regions. Positions of the cysteine residues are indicated. Open circles, cysteines dispensable for particle secre-tion; closed circles, cysteines essential for particle secretions; shaded circles, cysteines essential for infectivity (this study).
13058 ABOU-JAOUDE´ AND SUREAU J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.585.302.541.62.265.2]sessed after inoculation of mutant HDV to susceptible HepaRG cells and measurement of intracellular accumulation of HDV RNA at day 7 postinoculation to serve as a marker of infection (3, 4).
Effect of HBV envelope protein cysteine substitutions on the production of SVPs and HDV virions.Cysteines at positions 90, 121, 124, 137, 138, 139, 147, 149, and 221 were changed to serine or alanine residues, and each mutation was evaluated for its impact on envelope protein secretion and HDV produc-tion (Fig. 1). Substituproduc-tion of cysteine 107 was not considered, since it was previously shown to block secretion (27). Cysteines were changed into serine or alanine as single (C90S, C121S, C124S, C137S, C139S, C147S, C149S, C221S, C137A, C138A, and C149A), double (C121S-C124S and C147S-C149S), or tri-ple (C137S-C138S-C139S) mutations. As shown in Fig. 2, all mutations were tolerant, though to various degrees, of the secretion and maturation of HDV. Note that the detection of HBV envelope proteins (Fig. 2A) was achieved using a rabbit anti-S antibody (R247) that recognizes a linear epitope in the cytosolic domain of the three envelope proteins and a rabbit anti-pre-S2 antibody for specific detection of L- and M-HBsAg proteins (4). As observed previously (27), C138S mutation was the most inhibitory to secretion. Mutations of Cys-121, -137, and -149 induced a slight reduction of particle secretion (Table 1),
whereas those affecting of Cys-90, -124, -139, -147, and -221 had no significant impact on subviral or HDV particle secretion.
The variations in envelope protein secretion between mu-tants paralleled those recorded for viral RNA (Fig. 2B), indi-cating that the ratio of SVPs to HDV virions was not affected by the mutations. For each mutant, we measured the ratio of L-to S-HBsAg by performing an immunoblot analysis of enve-lope proteins that had been treated with PNGase F to remove Asn-linked sugars prior to SDS-PAGE. This treatment allowed better detection of weakly expressed envelope proteins by Western blotting, because each L-, M-, and S-HBsAg protein was detected as a single nonglycosylated form (Fig. 2A). For a more precise quantification, a125I-labeled antibody was
sub-stituted for the HRP-labeled antibody, and radioactive signals were measured using a phosphorimager. The phosphorimager values shown in Table 1 indicate, for each mutant, the percent-age of mutant S-HBsAg relative to wt S-HBsAg and the per-centage of the mutant L-HBsAg/S-HBsAg ratio relative to that of the wt. The results indicate that the mutants most affected for secretion were C137S and C138S (30 and 34% of the wt level, respectively), and the mutant displaying the lowest ratio of L-/S-HBsAg was C138S (15% of the wt level).
We concluded that all cysteine mutations were permissive
FIG. 2. Production of HDV particles coated with HBV envelope proteins with cysteine substitutions in the AGL. (A) Culture fluids from HuH-7 cells were harvested on days 5, 7, and 9 after transfection of 106cells with a mixture of 1g of pSVLD3 coding for HDV RNPs and 1g of pT7HB2.7 or derivatives coding for wt or HBV envelope protein mutants, respectively. Particles from 1 ml of culture fluids were concentrated and assayed for the presence of HBV envelope proteins before and after incubation with PNGase F as indicated. After SDS-PAGE and transfer to a PVDF membrane, proteins were probed with a mixture of rabbit anti-S antibody (1:500 dilution) and rabbit anti-pre-S2 antibody (1:1,000). (B) Particles from 140l of culture medium were assayed for the presence of HDV RNA by the Northern blot hybridization procedure using a genomic strand-specific32P-labeled HDV RNA probe. The size (in kilobases) of HDV genomic RNA is indicated. wt SML, HDV particles coated with wt S-, M-, and L-HBsAg. S, M, and L indicate the positions of the S-, M-, and L-HBsAg, respectively. The glycosylated (gp) and nonglycosylated (p) forms of S-HBsAg, M-HBsAg, and L-HBsAg proteins are indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
for production of HDV particles that were thus amenable to in vitro infection assays.
Effect of HBV envelope protein cysteine substitutions on the antigenicity of viral particles.Prior to evaluation of mutant HDV particles for infectivity, the particles were examined for antigenicity as an attempt to identify changes induced by the cysteine mutations in the “a” determinant. It was previously shown that reactivity of HBV particles with HBsAg anti-bodies was dependent upon envelope protein disulfide bonds (39) and that antigenicity of SVPs carrying AGL cysteine mu-tations was drastically affected (27). Here, we measured the reactivity of each cysteine mutant in two commercial immuno-assays (Monolisa HBsAg Ultra from Bio-Rad and ETI-MAK-4 HBsAg from Dia-Sorin). These assays utilize monoclonal an-tibodies (MAbs) directed to the immunodominant “a” deter-minant. The results presented in Table 1 indicate the percent-age of mutant HBsAg reactivity relative to that of the wt for each assay. A specific antigenicity was then defined as the ratio of HBsAg ELISA values to the S-HBsAg signal values ob-tained by phosphorimager quantification of immunoblots probed with125I-labeled antibodies. The results show that
mu-tation of any of the AGL cysteines drastically reduced antige-nicity according to both immunoassays. In comparison, muta-tion of non-AGL Cys-90 or Cys-221 had a limited impact on antigenicity. Hence, as reported previously (27), AGL cys-teines are essential to the structure of epitopes that define the “a” determinant.
Effect of AGL cysteine substitutions on infectivity of HDV particles.For infectivity analysis, each preparation of mutant HDV was normalized to 108GE/ml prior to inoculation to 3.3
105HepaRG cells. Noninfectious HDV particles coated with
the S-HBsAg protein only (wt S) were used as a negative control. Normalization of the inocula titers was controlled by measuring the levels of HDV RNA in the inocula that were recovered after the 16-h virus-cell adsorption period (Fig. 3A). This was carried out to ascertain that the stability of mutant
virions was not affected by the mutations (HDV RNA would be rapidly degraded and hence undetectable if particles were to be disrupted as a result of an unstable envelope). As shown in Fig. 3A, viral RNA in the postinoculation samples was detected in amounts equivalent to that of the wt and identical to those measured prior to inoculation (data not shown). This was an indication that cysteine mutations did not destabilize the viral envelope and that the target cells adsorbed a very small percentage of HDV RNA-containing particles. Seven days after inoculation, cells were assayed for intracellular HDV RNA (Fig. 3B). Evidence of infection was observed for hepatocytes exposed to wt HDV and to C90S and C221S mu-tants. In contrast, substitutions of cysteine at positions 121, 124, 137, 138, 139, 147, and 149 had a pronounced inhibitory effect on infectivity. The values indicated in Fig. 3B are amounts of HDV RNA as percentages relative to that of the wt. Overall, our findings establish a correlation between AGL cysteine residues, AGL antigenicity (i.e., structure of the “a” determinant), and infectivity. Whether cysteines are involved in defining a binding structure in the AGL and/or in partici-pating in the envelope disassembly process remains to be in-vestigated.
[image:4.585.43.542.80.269.2]Effect of mutations in the AGL CxxC motif on infectivity of HDV particles.Among the AGL cysteines, Cys-121 and -124 together form a CxxC motif that generally constitutes the cat-alyst of PDI-related proteins (9, 38). Hence, mutation of Cys-121 or -124 could inhibit infectivity either by inducing a con-formational change in the AGL or by blocking the AGL-borne PDI activity. The former effect could prevent the binding of an AGL epitope to a receptor; the latter could create a block to envelope disassembly. As an attempt to investigate whether there was a strict requirement for the CxxC motif, we chose to modify its sequence by creating deletions or insertions between positions 121 and 124 so as to block a potential enzymatic activity without removing any of the cysteines. Mutations in-cluded (i) a single-amino-acid insertion that changed the
TABLE 1. Specific antigenicity of mutant HDV particlesa
Designation
Western blot ELISA Specific antigenicity
S-HBsAg L-/S-HBsAg Monolisa ETI-MAK-4 Monolisa ETI-MAK-4
wt SML 100 100 100 100 100 100
wt S 82 1 134.7 112.6 164 138.2
C90S 95 162 115.3 126.4 120 133.6
C121S 50 68 4.2 5 8.3 10
C124S 82 117 1.7 17.5 2.1 21.3
C137S 30 33 0.1 2.9 0.3 9.4
C138S 34 15 Neg 0.3 Neg 0.9
C139S 87 111 Neg 17.2 Neg 19.9
C147S 89 111 0.6 24.4 0.7 27.2
C149S 64 78 Neg 2.4 Neg 3.7
C221S 76 97 47.4 53.7 62.7 70.3
C121S-C124S 69 107 Neg 1.6 Neg 2.3
C137S-C138S-C139S 45 44 Neg Neg Neg Neg
C147S-C149S 64 41 Neg 3.1 Neg 4.8
C137A 68 90 Neg 7.1 Neg 10.4
C138A 49 31 0.03 0.6 0.06 1.3
C149A 54 41 0.03 1.2 0.06 2.3
aQuantification of L- and S-HBsAg was achieved by Western blot analysis using125I-labeled secondary antibodies and a phosphorimager. Values are given as
percentages relative to the wt. Specific antigenicity was defined as the ratio of HBsAg ELISA values to S-HBsAg values as measured by Western blot analysis. The Monolisa HBsAg Ultra ELISA kit was from Bio-Rad, and ETI-MAK-4 HBsAg was from Dia-Sorin. wt SML, HDV particles coated with wt S-, M- and L-HBsAg. Neg, negative.
13060 ABOU-JAOUDE´ AND SUREAU J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
CRTC motif to CRATC, CRTAC, CNRTC, CSRTC, or CQRTC; (ii) a deletion of Arg-122 (CTC); and (iii) a substi-tution of residue 122 or 123 to create CATC, CKTC, CRAC, and CRSC sequences. Production of mutant virions was achieved as described above, and supernatants of transfected cells were analyzed for the presence of envelope proteins and viral RNA. As shown in Fig. 4A, envelope proteins and viral RNA were detected at levels similar to those detected for the wt particles. Note, however, that CNRTC mutant proteins carry an Asn-linked glycosylation signal (NRT) at position 122, which explains the presence of additional slower migrating bands in the immunoblot analysis. Infection assays were con-ducted by inoculating approximately 108GE of HDV virions to
3.3⫻105HepaRG cells in a 20-mm-diameter well. Cells were
harvested at day 7 postinoculation before being assayed for HDV RNA (Fig. 4B). All HDV mutants demonstrated a near-wt level of infectivity, with the exception of CATC, CTC, and CNRTC, for which infectivity was estimated at 36%, 39%, and 55%, respectively, of that of the wt. The greatest effect recorded for CATC and CTC was associated with the mutation of Arg-122, which indicates a preference for a basic amino acid at this position. Note that the conservative mutation R122K (CKTC), which corresponds to the sequence of the adw sub-type of HBV envelope proteins, did not alter infectivity, as expected. The addition of Asn-linked carbohydrates at position 122 (CNRTC) (Fig. 4A) had only a partial inhibitory effect (compare the CNRTC mutant to the CQRTC or CSRTC mu-tant).
Clearly, the results do not support the requirement for a CxxC sequence in the AGL to confer infectivity to HDV par-ticles, suggesting that Cys-121 and -124 do not form a typical redox active motif (i.e., disulfide-bond isomerase catalytic mo-tif) similar to the one borne by the peripheral subunits (SU) of the murine leukemia virus envelope protein (40).
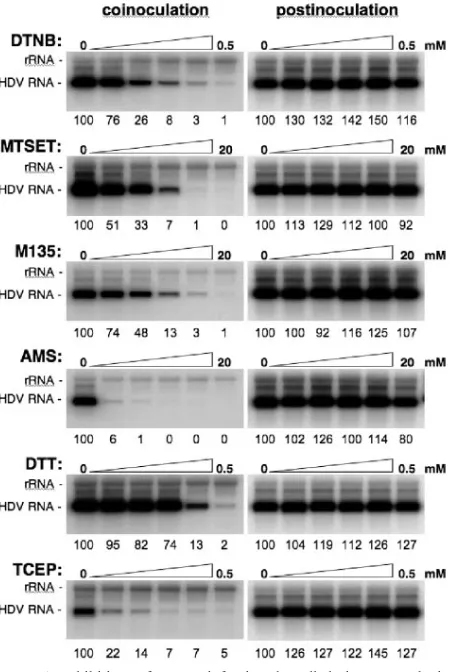
Inhibitory effect of membrane-impermeable alkylating or reducing agents on HepaRG infection with HDV.As shown previously (25–27, 42), the AGL disulfide network that cross-links the HBV envelope proteins is essential to structure the surface-exposed “a” determinant. However, the latter is not an absolute requirement for particle morphogenesis and secre-tion, which suggests that its strict conservation among all HBV genotypes is linked to a function at viral entry and, eventually, to a binding event. In addition, the intermolecular disulfide bonds mediated by the AGL cysteines, which are thought to confer structure and stability to the viral envelope, likely need to be reduced upon entry in order to release the virion’s cargo at a postattachment step. But as shown above, it is unlikely that a PDI activity is borne by the envelope proteins, suggesting that a cellular PDI activity might instead be recruited.
To address the implication of a thiol/disulfide exchange at viral entry, infection assays were conducted in the presence of PDI-interfering agents (33, 40). We chose to restrict the anal-ysis to surface-exposed thiol-disulfides by selecting membrane impermeable drugs (except for DTT), such as TCEP (a re-ducer) or AMS, MTSET, DTNB, and M135 (alkylators), at concentrations that were verified beforehand not to cause cy-totoxicity when incubated with the cells for 16 h. Cells and/or virus were mock treated or treated with 1:2 dilutions of drugs under various conditions. (i) Control experiments in which drugs were added to the culture medium after the 16-h cell-virus exposure period and left for 24 h were conducted. As shown in Fig. 5 (right panels), postinoculation treatment with any of the selected drugs had no inhibitory effect, demonstrat-ing that drugs were not interferdemonstrat-ing with cell metabolism or HDV RNA replication. (ii) Drugs were added to the cell su-pernatant with the inoculum and left for the 16-h duration of the virus-cell interaction. When membrane impermeable alky-lators (DTNB, MTSET, M135, or AMS) were used, a
dose-FIG. 3. Infectivity of HDV particles coated with HBV envelope proteins bearing cysteine substitutions.Results of infection assays are based on a Northern blot analysis of HDV RNA extracted from HepaRG cells exposed to wt or mutant HDV particles. In this experiment, 3.3⫻105 cells were exposed to approximately 108GE of HDV particles. (A) Inocula were recovered after their exposure to the cells, and their HDV RNA content was controlled by Northern blot hybridization using a genomic strand-specific,32P-labeled RNA probe. Signals are from 0.5 ml of postexposure inocula. (B) At day 7 postinoculation, cellular RNA extracted from 105cells was analyzed for the presence of HDV RNA. Signals were quantified using a phosphorimager. Numbers below each panel are percentages of the wt value. The size (in kilobases) of HDV RNA is indicated. wt SML, HDV particles coated with wt S-, M-, and L-HBsAg.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.135.449.69.267.2]dependent inhibition of infection was observed as shown in Fig. 5 (left panels). The strongest effect was observed with DTNB and AMS, for which concentrations of 0.5 mM and 2.5 mM, respectively, decreased infection to 1% of that of the control.
A similar dose inhibition relationship was observed when membrane-permeable (DTT) or impermeable (TCEP) reduc-tants were provided. Treatment with 125M TCEP was suf-ficient to reduce infection efficiency to approximately 7% of that of the control. The fact that HDV infection is inhibited when the drugs are present during the period of virus-cell exposure could indicate that free thiols and disulfide bonds are accessible during the entry process at the surface of cells and/or virions. This accessibility to membrane-impermeable drugs could be a consequence of the virus-cell interaction. The infection inhibition could also result from an effect of the drug prior to virus-cell interaction, leading to a decrease of HDV
binding capacity or to a block in virus internalization or un-coating.
Inhibitory effect of AMS and TCEP on HDV infectivity.To determine whether the surface of HDV particles displays free thiol groups critical for infectivity, particles were mock treated or treated with 1:2 dilutions of 20 mM AMS for 1 h at 37°C. After AMS treatment, virus was diluted 100-fold in culture medium and inoculated to HepaRG cells. After a 16-h incu-bation period, the inoculum was removed and replaced with fresh medium. As shown in Fig. 6, the inhibitory effect was dose dependent, and infectivity was drastically inhibited with 2.5 mM AMS (4% of that of the wt).
To investigate the possibility that surface-exposed disulfide bonds might be engaged in the viral entry process, virions were mock treated or treated with 1:2 dilutions of 0.5 mM TCEP for 2 h at 37°C before 1:100 dilution and inoculation to HepaRG cells. As shown in Fig. 6, TCEP demonstrated a clear
[image:6.585.47.275.62.359.2]dose-FIG. 4. Infectivity of HDV particles coated with HBV envelope proteins bearing mutations in the CxxC motif. (A) Production of mu-tant HDV particles was achieved as described in the legend to Fig. 2. Particles from 1 ml of culture fluids were concentrated and assayed for the presence of HBV envelope proteins as described in the legend to Fig. 2. Particles from 140l of culture medium were assayed for the presence of HDV RNA. (B) Infection assays were conducted as indi-cated in the legend to Fig. 3. Results are based on Northern blot analysis of HDV RNA extracted from HepaRG cells at day 7 postin-oculation. HDV RNA signals from 0.5 ml of postexposure inocula are shown in the upper panel. At day 7 postinoculation, cellular RNA extracted from 105cells was analyzed for the presence of HDV RNA (lower panel). Signals were quantified using a phosphorimager. Num-bers below each panel are percentages of the wt value. The size (in kilobases) of HDV RNA is indicated. wt SML, HDV particles coated with wt S-, M-, and L-HBsAg. S, M, and L indicate the positions of the S-, M-, and L-HBsAg, respectively.
FIG. 5. Inhibition of HDV infection by alkylating or reducing agents. Infection assays were conducted in the absence or presence of 1:2 dilutions of membrane-impermeable alkylators (DTNB, MTSET, M135, AMS) or reducers (DTT, TCEP) at the indicated concentra-tions. Inhibitors were added to the cell supernatant with the inoculum and left for 16 h (coinoculation). Control experiments were conducted with cells exposed the drugs for 24 h at day 1 postinoculation (posti-noculation). Infection assays were conducted as described in the leg-end to Fig. 3. At day 7 postinoculation, cellular RNA was extracted for measurement of intracellular HDV RNA by Northern blot analysis. Numbers below each panel are percentages of the control value. The size (in kilobases) of HDV RNA is indicated.
13062 ABOU-JAOUDE´ AND SUREAU J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.304.537.68.404.2]dependent inhibitory effect on infectivity; 125M TCEP for 1 h at 37°C was sufficient to reduce infectivity to approximately 7% of that of the control.
To determine whether the plasma membrane of HepaRG cells possesses accessible free thiol groups or disulfide bonds necessary for viral entry, cells were mock treated or treated with 1:2 dilutions of AMS or TCEP (20 mM and 0.5 mM, respectively) for 2 h at 37°C. Drugs were washed out with fresh culture medium before exposure of the cells to HDV. The results of the infection assays did not reveal any significant difference between treated and mock-treated cells, suggesting that alkylation of free thiol groups with AMS or reduction of disulfide bonds with TCEP at the surface of HepaRG did not interfere with viral entry (Fig. 6). Yet, there exists the possi-bility that thiol groups or disulfide bonds on the cell membrane could be engaged at viral entry without being accessible to the impermeable drugs. Alternatively, they could become exposed upon binding of HDV to its receptor.
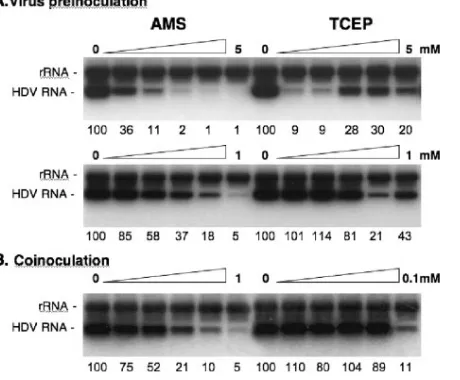
To ascertain that the infection inhibition observed when particles were treated with AMS or TCEP prior to inoculation (Fig. 6) was not due to an effect of the drugs (at 100-fold dilution) during the virus-cell exposure period, additional ex-periments were conducted. (i) Drugs were provided at low doses during the inoculation period (1:2 dilutions of either 1 mM AMS or 0.1 mM TCEP) to establish the precise concen-tration at which 50% inhibition is observed in a coinoculation treatment (Fig. 7B), and (ii) virions were treated prior to inoculation (preinoculation), with doses expected to be inef-fective after a 1:100 dilution during virus-cell interaction (Fig. 7A). The results of the coinoculation treatments (Fig. 7B) showed that a 50% inhibition of infection was observed upon treatment with 125M AMS or 50 to 100M TCEP.
Treatment of particles with 250M AMS prior to inocula-tion demonstrated a 50% inhibiinocula-tion of infectivity (Fig. 7A). In comparison, the effect of virus treatment with TCEP prior to inoculation was peculiar: we observed a⬎90% inhibition with
300 and 600 M TCEP and a rebound of infectivity when particles were treated withⱖ1 mM concentrations. Identical results were obtained in four independent experiments per-formed using different preparations of TCEP solutions.
These results thus demonstrate that the inhibition of infec-tivity observed for virions treated prior to inoculation (Fig. 7A) was not due to the activity of the drugs (at 1:100 dilutions) during the virus-cell exposure period (Fig. 7B). For instance, a 500M AMS treatment of virions prior to inoculation (pre-inoculation), corresponding to a 5 M concentration after 1:100 dilution (coinoculation), led to an 82% inhibition of infectivity. For comparison, a 250 M AMS coinoculation treatment was required to reach this level of inhibition. A similar conclusion was made regarding the effect of TCEP: a 300M treatment of virions prior to inoculation led to⬎90% inhibition of infection. In comparison, 3 M TCEP (1:100 dilution) during cell-virus incubation (coinoculation) had no effect (Fig. 7B).
Overall, the results indicate that free thiols and disulfide bonds are accessible to AMS or TCEP at the surface of HDV virions. Upon treatment with either drug, HDV particles lose infectivity.
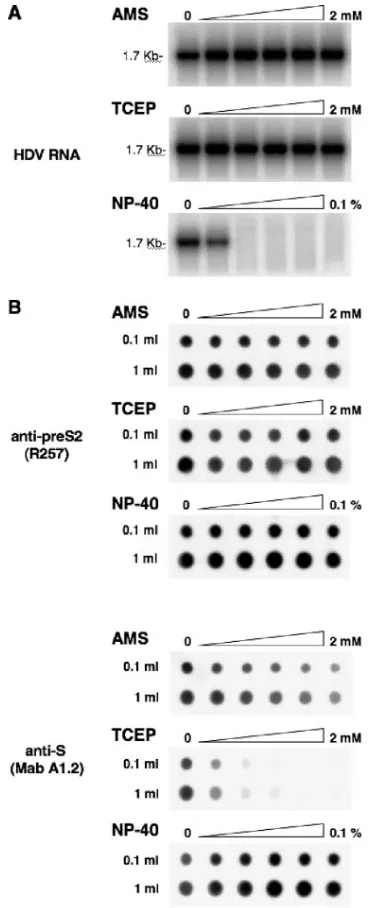
[image:7.585.49.280.69.192.2]Effect of AMS and TCEP on stability and antigenicity of viral particles. To determine whether AMS or TCEP could have altered infectivity by reducing the stability of the viral particles prior to receptor interaction, preparations of HDV virions were mock treated or treated with 1:2 dilutions of 2 mM AMS or TCEP. After treatment, viral RNA was analyzed to serve as a marker of viral particle integrity. As mentioned above, HDV RNA is rapidly degraded when the integrity of
FIG. 6. Infection assays with cells or HDV particles treated with AMS or TCEP prior to inoculation. HDV particles (virus) were mock treated or treated with 1:2 dilutions of 20 mM AMS or 0.5 mM TCEP for 1 h at 37°C. The drug/particle suspensions were then diluted 100-fold in fresh medium prior to inoculation to HepaRG cells. HepaRG cells (Cells) were mock treated or treated with 1:2 dilutions of 20 mM AMS or 0.5 mM TCEP for 2 h at 37°C. Cells were then washed extensively with fresh medium prior to exposure to inoculum. At day 7 postinoculation, cellular RNA was extracted for measurement of in-tracellular HDV RNA by Northern blot analysis. Numbers below each panel are percentages of the control value. The size (in kilobases) of HDV RNA is indicated.
FIG. 7. Infection assays with cells or HDV particles treated prior to inoculation with low doses of AMS or TCEP. (A) HDV particles were mock treated or treated with 1:2 dilutions of 5 mM AMS, 1 mM AMS, 5 mM TCEP, or 1 mM TCEP for 1 h at 37°c prior to 1:100 dilution in fresh medium and inoculation to HepaRG cells. (B) Infection assays were conducted in the absence or presence of 1:2 dilutions of 1 mM AMS or 0.1 Mm TCEP. Drugs were added to cells with the inoculum and left for the duration of the cell-virus exposure (16 h). At day 7 postinoculation, cellular RNA was extracted for measurement of in-tracellular HDV RNA by Northern blot analysis. Numbers below each panel are percentages of the mock value. The size (in kilobases) of HDV RNA is indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.306.536.72.262.2]the viral envelope is affected. In fact, a degradation of HDV particles was observed upon treatment with as little as 0.0125% NP-40 for 1 h at 37°C (Fig. 8A). This was sufficient to expose the viral RNA to degradation by RNases, showing that the
membrane of HDV RNP-containing particles had been dis-rupted. However, when particles were subjected to treatments with AMS or TCEP at concentrations shown to block infectiv-ity (Fig. 7), viral RNA was detected at levels comparable to that of the mock-treated virions, indicating that viral mem-brane integrity was preserved. Therefore, the loss of infectivity observed upon treatment of an inoculum with AMS or TCEP was not due to a disruption of the viral particles prior to binding. Note, however, that particle stability was affected upon treatments with⬎10 mM AMS (data not shown).
To determine whether treatments with AMS or TCEP could have affected the conformation of the AGL (or “a” determi-nant), particles were mock treated or treated with 1:2 dilutions of 2 mM AMS or TCEP (Fig. 8) before they were spotted on a PVDF membrane and probed with a MAb (A1.2) directed against the conformational “a” determinant (23, 34). An anti-pre-S2 (R257) antibody specific for a linear epitope of the pre-S2 domain was used as a control. As shown in Fig. 8B, binding of anti-pre-S2 to AMS- or TCEP-treated particles was not affected. By contrast, binding of MAb A1.2 to TCEP-treated particles was clearly inhibited in a dose-dependent manner, whereas AMS-treated particles demonstrated a lesser binding inhibition. In comparison, treatment of viral particles with 1:2 dilutions of 0.1% NP-40 had no inhibitory effect on their binding to MAb A1.2. Note that, as shown above, NP-40 concentrations of⬎0.006% disrupted the viral membrane, ex-posing the viral RNA to degradation (Fig. 8A).
From these results, we concluded that a reduction of disul-fide bonds with TCEP and, to a lesser extent, alkylation of free thiols with AMS, at the surface of viral particles modified the AGL conformation. Overall, AMS or TCEP is effective in blocking HDV infectivity at doses that are proven to denature the AGL epitopes through a modification of the redox status of surface-exposed cysteines.
DISCUSSION
The current model for the HBV (or HDV) entry mechanism ascribes to the pre-S1 domain of the L-HBsAg protein the initial function of attachment to a specific receptor at the surface of human hepatocytes (15), whereas a second infectiv-ity determinant maps to the AGL (21). Here, we have dem-onstrated that seven of the eight cysteines that are critical for the structure of the “a” determinant in the AGL (C107 could not be tested) are important for the activity of the HBV en-velope proteins at viral entry.
[image:8.585.68.255.67.521.2]In general, cysteines are of functional importance because they can support posttranslational modifications, establish structural disulfides, or act as a catalytic amino acid in enzyme-active sites. Hence, mutations of AGL cysteines could affect different aspects of the envelope proteins functions, including particle morphogenesis and infectivity. The AGL cysteines have been described as instrumental for the morphogenesis and stability of the particles (25–27, 42), and it is thought that the protein-rich and compact structure of the viral envelope is stabilized by a network of intermolecular disulfide bonds (7, 20). But as shown here, mutations of AGL cysteines were, to some extent, tolerant of the assembly of viral particles, sug-gesting that a precise network of disulfide bonds is not oblig-atory for morphogenesis or stability of viral particles. In fact,
FIG. 8. Effects of AMS and TCEP on stability and antigenicity of HDV particles. (A) To test the sensitivity of HDV virions to AMS, TCEP, or NP-40, 100-l aliquots of HDV particles (108GE) were mock treated or treated with 1:2 dilutions of 2 mM AMS, 2 mM TCEP, or 0.1% NP-40 for 1 h at 37°C. Viral RNA was then extracted and analyzed by Northern blotting. The size (in kilobases) of HDV RNA is indicated. (B) To analyze the effect of AMS, TCEP, or NP-40 on antigenicity of HDV, viral particles concentrated from 1 ml or 0.1 ml of supernatant (108or 107GE, respec-tively) were treated with 1:2 dilutions of 2 mM AMS, 2 mM TCEP, or 0.1% NP-40 and spotted on a PVDF membrane by using a microfiltration apparatus. Membrane was blocked with TBS-casein and then incubated in the presence of rabbit anti-pre-S2 (R257) or mouse anti-S (MAb A1.2) antibodies. Immunoblots were developed using ECL reagents (Amer-sham Pharmacia Biotech).
13064 ABOU-JAOUDE´ AND SUREAU J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
we were able to produce particles bearing substitutions for AGL cysteines 121 to 149. Each mutant, however, displayed conformational changes that translated into a loss of the “a” determinant. This was an indication that the “a” determinant, to which no precise function had been assigned thus far, is not a functional element of viral envelope morphogenesis or sta-bility. In contrast, we clearly established a correlation between the “a” determinant and infectivity. (i) The AGL cysteine mutations that disturbed the “a” determinant also altered HDV infectivity, and (ii) treatment of viral particles with mol-ecules that are known to modify the redox state of surface-exposed cysteines impaired both antigenicity and infectivity.
The loss of infectivity observed upon treatment of particles with membrane-impermeable TCEP prior to inoculation could be due to conformational changes that are detrimental to a binding event or to a block of the thiol/disulfide exchange reaction necessary for a postbinding event, such as the disas-sembly of the viral envelope. Surprisingly, exposing particles to
ⱖ1 mM TCEP was less inhibitory to infectivity than 0.3 or 0.6 mM treatment. One explanation for this phenomenon might be that 1 mM TCEP would generate a redox potential at which critical disulfide bridges are reduced, allowing the AGL to display an intermediate conformation that is normally engaged at a postbinding stage during the viral entry process. This specific and transient conformation could, for instance, be gen-erated upon pre-S1 domain binding to its receptor.
Regarding the inhibitory effect of AMS on HDV infectivity (drug applied to particles prior to inoculation), the explanation is not obvious, considering the fact that previous studies pointed to the absence of free thiols in the AGL of viral particles (26, 42). One possibility is that most, but not all, of the AGL cysteines would indeed be engaged in disulfide bridging, but a small percentage (presumably too low to be detected by conventional biochemical techniques) would display a reduced form. Upon alkylation with a large reagent, such as AMS (500 Da), the AGL conformation could be modified and the thiol/ disulfide exchange reaction that is required for envelope dis-assembly could be blocked.
Genetic or biochemical modifications of the AGL structure might thus interfere with infectivity in at least two ways: (i) by eliminating a specific conformation necessary for binding to a receptor, in association with or independent of the pre-S1 domain, with the pre-S1 domain being the tissue-specific bind-ing determinant; and (ii) by reshufflbind-ing the envelope proteins disulfide network and creating illegitimate bonds that would be inhibitory to the envelope disassembly process. Our results are clearly compatible with both a receptor binding function and a role in envelope disassembly, and experiments are under way to directly address these two hypotheses.
After internalization of HDV particles by the hepatocytes, disassembly of the viral envelope appears as a necessary event that is likely to involve the AGL disulfide bonds and a PDI activity for thiol/disulfide exchange reactions. That this activity could be borne by the envelope protein itself was suggested by the presence of a potential disulfide bond isomerase motif (CxxC) at positions 121 to 124 in the AGL. Such a motif could, for instance, be activated following attachment of the virus to the host cell receptor. An example of this phenomenon on the peripheral (SU) subunit of the murine leukemia virus envelope proteins has been described (40). In that model, the enzymatic
activity is engaged at viral entry by a conformational change induced by the binding of SU to the receptor, which leads to isomerization of disulfide-bonds between SU and the trans-membrane subunit and to fusion activation. However, the re-sults of our genetic analysis are not in agreement with the AGL CRTC acting as a catalytic motif. Yet they do not rule out the possibility that envelope proteins would bear a PDI activity with an atypical redox switch motif.
Another possibility is that a cell-surface PDI-related protein is involved, as is the case in the entry pathway of human immunodeficiency virus type 1 (1, 10, 11, 28, 31, 32). However, our data do not support this hypothesis for the HBV/HDV entry pathway, since treatment of cells with membrane-imper-meable drugs prior to virus exposure had no effect on infection efficiency. In addition, treatment of cells with two PDI inhibi-tors (bacitracin and anti-PDI antibodies) also failed to inhibit infection (data not shown).
A third possibility is that thiol/disulfide exchange occurs not at the cell surface but in an internal cellular compartment. In that case, membrane-impermeable AMS or TCEP would not be expected to inhibit infection when provided to cells prior to inoculation, because the cell PDI activity necessary for disas-sembly would be engaged after internalization of virus particles (by endocytosis, for instance).
In conclusion, our findings establish a correlation between AGL cysteines, their redox state, the conformation of the AGL, and the activity of HBV envelope proteins at viral entry. It is thus conceivable that the conservation of the conforma-tional “a” determinant among all HBV genotypes is related to an essential function at viral entry, assuming that the results obtained here with the HDV model are pertinent to the entry mechanism of HBV. Our findings emphasize the functional importance of the AGL, which is potentially active at two steps of the viral entry pathway, (i) the “a” determinant being in-volved in a binding function and (ii) the intermolecular disul-fide network controlling the envelope disassembly process. If these functions were to be demonstrated, it could open the way to the development of new antiviral strategies.
ACKNOWLEDGMENTS
We acknowledge C. Tre´po and O. Hantz for providing the HepaRG cell line and Ronald Kennedy for MAb A1.2. We are grateful to Re´mi Caparros for help with the HBsAg ELISA.
This work was supported through grants from ANRS and INTS. C.S. is a CNRS investigator.
REFERENCES
1.Barbouche, R., R. Miquelis, I. M. Jones, and E. Fenouillet.2003. Protein-disulfide isomerase-mediated reduction of two Protein-disulfide bonds of HIV en-velope glycoprotein 120 occurs post-CXCR4 binding and is required for
fusion. J. Biol. Chem.278:3131–3136.
2.Barrera, A., B. Guerra, L. Notvall, and R. E. Lanford.2005. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol.
79:9786–9798.
3.Blanchet, M., and C. Sureau.2006. Analysis of the cytosolic domains of the hepatitis B virus envelope proteins for their function in viral particle
assem-bly and infectivity. J. Virol.80:11935–11945.
4.Blanchet, M., and C. Sureau.2007. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues.
J. Virol.81:5841–5849.
5.Bonino, F., K. H. Heermann, M. Rizzetto, and W. H. Gerlich.1986. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B
virus-derived envelope. J. Virol.58:945–950.
6.Bruss, V.1997. A short linear sequence in the pre-S domain of the large hepatitis B virus envelope protein required for virion formation. J. Virol.
71:9350–9357.
on November 8, 2019 by guest
http://jvi.asm.org/
7.Bruss, V.2007. Hepatitis B virus morphogenesis. World J. Gastroenterol.
13:65–73.
8.Bruss, V., J. Hagelstein, E. Gerhardt, and P. R. Galle.1996. Myristylation of the large surface protein is required for hepatitis B virus in vitro infectivity.
Virology218:396–399.
9.Edman, J. C., L. Ellis, R. W. Blacher, R. A. Roth, and W. J. Rutter.1985. Sequence of protein disulphide isomerase and implications of its relationship
to thioredoxin. Nature317:267–270.
10.Fenouillet, E., R. Barbouche, and I. M. Jones.2007. Cell entry by enveloped viruses: redox considerations for HIV and SARS-coronavirus. Antioxid.
Re-dox Signal.9:1009–1034.
11.Gallina, A., T. M. Hanley, R. Mandel, M. Trahey, C. C. Broder, G. A. Viglianti, and H. J. Ryser.2002. Inhibitors of protein-disulfide isomerase prevent cleavage of disulfide bonds in receptor-bound glycoprotein 120 and
prevent HIV-1 entry. J. Biol. Chem.277:50579–50588.
12.Ganem, D., and A. M. Prince.2004. Hepatitis B virus infection—natural
history and clinical consequences. N. Engl. J. Med.350:1118–1129.
13.Gerin, J. L., J. L. Casey, and R. H. Purcell.2001. Hepatitis delta virus, p.
3037–3050.InD. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed.
Lippincott Williams & Wilkins, Philadelphia, PA.
14.Glebe, D., M. Aliakbari, P. Krass, E. V. Knoop, K. P. Valerius, and W. H. Gerlich.2003. Pre-S1 antigen-dependent infection ofTupaia hepatocyte
cultures with human hepatitis B virus. J. Virol.77:9511–9521.
15.Glebe, D., and S. Urban.2007. Viral and cellular determinants involved in
hepadnaviral entry. World J. Gastroenterol.13:22–38.
16.Glebe, D., S. Urban, E. V. Knoop, N. Cag, P. Krass, S. Grun, A. Bulavaite, K. Sasnauskas, and W. H. Gerlich.2005. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia
hepatocytes. Gastroenterology129:234–245.
17.Gripon, P., I. Cannie, and S. Urban.2005. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface
protein. J. Virol.79:1613–1622.
18.Gripon, P., J. Le Seyec, S. Rumin, and C. Guguen-Guillouzo.1995. Myristy-lation of the hepatitis B virus large surface protein is essential for viral
infectivity. Virology213:292–299.
19.Gripon, P., S. Rumin, S. Urban, J. Le Seyec, D. Glaise, I. Cannie, C. Guyomard, J. Lucas, C. Trepo, and C. Guguen-Guillouzo.2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA
99:15655–15660.
20.Heermann, K., and W. Gerlich. 1992. Surface proteins of hepatitis B
viruses.InA. Maclachlan (ed.), Molecular biology of HBV. CRC Press,
Boca Raton, FL.
21.Jaoude´, G. A., and C. Sureau.2005. Role of the antigenic loop of the hepatitis B virus envelope proteins in infectivity of hepatitis delta virus.
J. Virol.79:10460–10466.
22.Jenna, S., and C. Sureau.1999. Mutations in the carboxyl-terminal domain of the small hepatitis B virus envelope protein impair the assembly of
hep-atitis delta virus particles. J. Virol.73:3351–3358.
23.Kennedy, R. C., I. Ionescu-Matiu, K. Adler-Storthz, R. D. Henkel, Y. Sanchez, and G. R. Dreesman.1983. Characterization of anti-hepatitis B
surface antigen monoclonal antibodies. Intervirology19:176–180.
24.Le Seyec, J., P. Chouteau, I. Cannie, C. Guguen-Guillouzo, and P. Gripon.
1999. Infection process of the hepatitis B virus depends on the presence of
a defined sequence in the pre-S1 domain. J. Virol.73:2052–2057.
25.Mangold, C. M., and R. E. Streeck.1993. Mutational analysis of the cysteine
residues in the hepatitis B virus small envelope protein. J. Virol.67:4588–
4597.
26.Mangold, C. M., F. Unckell, M. Werr, and R. E. Streeck.1997. Analysis of intermolecular disulfide bonds and free sulfhydryl groups in hepatitis B
surface antigen particles. Arch. Virol.142:2257–2267.
27.Mangold, C. M., F. Unckell, M. Werr, and R. E. Streeck.1995. Secretion and antigenicity of hepatitis B virus small envelope proteins lacking cysteines in
the major antigenic region. Virology211:535–543.
28.Markovic, I., T. S. Stantchev, K. H. Fields, L. J. Tiffany, M. Tomic, C. D. Weiss, C. C. Broder, K. Strebel, and K. A. Clouse.2004. Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated
T-cell fusion during viral entry. Blood103:1586–1594.
29.Neurath, A. R., B. Seto, and N. Strick.1989. Antibodies to synthetic peptides from the preS1 region of the hepatitis B virus (HBV) envelope (env) protein
are virus-neutralizing and protective. Vaccine7:234–236.
30.Norder, H., A. M. Courouce, P. Coursaget, J. M. Echevarria, S. D. Lee, I. K. Mushahwar, B. H. Robertson, S. Locarnini, and L. O. Magnius.2004. Ge-netic diversity of hepatitis B virus strains derived worldwide: genotypes,
subgenotypes, and HBsAg subtypes. Intervirology47:289–309.
31.Ryser, H. J., and R. Fluckiger.2005. Progress in targeting HIV-1 entry. Drug
Discov. Today10:1085–1094.
32.Ryser, H. J., E. M. Levy, R. Mandel, and G. J. DiSciullo.1994. Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc. Natl. Acad. Sci.
USA91:4559–4563.
33.Senkevich, T. G., B. M. Ward, and B. Moss.2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with in-tramolecular disulfide bonds formed by the viral cytoplasmic redox pathway.
J. Virol.78:2348–2356.
34.Shearer, M. H., C. Sureau, B. Dunbar, and R. C. Kennedy.1998. Structural characterization of viral neutralizing monoclonal antibodies to hepatitis B
surface antigen. Mol. Immunol.35:1149–1160.
35.Sureau, C., B. Guerra, and R. E. Lanford.1993. Role of the large hepatitis B virus envelope protein in infectivity of the hepatitis delta virion. J. Virol.
67:366–372.
36.Sureau, C., J. R. Jacob, J. W. Eichberg, and R. E. Lanford.1991. Tissue
culture system for infection with human hepatitis delta virus. J. Virol.65:
3443–3450.
37.Sureau, C., A. M. Moriarty, G. B. Thornton, and R. E. Lanford.1992. Production of infectious hepatitis delta virus in vitro and neutralization with
antibodies directed against hepatitis B virus pre-S antigens. J. Virol. 66:
1241–1245.
38.Vuori, K., T. Pihlajaniemi, R. Myllyla, and K. I. Kivirikko.1992. Site-directed mutagenesis of human protein disulphide isomerase: effect on the assembly, activity and endoplasmic reticulum retention of human prolyl
4-hydroxylase inSpodoptera frugiperdainsect cells. EMBO J.11:4213–4217.
39.Vyas, G. N., K. R. Rao, and A. B. Ibrahim.1972. Australia antigen (hepatitis B antigen): a conformational antigen dependent on disulfide bonds. Science
178:1300–1301.
40.Wallin, M., M. Ekstrom, and H. Garoff.2004. Isomerization of the
intersub-unit disulphide-bond in Env controls retrovirus fusion. EMBO J.23:54–65.
41.Wang, C. J., P. J. Chen, J. C. Wu, D. Patel, and D. S. Chen.1991. Small-form hepatitis B surface antigen is sufficient to help in the assembly of hepatitis
delta virus-like particles. J. Virol.65:6630–6636.
42.Wounderlich, G., and V. Bruss.1996. Characterization of early hepatitis B
virus surface protein oligomers. Arch. Virol.141:1191–1205.
13066 ABOU-JAOUDE´ AND SUREAU J. VIROL.