0022-538X/80/01-0366/11$02.00/0 1
Role of the Host Cell in Bacteriophage
T4
Development
I.
Characterization
of
Host Mutants That Block T4 Head Assembly
HELEN R.REVEL,t BARBARA L.
STITT4,
ILGA LIELAUSIS, ANDWILLIAM B. WOOD§*DivisionofBiology,California Institute of Technology, Pasadena, California 91125
Tostudy
the role
ofthe hostcell
in bacteriophage T4 infection, we selectedmore
than
600 mutanthost-defective
bacteriathat adsorbed and werekilled
byphage
T4+ but wereunable to support its growth. The mutants were grouped intoseven
classes
by the growthpatterns
of T4phages carrying compensatingmuta-tions
(go
mutants[,grows
on]),selected
on fourprototypehost-defective
strains.Lysis and
DNAsynthesis
experiments indicated that classes A, AD, D, and B(the
majority of the host-defective
mutants) block T4+ development at anassembly
step,class C
mutantsaffect
anearly
stage in phage development, andclass
Fmutants appear to act at more than one stage. Analysis of T4+ infection intheassembly-defective
mutantsby in vitrocomplementation,
electronmicros-copy,
and sodium
dodecyl
sulfate-polyacrylamide
gelelectrophoresis
showed thatthe host-defective mutations interfere with T4+
capsidformation
atthe level ofphage
gene 31function, before
assembly of any recognizable capsid structure. Themutations
map nearpurA, but
at two or possibly threedifferent
sites. The gomutant
phages able
toovercome thehost defect
carrymutations in either gene31, as found
by
othersforsimilar
defective hosts, or in the gene for the majorcapsid protein (gene 23). The
gene 23go mutations do not bypass the requirementfor
gene 31function. These results
suggest that at least three components mustinteract
toinitiate
T4head
assembly: gp3l,
gp23, and one or more host factors.The
compensatoryeffects of mutational alterations
inthese
components arehighly
allele specific, consistent with the view that phage
and host componentsinteract
directly
in
protein
complexes.
The
morphogenesis of bacteriophage
T4is
known
toproceed in
stepwise
fashion
by the
subassembly
of
heads,
tails,
and tail
fibers,
which
are
subsequently joined
in
sequence toform
active
phage
particles (40). Although
mostof the
assembly
steps canproceed in
infected-cell
ex-tracts
(5), the earliest
stepsin these
subassembly
pathways have
not yetbeen demonstrated in
vitro
(14,
22,28).
Consequently,
wewondered
whether the host cell
might play
anobligatory
role in
initiating
someassembly
processes.
Vi-ruses
utilize much of the
existing host
cell
syn-thetic
machinery,
specifically redirecting
it tomake virus
particles.
Perhaps
thehost also
sup-plies
scaffolding,
templates,
orother componentsessential
forinitiating
viral
assembly.
Electron
microscopic
evidencesuggests
that the firststeps
inthe
assembly
of T4heads,
andperhaps
thattPresentaddress:DepartmentofMicrobiologyand Molec-ularBiology,TuftsMedicalSchool, Boston,MA 02111.
tPresentaddress: Public Health Research Institute of the
Cityof NewYork, Inc.,NewYork,NY 10016.
§Present address:DepartmentofMolecular, Cellular and
DevelopmentalBiology, UniversityofColorado,Boulder,CO 80309.
of
baseplates, proceed
onthe
host membrane
(29,
30).
We
sought
toinvestigate
these
possibilities by
the
study
of host
mutantsthat
specifically
block
T4
assembly. When this work
wasinitiated,
twohost
mutantsthatinterfere with
T4head
assem-bly had been described.
Georgopoulos
etal.
(9)
had
reported that
groEA44,
aK-12mutantse-lected for its
inability
tosupportphage
X
growth
and
found
toblock X head
assembly,
also blocked
T4
morphogenesis
early
incapsid
formation,
atthe
stepcontrolled
by
gene 31. Asingle
mutantderivative of
Escherichia
coli Bcalled
mop(37)
gave a
similar
phenotype
after T4 infection.Furthermore,
Pulitzer andYanagida (20)
hadreported
that amutantof W3350wasdeficient
inthe
assembly
offunctional
tailfibers,
andwehad inferred from
genetic
experiments
that host
involvement
in T4tailfiber
assembly
occursat thelevel
of thegene
57-controlled step
inpo-lymerization
of thetail
fibercomponents
(22).
These
results
encouraged
us tosearch formoreassembly-defective
mutanthosts.Accordingly,
weselected
host-defective(HD)
bacteria unable to propagate T4 and
grouped
366
on November 10, 2019 by guest
http://jvi.asm.org/
them into classes basedontheirabilityto
prop-agate mutant T4 go (grows on) phagesselected
for growth on various HD strains. We report
here genetic and physiological studies on four
classes of HD mutantsthat block T4 assembly
and on the T4 go phagemutantsthatovercome
these blocks. All of these HD mutant hosts
appear to interfere withT4 capsid assemblyat
the step controlled by gene 31, in agreement
with results found for similar mutants by other
investigators (3,
9, 36,37).
Geneticanalysis
shows that
the HD mutations define
atleast
twoandperhaps three host genes and that
compen-sating phage go mutationscan occurin either of
twophage genes. The patterns of compensation
among these mutants are discussed with
refer-ence to the detailed model of Takahashiet al.
(36)and the possible nature of phage-host
inter-actions in the initiation of T4
capsid assembly.
HD mutants of a fifth class affect
T4
assembly
but also exhibit additional pleiotropic effects
onT4development. We shall describe the
charac-terization of these mutantsin asubsequent
pa-per
(B.
L.Stitt,
H. R.Revel, I. Lielausis,
and W. B.Wood,
J.Virol., submitted
forpublication).
We also found mutants
of
asixth
class, which
affect
anearly
step in T4growth;
wehave
notfurther
investigated these
mutants.(A
preliminary
report of someof
the workdescribed
here has beenpublished
[39]. Thesestudies
areincluded
in the doctoral dissertation ofB.L.S., submitted
in partialfulfillment of therequirements for the
Ph.D. degree,California
Institute of Technology,
Pasadena, 1978.)MATERIALS AND METHODS
Media. H broth forphage and bacterial growth and
EHA top and bottom agarsfor plating were prepared
asdescribedpreviously (33). LB broth and top and
bottomagars(21)weresupplemented with 2.5x 10-3
MCaCl2 for growth of P1 or0.3% maltose for growth
ofphageA. M9, asynthetic, phosphate-buffered
me-dium (13), wasused when phage-infected cellswere
labeled with'4C-amino acids. M9 top and bottom agars
contained6.5and 10.0g of agar (DifcoLaboratories)
per liter, respectively. M9 was supplemented as
re-quired with amino acids, purines, or pyrimidines at 20
to 50,ug/ml and vitaminsat 0.1,ug/ml for the growth
ofauxotrophic bacterial strains inP1transduction and
F'transfer experiments. Buffers used were the dilution
medium (DM) described by King (15); Tris-maleate
(TM) buffer, pH 6.0 (1); andTris-magnesium buffer
(TMg), pH 6.8 or pH 7.4, which was 0.05 M
Tris-hydrochloride-0.02MMgSO4.
Chemicals and enzymes.CrystallineDNaseIand
RNase Aand2'-deoxyadenosine were obtained from
Sigma ChemicalCo.Acrylamide and
N,N'-methylene-bisacrylamidewerefrom Eastman Organic Chemicals.
TEMED (N,N,N',N'-tetramethylethylenediamine)
was from Matheson, Coleman & Bell.
Nitrosoguani-dine (N-methyl-N'-nitro-N-nitrosoguanidine) was
from AldrichChemical Co. Liquifluorwasfrom New
England Nuclear Corp. Casamino Acids were from
DifcoLaboratories. All otherchemicalswerereagent
grade.
Radioactive compounds. '4C-amino acids were
reconstituted protein hydrolysate no. 3122-10 from
Schwarz/Mann;[2-'4C]thymidine(61mCi/mmol)was
from AmershamCorp.
Bacteriaandbacteriophages.E.coli K-12CR63,
permissive for amber (am) mutants,wasthe host for
growth of phage stocks. E. coli BS/6/5, nonpermissive
forammutants,wasused forselectiveplatingofam+
phage.SKB178,anE.coli K-12 strain that is F-galE
and nonpermissive foram mutants, is the parent of
the HD strains tobe described (9). WH-1 (F' [F196
supD32] his-45trp-37phoA4recAlstrA144 "var-116"
[12;CGSC 4612])wasusedtomake HD strainssupD.
C600 (thr-1 leu-6 thi-IsupE44 lacY) tonA2A-) was
the donor in P1 transductions of supE. F' strains
KLF17/KL132 (F' [F117] pyrB31 thi-1 thyA25 his-i
pro-27 leu-6 thr-1 recAl xyl-7 malAU ara-13 gal-6
lacYl tonA2 str-9 rel-I Ar A- [CGSC 4255]) and
KLF18/KL132 (F' [F118]pyrB31 thi-1 thyA25his-i
pro-27 leu-6 thr-1 recAl xyl-7 maIAl ara-13 gal-6
lacYl tonA2str-9rel-1ArA-[CGSC4259]) (18) were
used formapping mutations in HD strains. K-12 strain
T832(argH his pro thi-1purA ampA maltsx-9Strr
A' A-)wasobtained from T. Takano(37) and usedas
arecipient in transductionexperiments. Our isolate of
this strain carries an unidentified am suppressor.
groEA44 andgroEB515wereobtained from C.
Geor-gopoulos (9).
T4D+ and T4 (am) andtemperature-sensitive (ts)
mutants werefrom theCalifornia Institute of
Tech-nology (Caltech) collection (now maintained at the
University ofColorado, Boulder). am mutants used
were amB8 (gene 20), amE209 (gene 22), amB270
(gene 22), amHll (gene 23), amB17 (gene 23),
amB272 (gene 23), amE506(gene 23), amN67(gene
25), amN120 (gene 27), amN54 (gene 31), amNG71
(gene 31), amN52 (gene 37), amN122 (gene 42), and
X4E [amB25 (gene 34):amA455 (gene 34):amN52
(gene 37):amB262 (gene 38):amB252 (gene 35)]. ts
mutantsusedweretsA70 (gene 31) and tsA56 (gene
31). r67 in gene rIIIwasincludedas anoutside marker
incrossesbetween gene31 mutants.T2L, T3, T5, T6,
and T7were from theCaltech collection. T4e (aT4
mutant that grows on groEA44) AcIb2, and AEA30
were received from C. Georgopoulos (9). P1 (Plkc)
was used for transduction experiments; fl phage is
male specific and was used to identify F'-carrying
strains.
HDmutant bacteria. SKB178cells grown in H
broth, washed threetimes,andsuspendedat 4 x108/
ml inTM bufferweretreated withnitrosoguanidine
(100
Ag/mi)
for 15min at 37°C bythe procedureofAdelbergetal. (1). The survivalrate was40%.
Nitro-soguanidinewasremovedby washingthree times with
cold TM buffer, and 26 subcultures (numbered 0
through 25)weremadebyinoculating5mlof H broth
with0.1ml ofmutagenizedcells andgrowingovernight
at370C.
Toselect HD mutants, subcultureswerediluted 1:
25, grown tolog phaseat 37°C,and0.05-mlsamples
containing about 106 cells werespread on relatively
VOL. 1980
on November 10, 2019 by guest
http://jvi.asm.org/
ET AL.
dryEHAplates.The plateswereincubated for60to
90minatthe desired selectiontemperatureandthen
sprayedwith 108phage in1/A,usinganaerosolsprayer
androtatingtheplatetoensureuniformcoverage.A
mixture ofT4and T6, relatedphages with different
hostranges, wasusedtoreduce therecoveryof
phage-resistantcells.InSKB178,which isr6- (permissive for
T6withnonglucosylatedDNA[21,24]), thisprocedure
also prevented the selection of galU-defective host mutants which produce nonglucosylated T4 and T6
progeny. Suchmutantscomprised about 25% of the
recovered colonies when selectionwaswithT4 alone.
After overnight incubation, therewere50to100
sur-viving colonies per plate. Cells from these colonies
werepurified by streakingonEHA plates.Nomutants
werefound when the selection procedurewasapplied
to nonmutagenized cells. Three separate selections
werecarriedout(see below).
Mutants from the three selections were called
HDXO.1-HDXO.29 and HDX1.0-HDX1.9 through
HDX25.0-HDX25.9 (selection I), HDX3.01-HDX3.26
(selection II), andHDX26.01-HDX26.30,
HDX27.01-HDX27.30, andso onthrough HDX35.01-HDX35.30
(selection III). HD isaphenotype designation;"X" is
aclassification letter (see below). The digitspreceding
the decimalpointindicate thesubculture oforigin. In
the genetic analysis of the HD strains and in the
Discussion, we usethegenotypic designation hdhfor
the HD strains blocked in T4 headassembly.A
par-ticular mutation is designated bythe number of the
HD strain; e.g., HDA17.5 carries the mutation
hdh-17.5, thehyphentobereplaced byaletter whenthe
identityof the hostgenesis better defined.
gomutantphage.Thegophageswereselectedby
plating 107to 108 T4+ particles perplate oneachof
the first 29 HDstrains isolated in selection I
(HDX0.1-HDXO.29). Mutant phage werepurified by stabbing
andreplatingfromlarge plaques thatappeared with
frequencies of 106 to 10-7. An analysis of plating
patternsbyspottestingwithpurified phageonthe29
HDhost strains resulted in the identification of four
different classes ofgomutantphage, designated goA,
goC, goD, andgoF (see below).Thefollowing
repre-sentatives of each of the four classeswereusedasthe
standard go phage for typing of HDstrains: goA1
(selectedonHDADO.1), goCl (selectedonHDCO.13),
goDl (selected onHDDO.18),andgoFl (selectedon
HDF0.26). Additionalgomutantswerepurified
simi-larly from the smallerplaques that appearedat
fre-quenciesof10' to105 whenT4+wasplatedonsome
HD hosts (see below). am:go double mutants were
constructed by appropriatecrosses and screened for theirabilitytogrowonHD hosts thatcarriedsupDor
supEand for theirinabilitytogrowonthe
correspond-ing nonsuppresscorrespond-ingHDhosts.
Streaktests. About 10
IlI
of two T4phagestockscontaining 108and 10"particlesperml, respectively,
wasstreakedacross EHAplatesand allowedto dry.
Bacterial cultures(-4 x
10'
cellsperml)werecross-streaked separately overeachphage streak, and the
plates wereincubated overnight.Cell lysisorits
ab-sencein the streakoverlapareasdifferentiates
sensi-tive, resistant,and HDcells: sensitivecells arelysed
atboth phage concentrations, resistant cellsare not
lysed at either concentration, and HD mutants are
J. VIROL.
lysed at the high phage concentration but not at the low concentration.
Spottests.Smalldrops (-51l) of phage solutions
atconcentrations of 108, 106, and 104 particles per ml
were spotted on agar plates seeded with 108 cells of
thebacterial strain to be tested, allowed to dry, and
incubated overnight atthe appropriate temperature.
Mapping of phage mutations. Phage crosses
wereperformedasdescribed by Wilson and Kells (38).
Special conditions used to distinguish recombinant
progeny are described in the appropriate figure
leg-ends.
Burstsize measurements. Burst sizes were
deter-mined as described previously (22).
Measurement of infected-cell lysis. Bacterial
cellsweregrown in20mlof H broth to2 x 108 to 3
x 108/ml (optical density at 660 nm= 0.50 to 0.60)
and infected withphageat amultiplicity of infection
of6 to 7. Samples (1 ml each) werewithdrawn at
intervals, and the optical density was measured at 660
nm. Surviving bacteria measuredat 7minwereless
than 1%. A few drops of CHCl3 were added to the
HDC and HDF culturesat 80minafterinfection, and
theoptical densitywasmeasuredonce more at 90min.
The time oflysis was also determined visually for
manyphage-infectedHDstrains.
Labeling phage proteins with"C-aminoacids.
Cellswere grown in M9at370C to 5 x 107/ml and
concentratedto 4 x 108/ml;2.0-mlcultureswere
in-fected withphageat amultiplicityof infection of5.0
att=0andweresuperinfectedwiththesame amount
ofphageatt =7min. '4C-aminoacidswereaddedto
a final concentration of 1 to 2 jLCi/ml to infected
culturesatthe desired time.After thelabeling period,
cultureswereusuallychased foratleast1minby the
addition of0.5ml of 10% Casamino Acids.Chilled cells
were pelleted, suspendedin 0.1 ml ofTMg, pH6.8,
containing 1,igofDNase,and frozen in asolid
CO2-ethanol bath. Upon thawing, the preparation was
made 2% in sodiumdodecylsulfate and
/3-mercapto-ethanolandwasboiled for 1 to 3min. Incorporated
radioactivity wasdetermined bytrichloroacetic acid
precipitation ofa
2-/A
portion, andsamples containingequal numbers ofcounts (and equal volumes when
identical labeling conditionswereused) wereloaded
ontogels.
Sodium dodecyl
sulfate-polyacrylamide gel
electrophoresis. Samples
prepared
as describedabovewere electrophoresedinthediscontinuous
so-diumdodecylsulfate buffersystem described
by
La-emmli (16),asmodifiedbyDickson (4),andadapted
forusewith slabgels bythemethod of Studier (35).
Gelswere runat a constant currentof 10 mAuntilthe
bromophenolbluemarkerdyeenteredthe
separating
gel and thenat20 mAuntil the marker
dye
reachedthe bottomof thegel.Gelswerefixedand stained for
1 to 2h inanaqueous solution of50%
(wt/vol)
trichlo-roacetic acid and 0.2% Coomassie brilliant blue(Schwarz/Mann).Destainingwasfor 10to20h in10%
methanol-10% acetic acid. Thegelsweredriedonto
Whatman3MMchromatographypaper undervacuum
andexposedtoKodakNo-Screen
X-ray
film.Invitrocomplementationtests.In vitro
comple-mentation assayswereperformed bythe
procedure
ofEdgar andWood(5).Forthe
preparation
of defectiveon November 10, 2019 by guest
http://jvi.asm.org/
extracts,nonpermissive cells weregrownto4x 108/
mlin Hbroth with vigorous aeration, infectedat t=
0withanappropriateammutant atamultiplicity of
infection of 7, superinfected at 7 min at the same
multiplicity of infection, chilled at t = 35 min, and
pelletedat45min.Thepelletsfroma250-mlculture
were suspended in 0.5 to 1.0 ml of TMg, pH 7.4,
containing 10,tg of DNase, frozen at -70°C, and
thawedonce.Samples (20p1 each)weremixedat4°C
with either 20p1 ofTMgor20
pl
ofadifferentextract,incubatedat30°C foraminimumof 2h, and assayed
forphage.
Electronmicroscopy. Samplesofinfected-cell
ex-tracts prepared as described above were placed on
carbon-coated grids and negatively stained with 1%
uranylacetate.Gridswereexamined inaPhilips201
electronmicroscopeat60kV.
P1 transductions. Transductions using phage
PlkcwereaccordingtoMiller (19)orRothman (25).
F'transferexperiments.Cellsgrownin Hbroth
werematedataratio of 10donorcellsto 1recipient
cell for60minat37°Cwithout aeration. The mixture
was diluted and plated onM9 agarplates toselect
against themultiply auxotrophic donors. F-ductants
werepurifiedby streakingonM9platesandtestedby
thestreaktestfor theirabilityto supportfl andT4
growth.
RESULTS
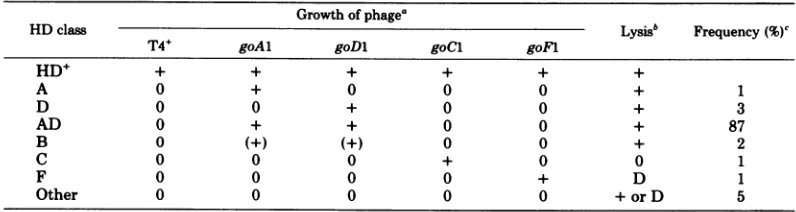
Selection and classification ofHD
bacte-rialmutants.HDmutants,isolatedascolonies
unabletopropagateT4orT6,weregrouped into
atleast sixclassesaccordingtothe plating
prop-erties of four standard go phage mutants that
wereselected forgrowthonfourprototype HD
mutant hosts (Table 1). Lysis measurements,
shown by Epsteinetal.
(6)
todifferentiatebe-tweenT4earlyand latenonsensemutants,were
usedtodistinguishthose bacteriathatcausedan
earlyarrestofT4development from those that
blocked morphogenesis. Mutants of classes A,
AD, D, and B showed normal lysis, suggesting
an assembly defect. Lysis didnotoccur in
T4-infected class C mutants and was delayed in
class F mutants. T4 DNA synthesis
measure-mentssupported these tentativeassignments of
phenotype: DNA synthesis was normal in the
putative assemblymutantsofclasses A, AD, D,
and B butdelayed and depressed inmutantsof
classes C and F, which exhibited defective lysis
(datanotshown).
When physiological and genetic studies
re-vealed thatmutantsfromallfour
assembly-de-fective classes blocked T4 development at the
samestage, twofurthermutantisolationswere
done, using either goA 1 (selection II) orgoDl:
goFl (selection III) in combination with T6as
selecting agents to favorthe detection ofrarer
classes of altered bacteria thatmight affect other
morphogeneticpathways. After these selections,
the majority AD class was eliminated; other
defined classes were enriched, and many
mu-tants (17to64%) appeared in the "other"
cate-gory(datanotshown). These mutants,
however,
didnot represent blocks in different assembly
pathways. Measurements of lysis,invitro
com-plementation analysis, and genetic studies
re-vealed thenewmutationstobe variations of the
samedefect(seeHD3.10,arepresentative of the
"other" class, below).
Growthandplating propertiesof
assem-bly-defective HD mutants. The ability to
propagateT4phagesand thegrowth properties
oftheassembly-defective mutantsdescribedin
thispaperareshowninTable 2.Althoughmost
HD strainsgrewnormally,someclass Dand all
class Bmutantsshowedtemperature-dependent
growthpatterns.Phage propagationinthesetwo
classes also appeared tobe influenced by
tem-perature: some strains were defective at high
temperatures but supported phage growth at
300C
(HDDO.18, HDD25.9, and HDD4.3),whereas others showed the converse behavior
(HDD3.6, HDD7.1, and HDB4.5).
Several observations indicated that the four
TABLE 1. Classification of HD
mutants
Growthofphage'
HDclass Lysisb Frequency (%)c
T4+ goAl goDi goCl goFl
HD+ + + + + + +
A 0 + 0 0 0 + 1
D 0 0 + 0 0 + 3
AD 0 + + 0 0 + 87
B 0 (+) (+) 0 0 + 2
C 0 0 0 + 0 0 1
F 0 0 0 0 + D 1
Other 0 0 0 0 0 + or D 5
a The fourstandard go phage mutants were isolated as large-plaque
formers
on the appropriate HD hoststrainsasdescribedin the text.Phage growth was determined by spot tests. Symbols: 0, no phage growth; +,
phagegrowth; (+), phage growth is better than T4+ by a factor of about 1,000.
bLysis measurementsaredescribed in the text.Symbols: +, lysis; 0, no lysis; D, delayedlysis.
"Atotal of279mutantsderived from 26subcultures were classified to yield 75 independent HD strains.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.504.41.439.500.606.2]TABLE 2. Properties of HD strains that block T4assembly
Bacteria Growth of phage
EOPb Burst sizec
Strain' Growth
T4+ goAl goDl T4+ goAl goDl
HD+ 1.0 1.0 1.0 186 185 152
HDA17.5 <10-6 1.0 <10-6 0.002 122 0.2
HDADiJ <10-6 1.0 1.0 0.01 102 41
HDD3.6 lo-4-lo-6 <10-6 1.0 0.3 0.002 34
HDDUYI ts 10-4_10-6 <10-6 1.0 1.0 0.004 89
HDD7.1 cs 10-4_10-6
<10-6
1.0 1.0 0.35 63HDDO.18d
10-4_10-6 <10-6 1.0 <0.1 0.001 20HDD25.9d 10-4_10-6 <10-6 1.0 0.2 0.003 23
HDB4.5 cs 10-4 <10-6 10-2 <0.1 NTe NT
HDB8.4 cs+ 1.0 <10-3 10-3 36 0.4 1.0
HDB17.3 ts <10-6 10-3 <10-6 0.002 NT NT
HD3.10 Cs 10-4 <10-6 10-4 0.1 NT NT
groEA44 ts <10-6 <10-6 1.0 0.03 NT 251
groEB515 1.0 1.0 10-5 107 NT 0.3
a
The
courseof T4 infectionwascharacterized in detail in the underlined strains.bEOP,Efficiency ofplating;measurements were at37°C unless otherwise specified.
'Phageadsorptionand
killing
werenormal.dEfficiency ofplatingand burst size measurementsat
420C.
e NT, Nottested.
assembly-defective classes of
HD mutants arerelated.
(i) Both goA and goD phages
grew onHDAD hosts. However, goA phages showed
alower
efficiency
of
plating than did T4+
on HDDhosts. This
property was mostobvious
atper-missive
temperaturesin host strains with
tem-perature-dependent
defectiveness;
for
example,
at
370C
onHDDO.18 and
HDD25.9, T4+
grewwell, but goAl showed
anefficiency of plating of
i0-'
(data
notshown). (ii) Both goAl and goDl
showed
efficiencies of
plating
about
103times
higher than that of T4+
onHDB
hosts. However,
a
cold-sensitive
(cs+) derivative of HDB8.4 that
had
regained the ability
topropagate T4+still
inhibited the growth
of both
go mutants. (iii)After selection with
goDl:goFl
in combination
with
T6 at300C,
many ofthe
survivors that wereHD
for both
T4and
T6under the
selection
conditions
propagated T4+
at370C
but
werespecifically
defective for
goAl, goDl, and goD3.
These
observations
suggestthat
the four
HDmutant classes impose a common block to T4
development.
Additional evidence for thissug-gestion
ispresented
below.The
ability of
otherphages
to grow onthe
assembly-defective
HD mutants wasinvesti-gated.
T2 and T6 behaved like T4.T3, T5, T7,
andP1 grew on all strains withone
exception:
T5
failed
to grow on HDB17.3. HD strainsHDB4.5,
HDB8.4cs+,
HDB17.3,
and HDD4.3inhibited the
growth
ofXcIb2,
butallowed
thegrowth
of its derivativeXeA30,
aphage
thatcompensates for many
groE
host mutations(8).
Course of
T4infection
inassembly-defec-tive
HD
mutants.The
courseof
T4infection
was
studied in
amutanthost of
each class. First,
in
vitro
complementation
tests werecarried
out todetermine
whether active structural
inter-mediates in
assembly
accumulate in
T4+-in-fected
HDstrains. Extracts of
T4+-infected HD
cells
weremixed with
either
particles
lacking tail
fibers
orwith
anextract(prepared by using the
appropriate
mutantphage [see above]) that
pro-vided heads and tail fibers
(tail defective)
ortails
and
tail fibers
(head
defective). The production
of infectious
phage
particles
inthese mixtures
was
assayed.
As
shown in Table
3,T4+ infection
of
representatives
of the four classes of
assem-bly-defective
HDmutantsproduced
active tail
fibers and tails
comparable in quantity
tothose
in
the head-defective
control, but
yielded
noactive heads. These results show that
inthe
HDhosts,
assembly
of tail
baseplates
and
fibers is
normal, whereas
assembly
of heads is defective.
The
extractsof
T4+-infected
HDcells used for
in vitro
complementation
also were examinedbyelectron
microscopy
using negative-staining
techniques.
Inagreement with the in vitrocom-plementation data,
the HD extracts containednormal
numbers of tails butno headsorhead-related structures. As
controls,
thehead-defec-tive and
tail-defective
extractsused inthecom-plementation
testsshowed normal numbers oftails
andheads, respectively (data
notshown).
The
phage
protein
geneproducts (gp)
labeledduring the
latter half of the latentperiod
afterT4+
infection of HD hosts wereanalyzed by
sodium
dodecyl
sulfate-polyacrylamide
gel
elec-370
REVEL ET AL. J. VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.504.74.466.78.260.2]TABLE 3. In vitrocomplementationwithextractsof
T4+-infectedHDstrainsa
Complementing N Tail fiber Tail de- Head de-prepn one defective fective fective T4++HDA17.5 0.4 355 544 4.7
T4++HDAD1.1 0.5 408 488 4.1 T4+ +HDB4.5 0.6 242 294 5.0 T4++HDD3.6 5.0 456 370 5.0 T4++HD3.10 0.3 25b 105 5.6 Head defective 4.1 290 487
Tail defective 0.3 413 Tail fiber 0.3
defective
aTwoextracts(50
ttl
ofeach) weremixed,incubated for300minat30°C and thenassayedforplaque-formingphage.
Results areexpressedasthe titer(active phagepermilliliter) x109 in the reaction mixture. Extracts ofphage-infectedcells wereprepared as described in thetext.The tailfiber-defective preparation (headand taildonor)wasparticles lackingtail fiberspurifiedfrom X4E-infectedcells;5 x10"particlesper ml were used in thecomplementation reactions. The tail-defectivepreparation(headand tail fiberdonor)was an ex-tractofSKB178infected withamN120(gene 27).The head-defectivepreparation(tailand tailfiberdonor)wasanextract ofSKB178 infected withamB17(gene 23).
bThe maximumpossiblevalue in thisexperimentwas50 because only5 x 1010 particleswereaddedtothe reaction
mixture.
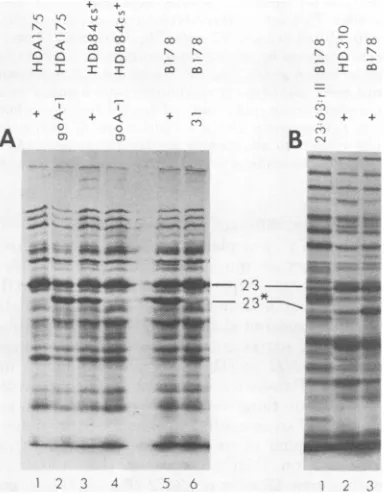
trophoresis (Fig. 1). In all
infections, the
major
capsid
protein (gp23
plus
its
cleavage
product
gp23*)
wasproduced
atapproximately
the
samelevel.
Inthe
permissive infections (T4+ infection
of
SKB178 and
HDB8.4cs+
andgoAl infection
of
HDA17.5), gp23
wascleaved
togp23*. In the
nonpermissive
infections [amN54 (gene 31)
in-fection
of
SKB178,
goAl
infection
of
HDB8.4cs+, and
T4+ infection of HDA17.5 and
HD3.10], all
of the
gp23remained uncleaved.
Similar results also
werefound in
T4+-infected
HDD3.6 and HDAD1.1. These
findings
supportthe conclusion that
all of the
HDstrains
pre-sumed
toaffect phage morphogenesis block
T4+development
at anearly
stagein
head assembly
at
the
level of
gene 31function (17). The data
also
suggestthat the mutation in
HDB8.4cs+
which
preventsthe
growth of
goAl probably
actsat
the
samelevel.
Genetic
analysis
of
go mutants.The
goAl
and goDl mutations
mapin
gene 31 ataninter-nal
site
neartsA70 and give
wild-type
recombi-nants
in
crossesof
goAl
by
goDl
(Fig.
2).Com-plementation
testswith
gene 31 ammutants
confirned
theassignment
togene 31 (data notshown).
Further crossesshowed thatgoDl
anda number of
spontaneous,
independently
iso-lated
gophage
mutants with identical platingpatterns, selected
onclassA, AD, or B mutanthosts, failed
to recombine withT4e.
This gomutant,
selected
previously ongroEA44
(9),hasgoDl
growth characteristics
at37°C
butdiffers
by
its
inability
to grow at 42°C.Similarly,
anumber of
independently isolated
mutantswith
goAl
plating
properties showed the
samefre-quency
ofrecombination with tsA70
asdid the
prototype
phage goAl.
Thus, goAl-like
andgoDl-like mutations
appear to recur atsites
identical
orclosely linked
tothose of the
original
mutations.
However,
notall
go mutantsselected
onHDAstrains
weresimilar
togoA1. Four
suchmutants,
isolated in selection
III, failed
toplate
onHDADO.1
orHDA17.5. One of these
novel
mu-tations
mapped
in gene 31but
gavewild-type
recombinants with both
goAl and goDl (percent
recombination
= 0.16and
0.17,respectively).
These results
areconsistent with
the view that
the different
plating
characteristics of
gomu-+ _ - cc cC < < co £
<0:Ca
= I I I
cc cc
c Co
* < + < + - + +
A
z
m
B2
a
_11P1
-,0_ _v,I - 23-- - - --23_2
- a.s3ii- -
-_
. 4.sw-. *_. 4d _
_
2 3 5%6 2 3
FIG. 1. Sodiumdodecylsulfate-polyacrylamidegel
electrophoresis ofextracts ofphage-infected cells.
Cells were infected withphage, labeled with
14C-amino acidsfrom13to24minafter
infection
at37°C,andprepared forsodium dodecyl
sulfate-polyacryl-amidegel electrophoresis as described in the text.
Samples containingabout40,000cpm were
electro-phoresedon10%polyacrylamideslabgelsand
proc-essed asdescribed in the text. Phageand bacteria
used in thepreparationofextractsarelisted above
each track: "+"means T4+; "31" refers tophage
carrying amN54 (gene31),and "23:63:rII"refersto
phage carrying amB17, amM69, and rEDdf4l;
"B178"referstoSKB178. 23and23*designate,
re-spectively, theproduct ofgene 23 and the cleaved
productof gene 23. (A) and (B) aregelsfrom two
different experiments.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.504.45.238.77.190.2] [image:6.504.250.442.251.500.2]372 REVEL ET AL.
gene 31 pseT
4) I
1.09
rm 30
4-1.50 1.95
0.3 _ 0.80
[image:7.504.121.415.63.197.2]0.80
FIG. 2. Genetic location of goAl and goDI. Themapof thegene31regionofthe T4genomeisfromthe
dataof Reveland Lielausis(23)and showsthepositions of amNG71 and amN54 ingene31relativetooutside
markerspseT (to the left) and rIII andgene30(totheright),aswellasrecombinationpercentagesbetween
amNG71 and amN54, amN54 andr67,andamNG71 and r67.Thetoplineindicates the relative order of the genes. Thesecond line isthegeneticmap showing the positions of phagemutations. Each number in the
figureis the observedpercentrecombination inacross betweenmutants atthe ends of thecorresponding
arrows. Twosetsof three-factorcrosses[(i)r67(rIII):amN54 with T4e, goDl, amNG71, and tsA70; and (ii)
r67(rHII):tsA70
(gene 31)withT4e, goD1,amN54 andamNG71]andatwo-factorcrossbetween T4eandgoD)wereanalyzed by plating totalprogenyonCR63 andwild-typerecombinantsongroEB515at42°C,conditions
under whichgoDl, T4e, tsA70andammutants cannotgrow.Inathree-factorcrossbetweenr67(rIII):tsA70
andgoAl, wild-type recombinantswereassayedonHDD4.3at30°C, wheregoAland tsA70failtoplate.In
a crossbetweengoAl andgoDl, wild-typerecombinantswereassayedonHDB8.4cs+ at37°C. Theratio of
wild-typeprogeny with therphenotypetototalwild-typeprogenypermitted ordering ofthemutantalleles
with respecttotheoutsidemarkerr67 ingenerIII. Recombinationpercentages arecalculatedasfollows:
(wild-typerecombinants/totalprogeny)x200. Nowild-typerecombinantswerefoundinthecrossbetweenT4E
andgoDl.
tantsreflect different mutationalsites.
When T4+was
plated
onHDD hoststrains,two distinctgo mutantplaque types were
ob-served. Large plaques similartothose ofgoDl
appeared at a frequency of -10-6, and small
plaques appearedatafrequency of
_10-4
(Table2). Plating patterns ofthree suchsmall-plaque
isolates, goD2, goD3, and goD4, are shown in
Table 4.Two-factor crosses showedthat these
newgomutationswerenotingene31 butwere
closely linkedtoamB17 ingene23. More
defin-itive mapping of one of these mutants, goD3
(selected on HDD3.6), placed this mutation
within gene 23near amB272 (Fig. 3). Thus,go
phage mutants with alterations affecting the
geneforthemajor
capsid protein
alsocanover-come the assembly block in some HDD host
strains. Two experiments showed that goD3
doesnotsimply bypass the requirement forgp3l
function: (i)goD3:amN54 (gene 31) double
mu-tants grew on HDD3.6 only when gp3l was
supplied bya
complementing
phage, and (ii) inmixed infection of HDD3.6 withgoD3:amN54
andgoD3invarying ratios, the phage yieldwas
afunction ofthelevelofwild-type gp3lpresent
(datanotshown).
Inability of T4 go mutants to grow on
some HD strains. As noted inTable 2, some
go phage mutants selected on specific HD
strains failtogrowoncertainother
temperature-dependent HD strains at low temperatures at
TABLE 4. Plating properties of various goD phage
mutants
Bacterial Growth of phagea
strain
T4+
goDl
goD2goD3b
goD4HD+ + + + + +
groEA44 0 + 0 0 0
HDDO.18c 0 + + + 0
HDD3.6 0 + 0 + +
HDD4.3 0 + + + +
HDB4.5 0 0 0 + 0
aPhage growthwas determined by aspottest at
37°C as described in the text. Symbols: +, phage
growth;0,nophagegrowth.
bThe burst size ofgoD3onSKB178(HD+)wasthe
same asthatof T4+(-150).OnHDD3.6, the burst size
ofgoD3wasreducedto -30.
cSpottest at42°C.
which
wild-type
T4 can grow.Similarly,
wefound
thattsA70
(gene 31)
did notplate
onHDDO.18,
HDD25.9,
and HDD4.3at30°C.
Thegrowth of
tsA56(gene
31)
wasinhibitedonly
onthe
last
host.Genetic
analysis
ofhdh strains.
P1trans-duction studies
showed
that most of the hostmutations
cotransduced
withpurA
withfre-quencies
ranging
from8.5 to 17.3%for differentmutations
(Table
5 andFig. 4).
Twostrains,
HDB4.5 and
HDD7.1,
wereclearly
different
fromtherest:therewasless than 1% cotransfer
of
the HD mutation with thepurA+
marker.on November 10, 2019 by guest
http://jvi.asm.org/
VOL. 33, 1980
20 2 22 23 24
Gene
1. , II I I I I
t
g0oD-3
I -- Mutant
I_
4.55 _ 2.95 _ 1.63 59-.93- .88 _5.43 - -2.21 .-738_
3.31 _ 1.28
_ 5.37 4 1.63 _
___
~ ~ ~
---.0
5.92
4 2.45-
-3.37 _
3.53
.04 .( .78
-1.6
3.5
6.6
13.6 I.5
FIG. 3. Geneticmapofthegene23region ofthe T4genome.Thetoplinegivesthe relative order andsizes ofgenes 21 to 23 (41). The second line is the genetic map showing theposition of am mutations. (a)
Recombination percentages betweenmutations ingenes20,22, 23,and 25. Geneticcrosses weredone inCR63
under standard conditionsdescribed in the text. Totalprogenywasdetermined by platingon CR63.
am'
recombinants wereassayedonS/6/5. Recombinationpercentages arecalculatedasfollows: (am+
recombi-nants/totalprogeny)x200 andareaveragesobtained in twoor more crosses.(b)Recombinationpercentages
from thecrosses betweengoD3:amXdoublemutantswithwild-type phage. goD3:amXdouble mutantswere
constructedasdescribedin the text. ThegoD3:amXdouble mutantswere backcrossed towild-type T4+ in
CR63 under standardconditions.TotalprogenyweredeterminedbyplatingonCR63.goD3:am+recombinants
wereassayedonHDD3.6at37°C,wherewild-typeandamphagecannotgrow.Recombinationpercentages
arecalculatedasfollows: (goD3:am+recombinants/totalprogeny) x200.
Transfer of either of the F' factors, F'117 or
F'118, into these strains or into HDD3.6
ren-dered thehosts abletosupportT4+growth. This
result indicates that the wild-type alleles are
probably dominant and that the HD mutations
inallthree of these strainsmustlie between the
ends ofthe F'117 factor. P1transduction
exper-iments with melA andpyrB (Fig. 4) markersto
locate these mutations moreprecisely havenot
been done.
DISCUSSION
Bacterial hdh mutations block an early
step inhead assembly. Our combined
physi-ological and genetic data show that the HD
mutations describedhere block T4
morphogen-esisatthelevel ofgene31action.After infection
ofthese strains byT4+, neitherheadsnor
head-relatedstructures are made, and headproteins
are not cleaved as in normal infection. This
phenotype is distinct from that ofanassembly
core defect (27), but is the same as observed
after infection of wild-type (nonsuppressing)
hostsbygene31ammutants(17). It is also the
same asthe phenotype reportedforT4+
infec-tion of the previously described mutant host
strainsmop(37), groEA44 (9),tabB(3, 36),and
hdB3(31).Aswasreportedforthesestrains,we
find that mutationsin T4 gene31can
compen-satefor thehostdefect,and that certaingene31
tsmutants(kmutants[3, 36])interactnegatively
withcertaintemperature-dependentHD mutant
hosts, in that the k mutantphagefail to
propa-gate underconditions whereT4+ phage cando
so. Therefore, the HD mutants, like those
de-scribedpreviously, appeartointerfere with the
normal function of T4 gp3l in head assembly.
However,we haveshown, in addition, thata
mutationingene23, goD3, alsocancompensate
for thehost defect in certain HD strains suchas
HDD3.6. Gene23codesforthemajor T4 capsid
protein, which presumably must interact with
gp3l in ordertoassemblecorrectly (17).In
pos-sibly analogousmutants ofphage X,mutations
C
I
a
L
b
N67(25)on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.504.49.440.48.322.2]374 REVEL ET AL.
in gene E (major capsid protein) restore phage
growth on certain HD groE
bacterial
mutantsapparently
bylowering
the amount of gpEpro-duced,
thereby restoring
arequiredbalance
be-tween the levels of
this
protein
andthe
hostgroE function
(8,
34).
However, it appears
un-likely that
the goD3mutation
in T4 acts byreducing the
amountof functional
gp23pro-duced.The
goD3 mutation
liesfar
from the
gene23 promoter (Fig. 3), and goD3 phage give a
normal
burst size
onwild-type host
strains (Ta-ble4).Furthermore,
severalgene 23am
mutantswere found not to grow in a
HDD3.6 supE
host(data
notshown),
inwhich
gp23should be
un-TABLE 5. Frequency of Pl cotransduction of the
HDphenotype of HD strains with purA+a
HDstrain HD
transductants/
Cotransduction(donor) PurA+transductants (%
tested
HDD3.24b 16/188 8.5
HDA17.5 19/200 9.5
HDD4.3 8/75 10.6
HDAD1.1 22/200 11.0
HDB17.3 24/200 12.0
HDB8.4cs+c 29/200 14.5
HDDO.18d 51/299 17.1
HDD3.6 52/300 17.3
HDD7.1 0/90 <1.0
HDB4.5 0/199 <0.5
groEA44 22/200 11.0
aPlkc was grown onthe HD strains and usedto
transduce T832purAasdescribedinthetext.purA
transductants were selectedonsupplemented minimal
medium in the absence ofpurines,
purified,
andtestedby astreak test at
370C
for theability togrowT4+(except HDB8.4cs+ [see footnote
c]).
Frequencies ofpurA+transductants rangedfrom5 x 10-7to 100 x
10-7indifferent experiments.
bThe testfor host defectivenesswas at
300C.
CHDB8.4cs+
is sensitive toT4+ butdefective forgoA 1;therefore, goA1wasusedtoscreenthepurA +
transductants for hostdefectiveness.
dThe testfor host defectivenesswasat
420C.
J. VIROL.
derproduced because
of thelow
efficiency ofthesupE
suppressor.We
have also
presentedevi-dence that the
goD3
mutationdoes
notsimply
bypass
the normal requirement for gp3lfunc-tion.
Therefore,
themostlikely interpretation
isthat the HDD3.6 host defect can be overcome
by
aspecific change in either gp23 or gp3l of T4,implying
interaction between these proteins andone ormorehost functions in the first step of T4
head assembly. This view is supported by
the
findings
that k mutations, as well ascompensat-ing mutations, can occur in gene 23 andthat the
corresponding k mutants can be
used
to selectspecifically for tabB-defective host mutants (36).
hdh mutations define at least two
hostfunctions. Most of the hdh
mutations
tested
canbe
cotransduced
with thepurAmarker,
ashas been
reportedfor
mop(37),
groE (7), and tabB(36).
Inourexperiments, thecotransduc-tion frequencies of various hdh mutants range
from 8.5 to 17.3%, but show two apparent
clus-ters
(HDD3.24,
HDA17.5, HDD4.3,HDAD1.1,
HDB17.3,
and groEA44 andHDB8.4,
HDDO.18, andHDD3.6)
with means ofabout 10 and 16%,respectively.
Data inthe
literature
for mop,groE, and tabB mutants show
similar
rangesand tendencies to cluster around 10 and 21%.
The finding of two apparent
frequencies
couldbe due to an effect of somehdh
mutations
onthe recovery of
purA+hdh transductants
togivea lowerapparent cotransduction
frequency.
Al-tematively,
the twofrequencies
could representtwodifferent sites at whichhdh mutations occur.
From thedifference in transduction
frequency,
the two sites would be about10i nucleotide pairs
apart
and,
therefore,
probably
indifferent
genes.This
hypothesis
could betested
by genetic
com-plementation
analysis. Of interest
inthis
con-nection is a recent report that a A
transducing
phage
carrying 8,000
basepairs of bacterial
DNA,
including
a mutantgroE
gene cannotgrow on somegroE hosts but can grow on others,
___, , , | , , § , I§ , , (Eco/i 90 91 92 93 94 95 chromosome
4 F117 F118 F
FIG. 4. Locationofhdh mutationsontheE.coli chromosome. The upper line shows the90-95-minsegment
ofthe E.coli chromosome withrelevantmarkers
(2).
Theheavier lines below the chromosome show theextentsofF'factorsF'117 andF'118basedonthe data
of
Low(18)
and Takano andKakefuda (37),
asadjusted
tocorrespondtothemost recentmap
of
the E.coli chromosome(2).
The bracket above the chromosome indicatesthe location
of
mostof
the mutations inourHD strainsasobtainedby
P1cotransductionof
hostdefectiveness
withthepurA + marker (Table 5). Cotransductionfrequencieshave been convertedtominutesontheE.coli
chromosomebytheequationofWu(42)asdescribed inreference2.
Iq
R
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.504.66.257.241.375.2] [image:9.504.145.385.512.587.2]HEAD
suggesting that
twocomplementing groE
genesmay
be
present(10).
In
addition
tothe
purA-linked
mutations,
we havefound
two hdh mutations that are notcotransduced with
purA,
although F' transfer
experiments
showthat
they
are inthe
sameregion
of the
bacterial
chromosome.The defects
instrains HDB4.5 and HDD7.1 can be overcome
by the phage mutations
goD3
andgoD1,
respec-tively.
Therefore,
hdh mutations
mayoccurin
three
or morehostgenes.To
complicate
thepicture still
further,
twoother
HDbacterial
mutantsthat block
T4head
assembly and
canbe
overcomeby
mutations in
T4 gene 31
have been
reported. These
mutantsdiffer from
ourcharacterized
hdh
mutantsand
from
mop,groE, and tabB
mutantsin that
oneof them
mapsnearpro onthe
E.coli
chromo-some
(31) and the other shows
adissimilar
T4-defective
phenotype (32).
We haveselected
gomutants on
the latter
strain,
hd590,
kindly
fur-nished
to usby L. D. Simon. Most of the
corre-sponding mutations do
notmap near gene 23 orgene 31. A
few
map in gene31,
butthese
mutantsfail
togrow on any of ourHDA, HDAD, HDB,
or HDD
strains
(Stitt, unpublished
observa-tions).
Acomprehensive
model for the
interac-tion of
gp3l with host functions
mustaccount forthese
HD mutants aswell
asthe
otherclasses
described here.
go
and hdh mutations
probably
define
specific
phage-host protein
interactions.
Two
kindsof models
canbe
proposed
toexplain
the
interactive
systems ofhost and
T4phage
mutations described here and
by
others
(3, 9,
31, 36,37).
Theinteraction could be indirect; for
example, host mutations might alter membrane
transport
properties
so as tocause achange in
the
intracellular
ionicenvironment that could
prevent
the
gp3l-mediated
stepin
T4head
as-sembly.
Alternatively,
the interaction
could be
direct; hdh mutations
might
block head
assem-bly
by
altering
ahost
protein
so as toprevent arequired specific association of
gp3l
and
gp23with
ahost
protein complex.
Ineither model,
compensating mutations
inphage
gene 23 or 31could
overcomethe block.
From
the evidence available
sofar,
wecannotrule
outeithermodel. Indirectinteraction
seemsto
be
supported
by
ourfindings
that: (i) a single phage mutation in gene 31 can overcome several host defects that may represent mutations inthree different host genes, and
(ii)
certainhostdefects can be overcome
by
mutations in eitherof two
phage
genes. Either model would beconsistent with the
findings
of bothcompensa-tory(go) and
killing (k)
mutations in both genes23and 31 of T4: in somecases, a phage mutation
cancompensate
directly
orindirectly
foradet-rimental host
mutation, whereas in other
cases, aphage mutation and
ahost
mutation, both
innocuous
alone,
caninteract
either
directly
or indirectly to give a lethal phenotype.However,one aspect
of
thedata
supportsthetype
of direct-interaction model
proposed by
Takahashi (36),
involving
aspecific
activecom-plex
ofphage
andhost
proteins
required for
phage head
assembly. Host
mutantsthat appearrelated on the
basis of similar
cotransduction
frequencies
with purA+
showdifferent patternsof
compensation
by different classes of
gomu-tant phage.
Conversely, different
gomutationsor
k mutations in
aparticular
geneshow
differ-ent
plating
patterns onthe
HDstrains.
Inother
words,
the
properties of hdh,
go,and k
mutations
appear to
be allele
specific
rather thanclass
specific
orgenespecific.
Thisfeature would
beless likely if the HD mutations
causealterations
in a
physiological
parameter forwhich
the gomutations
can compensate.Allele specificity
would
be morelikely
ifthese mutations
arecausing
compensating conformational changes
in an
interacting
complex of
proteins. Fromthe
observationsreported here, the
functional
com-plex might consist of
atleast three
host proteinsand the viral
proteinsgp3l and
gp23. Theap-parent
ability of compensating mutations
tooc-cur
in
anyphage-host pair of these
componentswould
seempuzzling. However,
apossibly
anal-ogous
observation
has been made with
themul-timeric regulatory
enzyme aspartatetranscar-bamylase: mutational changes
inthe regulatorysubunit affect
thespecificity
ofthe
catalytic
subunit
active
site, solely by interaction
betweenthe
heterologous subunits
(26).The
recentiden-tification
of ahost
protein defined by
agroE
mutation
(10,
11) should lead
to moredirect
tests
for
theexistence of
afunctional phage-host
protein complex in head assembly.
ACKNOWLEDGMENTS
These studieswere supportedby Public Health Service grantAI-09238 from the National Institutes of Health and by
special grant no. 573 from the American Cancer Society, CaliforniaDivision,toW.B.W.B.L.S.wassupportedby Public Health Servicetraining grant GM-00086 from the National Institute of Health.
The technical assistance of Marie Beall isgratefully ac-knowledged.
LITERATURE CITED
1. Adelberg,E.A.,M.Mandel,andG. C.C.Chen.1965. Optimal conditions for mutagenesis by N-methyl-N'-nitro-N-nitrosoguanidine in Escherichia coli K12. Bio-chem.Biophys. Res. Commun. 18:788-795.
2. Bachmann,B.J.,K.B.Low, and A. L. Taylor. 1976. Recalibrated linkage map of Escherichia coli K-12. Bacteriol. Rev. 40:116-167.
3. Coppo, A., A. Manzi,J. F.Pulitzer, andH.Takahashi. 1973.Abortivebacteriophage T4 head assembly in mu-tantsofEscherichia coli. J. Mol. Biol.76:61-87.
on November 10, 2019 by guest
http://jvi.asm.org/
4. Dickson, R. C. 1973. Assembly of bacteriophage T4 tail fibers. IV. Subunit composition of tail fibers and fiber precursors. J. Mol. Biol. 79:633-647.
5. Edgar, R.S.,and W. B. Wood. 1966. Morphogenesis of bacteriophage T4 in extracts ofmutant-infectedcells.
Proc. Natl. Acad. Sci.U.S.A. 55:498-505.
6. Epstein, R. H., A. Bolle, C. M. Steinberg, E. Kellen-berger, E. Boy de la Tour, R. Chevalley, R. S. Edgar, M.Susman, G. H. Denhardt, and I. Lielau-sis. 1963. Physiological studies of conditional lethal mutants ofbacteriophage T4D. Cold Spring Harbor Symp. Quant. Biol. 28:375-392.
7.Georgopoulos, C. P., and H. Eisen. 1974. Bacterial mutants which block phage assembly. J. Supramol.
Struct. 2:349-359.
8. Georgopoulos, C. P., R. W. Hendrix, S. R. Casjens, and A. D.Kaiser.1973.Hostparticipation in bacterio-phage lambda head assembly. J. Mol. Biol. 76:45-60. 9. Georgopoulos, C. P.,R. W.Hendrix,A. D. Kaiser, and W. B. Wood.1972.Role of thehostcellin bacte-riophage morphogenesis: effects of a bacterial mutation onT4head assembly. Nature (London) New Biol. 239: 38-41.
10. Georgopoulos, C.P., and B. Hohn. 1978.Identification
of a hostproteinnecessaryfor bacteriophage
morpho-genesis (groE).Proc. Natl. Acad. Sci. U.S.A. 75:131-135.
11. Hendrix, R. W.,andL. Tsui. 1978. Role of the host in virus assembly: cloning of the Escherichia coli groE gene and identification of its protein product. Proc. Natl. Acad. Sci.U.S.A.75:136-139.
12. Hoffman, E. P., and R. C. Wilhelm. 1970. Genetic
mappingand dominance of the amber suppressor,sul (supD), in Escherichia coli K-12. J. Bacteriol. 103:32-36.
13.Kellenberger, E., and J.Sechaud.1957.Electron
mi-croscopicalstudies ofphagemultiplication.II. Produc-tion ofphage-relatedstructuresduringmultiplicationof phages T2 and T4.Virology3:256-274.
14. Kikuchi,Y., and J.King.1975.Genetic control of
bac-teriophage T4baseplate morphogenesis. II. Mutants
unabletoform the central part of thebaseplate.J.Mol. Biol.99:645-672.
15. King, J. 1968.Assemblyof thetail ofbacteriophageT4. J. Mol. Biol. 32:231-262.
16.Laemmli, U. K. 1970. Cleavageof structural proteins duringtheassemblyof the head ofbacteriophageT4. Nature(London)227:680-685.
17.Laemmli,U.K.,F.Beguin,and G. Gujer-Kellenber-ger. 1970.Afactorpreventingthemajorheadprotein
ofbacteriophageT4from randomaggregation.J.Mol.
Biol. 47:69-85.
18. Low,K. B. 1972.Escherichia coli K-12F-primefactors,
old andnew.Bacteriol.Rev. 36:587-607.
19. Miller, J. H. 1972.Experiments inmolecular genetics.
ColdSpringHarborLaboratory, ColdSpring Harbor,
N.Y.
20. Pulitzer, J. F., and M.Yanagida. 1971. Inactive T4 progeny virus formation inatemperature-sensitive mu-tantofEscherichiacoli K12.Virology 45:539-554.
21. Revel, H. R.1967.Restriction ofnonglucosylatedT-even bacteriophage. Properties of permissive mutants of Escherichia coli B and K12.Virology31:688-701. 22. Revel,H.R.,R.Herrmann, and R. J.Bishop. 1976.
Geneticanalysisof T4 tail fiberassembly.II. Bacterial hostmutantsthat allowbypassof T4gene57function. Virology 72:255-265.
23. Revel,H.R.,andI.lielausis.1978.Revised location of therIII geneonthegeneticmap ofbacteriophageT4.
J.Virol. 25:439-441.
24. Revel, H. R., and S. E. Luria. 1970. DNA glucosylation in T-even phage: genetic determination and role in phage-host interaction. Annu. Rev. Genet. 4:177-192. 25. Rothman, J. L. 1965. Transductionstudies on the rela-tionbetween prophage and host chromosome. J. Mol. Biol.12:892-912.
26. Schachman, H. K. 1972. Structure, function and dynam-ics of a regulatoryenzyme-aspartate transcarbamylase, p. 17-56. In R. Jaenicke and E.Helnrich(ed.), Protein-protein interactions.Springer-Verlag, Berlin. 27. Showe, M. K., andL W. Black. 1973. Assembly core of
bacteriophage T4: an intermediate in head formation. Nature(London) New Biol.242:70-75.
28. Showe, M. K., and E. Kellenberger. 1975. Control mechanisms in virus assembly, p. 407-438. In D. C. Burke and W. C. Russell (ed.), Controlprocesses in virusmultiplication.Cambridge University Press, New York.
29. Simon,L. D.1969.The infection of Escherichia coliby T2 and T4bacteriophages as seen in the electron mi-croscope. III. Membrane-associated intracellular
bac-teriophages.Virology 38:285-296.
30. Simon,L. D. 1972. Infection ofEscherichiacoli by T2 and T4bacteriophages as seen in the electron micro-scope: T4head morphogenesis. Proc. Natl. Acad. Sci. U.S.A. 69:907-911.
31. Simon, L. D.,T. J. M.McLaughlin,D.Snover,J.Ou,
C. Grisham,and M. Loeb.1975.E. coli membrane
lipidalteration affecting T4 capsid morphogenesis. Na-ture(London)256:379-383.
32. Simon,L. D., D.Snover, andA. H.Doermann.1974. Bacterial mutation affecting T4phageDNAsynthesis andtailproduction. Nature(London)252:451-455.
33. Steinberg, C. M., andR.S.Edgar.1962.Acriticaltest of a currenttheory of genetic recombination in bacte-riophage. Genetics 47:187-208.
34. Sternberg, N. 1973. Propertiesofamutant of Esche-richiacoli defective in bacteriophage A head formation (groE). II. Thepropagationofphage A. J. Mol. Biol. 76:25-44.
35. Studier,W. F.1974.AnalysisofbacteriophageT7early
RNAsandproteinsonslabgels.J.Mol. Biol. 79:237-248.
36. Takahashi, H.,A.Coppo,A.Manzi, G. Martire,and J.Pulitzer. 1975.Designofasystem ofconditional lethalmutations(tab/k/com) affecting protein-protein
interactions inbacteriophage T4-infected Escherichia
coli. J.Mol.Biol.96:563-578.
37. Takano,T.,andT. Kakefuda. 1972.Involvement ofa
bacterialfactor inmorphogenesisofbacteriophage
cap-sid.Nature(London)New Biol.239:34-38.
38. Wilson,J. H., and S.KeIls. 1972.Bacteriophage T4 transfer RNA. I.Isolation andcharacterizationoftwo
phage-codednonsensesuppressors. J.Mol. Biol.
69:39-56.
39. Wood,W.B.,R.C.Dickson,R.J.Bishop,and H. R. Revel. 1973. Self-assembly and non-self-assembly in bacteriophage T4morphogenesis,p.25-58.InR. Mark-ham (ed.), The generation ofsubcellular structures, FirstJohnInnesSymposium.NorthHollandPublishing
Co., Amsterdam.
40. Wood,W.B.,R.S.Edgar,J.King,I.Lielausis,and
M. Henninger. 1968. Bacteriophage assembly. Fed.
Proc.Fed.Am.Soc.Exp.Biol. 27:1160-1166. 41. Wood, W.B., and H. R.Revel. 1976.Thegenome of
bacteriophageT4.Bacteriol. Rev.40:847-868. 42.Wu, T. T. 1966. A model for three-point analysis of
randomgeneraltransduction.Genetics54:405-410.