I-Dependent Innate Immune Response
Changzhi Xu,a,bXiang He,aZirui Zheng,a,bZhe Zhang,aCongwen Wei,aKai Guan,aLihua Hou,aBuchang Zhang,bLin Zhu,b Yuan Cao,cYanhong Zhang,aYe Cao,aShengli Ma,aPenghao Wang,aPingping Zhang,a,cQuanbin Xu,aYouguo Ling,aXiao Yang,a Hui Zhonga
Beijing Institute of Biotechnology, Beijing, Chinaa
; Institute of Health Science, School of Life Sciences, AnHui University, Hefei, Anhui, Chinab
; Department of Laboratory Medicine, General Hospital of Jinan Military Region, Jinan, Shandong, Chinac
ABSTRACT
Retinoic acid-inducible gene I (RIG-I) is an intracellular RNA virus sensor that induces type I interferon-mediated
host-protec-tive innate immunity against viral infection. Although cylindromatosis (CYLD) has been shown to negahost-protec-tively regulate innate
antiviral response by removing K-63-linked polyubiquitin from RIG-I, the regulation of its expression and the underlying
regu-latory mechanisms are still incompletely understood. Here we show that RIG-I activity is regulated by inhibition of CYLD
ex-pression mediated by the microRNA miR-526a. We found that viral infection specifically upregulates miR-526a exex-pression in
macrophages via interferon regulatory factor (IRF)-dependent mechanisms. In turn, miR-526a positively regulates
virus-trig-gered type I interferon (IFN-I) production, thus suppressing viral replication, the underlying mechanism of which is the
enhancement of RIG-I K63-linked ubiquitination by miR-526a via suppression of the expression of CYLD. Remarkably,
virus-induced 526a upregulation and CYLD downregulation are blocked by enterovirus 71 (EV71) 3C protein, while ectopic
miR-526a expression inhibits the replication of EV71 virus. The collective results of this study suggest a novel mechanism of the
regu-lation of RIG-I activity during RNA virus infection by miR-526a and suggest a novel mechanism for the evasion of the innate
immune response controlled by EV71.
IMPORTANCE
RNA virus infection upregulates the expression of 526a in macrophages through IRF-dependent pathways. In turn,
miR-526a positively regulates virus-triggered type I IFN production and inhibits viral replication, the underlying mechanism of
which is the enhancement of RIG-I K-63 ubiquitination by miR-526a via suppression of the expression of CYLD. Remarkably,
virus-induced miR-526a upregulation and CYLD downregulation are blocked by enterovirus 71 (EV71) 3C protein; cells with
overexpressed miR-526a were highly resistant to EV71 infection. The collective results of this study suggest a novel mechanism
of the regulation of RIG-I activity during RNA virus infection by miR-526a and propose a novel mechanism for the evasion of the
innate immune response controlled by EV71.
E
V71 is a positive-stranded RNA virus which belongs to the
picor-navirus family (
1
) and is the causative agent of
hand-foot-and-mouth disease (HFMD) in young children and infants. The genome
of EV71 is approximately 7.5 kb in length and contains a single open
reading frame encoding a polyprotein precursor, which is processed
into structural (VP1, VP2, VP3, and VP4) and nonstructural (2A, 2B,
2C, 3A, 3B, 3C, and 3D) proteins during viral infection (
2
). Despite
the protective role of type I interferon (IFN-I) in EV71 infection,
EV71 inoculation is unable to elicit production of these interferons.
Most members of the picornavirus family, including poliovirus,
rhi-novirus, echovirus, and encephalomyocarditis virus, use strategies to
inhibit IFN-I induction by interfering with melanoma
differentia-tion-associated gene 5 (MDA-5) and retinoic acid-inducible gene I
(RIG-I) (
3–5
) or by restricting IFN secretion through repression of
the cellular secretory pathway (
6
). Recent studies revealed that the 3C
protease of EV71 associated with RIG-I and cleaved TRIF
(TIR-do-main-containing adapter-inducing interferon beta) and IRF7
(inter-feron regulatory factor 7) (
7
,
8
); moreover, EV71 inhibited
IFN-I-induced ISGs (interferon stimulating genes) in host cells by reducing
IFNAR1 (type I interferon receptor 1) levels in host cells (
9
).
How-ever, additional work is required to understand the mechanisms by
which EV71 is able to escape from innate antiviral responses.
IFN-I, as the first line of host immune response, is critical in
mediating antiviral defense. The host senses viral and bacterial
pathogen invasion by recognition of pathogen-associated
molec-ular patterns with pattern recognition receptors, including
mem-brane-bound TLRs (Toll-like receptors) (
10
,
11
) and cytosolic
sensory molecules, such as the multidomain-containing NOD
(nucleotide-binding oligomerization domain) proteins, RIG-I,
and MDA-5 helicases (
12–14
). Both RIG-I and MDA-5 contain
caspase recruitment domains (CARDs) that interact with the
CARD domain-containing protein mitochondrial antiviral
sig-naling (MAVS) upon binding to uncapped RNA, resulting in
MAVS association with I
B kinase (IKK) proteins. While MAVS
association with IKK
␣
/

activates NF-
B (nuclear factor-
gene
Received14 May 2014Accepted8 July 2014
Published ahead of print23 July 2014
Editor:B. Williams
Address correspondence to Xiao Yang, xyang@nic.bmi.ac.cn, or Hui Zhong, towall@yahoo.com.
C.X, X.H., Z. Zheng, and Z. Zhang contributed equally to this work.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01400-14
on November 7, 2019 by guest
http://jvi.asm.org/
binding), its association with TBK1 (TANK-binding kinase 1) as
well as IKK
ε
leads to the activation of IRF-3/IRF-7; coordinated
activation of the NF-
B and IRF pathways further results in the
assembly of a multiprotein enhancer complex that drives the
ex-pression of IFN-

and the IFN-mediated antiviral immunity
(
15–19
).
RIG-I signaling is negatively regulated at multiple levels.
Pre-vious reports showed that the ubiquitination status of RIG-I is
controlled by CYLD, a tumor suppressor originally identified as a
genetic defect in familial cylindromatosis (
20
). Indeed, CYLD was
shown to interact with the CARDs of RIG-I and to remove
K63-linked polyubiquitin chains from RIG-I, which inhibits
down-stream signaling. DC (dendritic cells) lacking CYLD constitutively
polyubiquitinate RIG-I and show enhanced activity of TBK1 and
IKK
ε
, suggesting that CYLD regulates basal RIG-I activity by
modulating its K63-polyubiquitin status (
21
). CYLD also acts as a
negative regulator of NF-
B and Jun N-terminal kinase signaling
pathways by removing Lys-63-linked polyubiquitin from NEMO
(NF-
B essential modulator), IKK
ε
, TRAF2 (TNF receptor
asso-ciated factor 2), or BCL3 (B-cell CLL/lymphoma3) (
22–25
). These
findings thus establish CYLD as a critical regulator of antiviral
innate immune response.
MicroRNAs (miRNAs), an abundant class of highly conserved
noncoding RNA oligonucleotides (18 to 25 nucleotides [nt] long),
suppress gene expression by binding to the 3
=
untranslated region
(UTR) of target mRNAs. MiRNAs play key roles in the regulation
of diverse biological processes. Recently, a role for miRNAs in the
regulation of innate immune responses in monocytes and
macro-phages was proposed (
40
). Direct roles of miRNAs in innate
im-mune responses were discovered in a study that identified
miR-146a as a negative-feedback regulator in RLR signaling by
targeting IL-1R-associated kinase (IRAK) 1 and TNF
receptor-associated factor 6 (TRAF6) (
26
). Further reports showed that
both miR-155 and miR-132 were induced in a monocyte cell line
treated with the TLR4 ligand lipopolysaccharide (LPS) (
27
). Given
the important roles of the RIG-I signaling pathway in the innate
antiviral immune response, identifying more miRNAs that can
regulate RIG-I-dependent IFN-I production is of vital
impor-tance. In fact, many viruses have evolved strategies to interfere
with these innate signaling events and hence inhibit IFN-

pro-duction. However, to date, there are few reports about the
regula-tion of RIG-I signaling pathway by miRNAs, especially during
EV71 infection.
In the present study, we found that miR-526a was significantly
upregulated in macrophages upon viral infection in an
IRF-de-pendent manner. Then we demonstrated that miR-526a feedback
positively regulated vesicular stomatitis virus (VSV)-triggered
IRF3 activation by suppressing CYLD expression and subsequent
RIG-I ubiquitination. Furthermore, we found that miR-526a
up-regulation was blocked by EV71 3C protease, whereas ectopic
miR-526a expression inhibited the replication of EV71. Thus, the
present study demonstrates for the first time that miR-526a is a
positive feedback regulator of the RIG-I signaling and that EV71
targets miR-526a to suppress RIG-I-dependent IFN-I production.
MATERIALS AND METHODS
Cell culture and transfection.293T, Vero, RD, MDCK, and THP-1 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) and RPMI 1640 (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen), 100 U/ml penicillin, and 100 mg/ml
streptomy-cin. Vectors and epitope tagging of Flag-tagged IRF3, IRF7, p65, MDA-5, RIG-I, N-RIG-I, MAVS, and TBK1 were expressed by cloning the respec-tive genes into the pcDNA3-Flag vector. The miR-526a expression plas-mid was expressed by cloning miR-526a into the Pires 2-EGFP vector. Small interfering RNA (siRNA) oligonucleotides were ordered from GenePharma and transfected with Lipofectamine 2000 (Invitrogen) ac-cording to the manufacturer’s instructions. The sequences of primers for small interfering RNAs used are shown inTable 1.
miRNA mimics and inhibitors.MiR-526a mimics (double-stranded RNA [dsRNA] oligonucleotides) and miR-526a inhibitors (single-stranded chemically modified oligonucleotides) from GenePharma were used for the overexpression and inhibition of miR-526a activity, respectively. Nega-tive-control mimics and inhibitors (GenePharma) were transfected as matched controls. Primer sequences were as follows: miR-526a mimics, 5=-CUCUAGAGGGAAGCACUUUCUG-3=(sense) and 5=-GAAAGUGC UUCCCUCUAGAGUU-3=(antisense); miR-526b mimics, 5=-CUCUUG AGGGAAGCACUUUCUGU-3=(sense) and 5=-AGAAAGUGCUUCCC UCAAGAGUU-3=(antisense); control mimics, 5=-UUCUCCGAACGUG UCACGUTT-3= (sense) and 5=-ACGUGACACGUUCGGAGAATT-3= (antisense); miR-526a inhibitor, 5=-CAGAAAGUGCUUCCCUCUAGA G-3=(chemically modified by 2=-Ome), and control inhibitor, 5=-CAGU ACUUUUGUGUAGUACAA-3=.
[image:2.585.298.543.77.404.2]Viruses and viral plaque assay.The construction of the green fluores-cent protein-encoding Newcastle disease virus (NDV-GFP) used in this study was previously described (28) and was obtained from Cheng Cao (Beijing Institute of Biotechnology). Vesicular stomatitis virus (VSV) and herpes simplex virus (HSV) were also obtained from Cheng Cao. VSV and NDV-GFP were propagated in chicken embryo fibroblasts and chicken embryos, respectively, and titrated in MDCK cells. HSV was propagated TABLE 1Sequences of RNAi oligonucleotides used in the present study
Name Sequences
si-control 5=-AGACCCACUCGGAUGUGAAGAGAUA-3=(sense) 5=-UAUCUCUUCACAUCCGAGUGGGUCU-3=(antisense)
si-MDA5 5=-GUUAUAGUUCUUGUCAAUA-3=(sense) 5=-UAUUGACAAGAACUAUAAC-3=(antisense)
si-RIG-I 5=-GACUAGUAAUGCUGGUGUA-3=(sense) 5=-UACACCAGCAUUACUAGUC-3=(antisense)
si-IFNAR 5=-AGCUCAGAUUGGUCCUCCA-dTdT-3=(sense) 5=-UGGAGGACCAAUCUGAGCU-dTdT-3=(antisense)
si-p65 5=-GATGAGATCTTCCTACTGT-3=(sense) 5=-CAAGATCAATGGCTACACA-3=(antisense)
si-IRF3 1 5=-AACUCAUCCAGAAUGUCUUCCUGGG-3=(sense) 5=-CCCAGGAAGACAUUCUGGAUGAGUUTT-3=(antisense)
si-IRF3 2 5=-GGAGGAUUUCGGAAUCUUCTT-3=(sense) 5=-GAAGAUUCCGAAAUCCUCCTG-3=(antisense)
si-IRF7 1 5=-GGUCAGUAGUGGCCGGUACTT-3=(sense) 5=-GUACCGGCCACUACUGACCTT-3=(antisense)
si-IRF7 2 5=-CACUAUGGGCCCUGCGAAUTT-3=(sense) 5=-AUUCGCAGGGCCCAUAGUGTT-3=(antisense)
si-CYLD 5=-GUAUAGGACAGUAUAUUCATT-3=(sense) 5=-UGAAUAUACUGUCCUAUACTT-3=(antisense)
si-MAVS 5=-UAGUUGAUCUCGCGGACGA-dTdT-3=(sense)
5=-UCGUCCGCGAGAUCAACUA-dTdT-3=(antisense)
on November 7, 2019 by guest
http://jvi.asm.org/
and titrated in Vero cells. EV71 viral strain GDV-103 (purchased from China Center for Type Culture Collection [CCTCC]) was grown in RD cells and was propagated and titrated in RD cells.
Luciferase reporter assays. (i) CYLD 3=UTR luciferase reporter as-say.Four CYLD 3=UTR luciferase reporter constructs were made by am-plifying the human CYLD mRNA 3=UTR sequence by PCR and cloning into the pGL3-cm plasmid. The 293T cells were cotransfected with 200 ng of luciferase reporter plasmid (cm-1 to cm-4), 4 ng of pRL-TK-Renilla luciferase (pRL) plasmid, and various RNAs (final concentration, 20 nM). After 36 h, luciferase activities were measured using the dual-luciferase reporter assay system (Promega) according to the manufacturer’s instruc-tions. Data were acquired by determining the ratios of firefly luciferase activity to that of pRL luciferase.
(ii) IFN-, NF-B, and IRF3 luciferase reporter assay.Luciferase re-porter plasmids (200 ng) containing promoters for IFN-, NF-B
(IFN--luc, NF-B-luc) and IRF3-luciferase plasmids (Gal4-IRF3 and UAS-luc) were cotransfected with 4 ng of pRL plasmid into 293T cells. After various treatments, luciferase activities were measured as described above (29).
RNA quantification.Total RNA from cells was extracted with TRIzol reagent (Invitrogen) following the manufacturer’s instructions. For the quantification of miR-526a and miR-526b, RNA was reverse transcribed using the TaKaRa microRNA reverse transcription kit and miRNA-spe-cific stem-loop primers (Table 2). Similarly, U6A small nuclear RNA was quantified using its reverse primer for the reverse transcription reaction (Table 2). Real-time quantitative PCR (RT-PCR) analysis was performed using the multicolor real-time PCR detection system (IQ5; Bio-Rad) and SYBR RT-PCR kits (TaKaRa). RT-PCR primer sequences for miR-526a, miR-526b, and U6A are listed inTable 3, and the relative expression level of miRNAs was normalized to that of U6A by the 2⫺⌬⌬CTthreshold method (30). RT-PCR mixtures were incubated in a 96-well plate at 94°C for 2 min, followed by 40 cycles of 94°C for 20 s and 60°C for 30 s. All reverse transcriptase reactions, including no-template controls and re-verse transcription-negative controls, were run in duplicate. Sequences of RT-PCR primers for other genes used in the study are listed inTable 3. Data were normalized to the level of-actin expression in each sample as described above.
Immunoprecipitation and immunoblotting.Cells were harvested in cell lysis buffer (50 mM Tris-HCl [pH 7.5], 10 mM sodium fluoride, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 1 mM phenylmethylsulfonyl fluo-ride, 1 mM dithiothreitol, and 10 mg/ml pepstatin A) containing 1% Nonidet P-40. Whole-cell lysates was subjected to immunoprecipitation with anti-Flag agarose beads (Sigma-Aldrich). Immunoblot analysis was performed with anti-hemagglutinin (HA) and anti-Flag (Sigma-Aldrich). The prepared samples were detected with anti-VSV G (Santa Cruz Bio-technology), anti-CYLD, -IRF3, -IRF3-p, -IKK␣, -IKK, and -IRF7 (Epitomic), anti-IKK␣/-p (Cell Signaling Technology), anti-IB-p and -IB (Sigma-Aldrich), and anti-MDA-5 and -IFNAR1(Abcam) antibod-ies.
Flow cytometry.293T cells were cotransfected with miR-526a mimics or miR-526a inhibitors followed by NDV-GFP infection. At 24 h postin-fection, cells were subjected to flow-cytometric analysis on a FACSCalibur system, and data were analyzed with CellQuest software (both from BD Biosciences). The mean fluorescence intensity and percent green-fluo-rescing cells were determined.
Assay of IFN-secretion from THP-1 cells.To assay for IFN- se-cretion, THP-1 cells (1⫻106cells/ml) were infected with VSV. The
cul-ture medium was then used to quantify IFN-with an AlphaLISA IFN- kit following the manufacturer’s instructions (PerkinElmer Life Sciences).
In vivoubiquitination assay.Samples were harvested after transfec-tion and infectransfec-tion, and whole-cell lysates (WCL) were prepared in a 1% NP-40 lysis buffer supplemented with 0.1% protease inhibitor cocktail (Sigma-Aldrich) and the deubiquitinase inhibitor N-ethylmaleimide (10 mM; Sigma-Aldrich). Protein-protein interactions were disrupted by sonication (3 pulses of 10 s) using the 550 Sonic Dismembrator (Fisher Scientific Inc.) followed by boiling for 10 min in 1% SDS. WCL (250 to 500g) were immunoprecipitated with 1g of anti-Flag antibody. Polyu-biquitination was detected using a monoclonal anti-HA antibody (Sigma-Aldrich).
Statistical analysis.The difference between two groups was statisti-cally analyzed by a two-tailed Student’sttest. All data are the averages of triplicates and are representative of results from at least 3 independent experiments.
RESULTS
Viral infection upregulates miR-526a expression through the
IRF pathway.
To identify miRNAs selectively involved in the
[image:3.585.298.545.76.335.2]reg-ulation of innate immune response against viral infection, we
con-structed 168 miRNAs of unknown function by using the Pires
2-EGFP vector as a backbone and screened for IFN-

luciferase
activity in 293T cells transfected with a cDNA construct from a
sublibrary for 168 miRNAs together with an IFN-

luciferase
re-porter followed by VSV infection. MiR-526a was identified as 1
out of 21 potent IFN-

inducers among 168 miRNAs that we
analyzed (data not shown). We thus wanted to investigate whether
miR-526a expression was regulated by VSV challenge in human
immune cells. To this end, the expression of miR-526a from
VSV-infected THP-1 cells was determined at different times
postinfec-tion.
Figure 1A
indicates that miR-526a is a VSV
infection-re-sponsive gene in THP-1 monocytes. Its induction was upregulated
TABLE 2Sequences of RT primers used in the present study [image:3.585.40.288.78.145.2]Name Sequences RT-miR-526a 5=-CTCAACTGGTGTCGTGGAGTCGGCAATT CAGTTGAGCAGAAAGT-3= RT-miR-526b 5=-CTCAACTGGTGTCGTGGAGTCGGCAATT CAGTTGAGACAGAAAG-3= RT-U6A 5=-CGCTTCACGAATTTGCGTGTCAT-3=
TABLE 3Sequences of RT-PCR oligonucleotides used in the study
Name Sequence
CXCL-2 F 5=-TCACCTCAAGAACATCCAAAGTGTG-3=
CXCL-2 R 5=-CTTCAGGAACAGCCACCAATAAGC-3=
CYLD F 5=-TTCACTGACGGGGTGTACCA-3=
CYLD R 5=-CAGGACCTGCGTAATCACTTTC-3=
IFN-F 5=-GTCAGAGTGGAAATCCTAAG-3=
IFN-R 5=-ACAGCATCTGCTGGTTGAAG-3=
IL-8 F 5=-AGGTGCAGTTTTGCCAAGGA-3=
IL-8 R 5=-TTTCTGTGTTGGCGCAGTGT-3=
ISG54 F 5=-TGCAACCTACTGGCCTATCTA-3=
ISG54 R 5=-CAGGTGACCAGACTTCTGATT-3=
ISG56 F 5=-TAGCCAACATGTCCTCACAGAC-3=
ISG56 R 5=-TCTTCTACCACTGGTTTCATGC-3=
MAVS F 5=-CAGGCCGAGCCTATCATCTG-3=
MAVS R 5=-GGGCTTTGAGCTAGTTGGCA-3=
miR-526a F 5=-ACACTCCAGCTGGGCTCTAG-3=
miR-526a R 5=-TGGTGTCGTGGAGTCG-3=
miR-526b F 5=-ACACTCCAGCTGGGCTCTTG-3=
miR-526b R 5=-TGGTGTCGTGGAGTCG-3=
U6A F 5=-GCTTCGGCAGCACATATACTAAAAT-3=
U6A R 5=-CGCTTCACGAATTTGCGTGTCAT-3=
VP1 F 5=-GAGAGTTCTATAGGGGACAGT-3=
VP1 R 5=-AGCTGTGCTATGTGAATTAGGAA-3=
-actin F 5=-AAAGACCTGTACGCCAACAC-3=
-actin R 5=-GTCATACTCCTGCTTGCTGAT-3=
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 1Viral infection upregulates miR-526a expression through the IRF pathway. THP-1 cells were infected with VSV (multiplicity of infection [MOI]⫽1) or LPS (0.1g/ml) for the indicated times. The expression of miR-526a (A) and miR-526b (C) was measured by RT-PCR and normalized to the expression of U6A in each sample. (B) The titers of VSV harvested from the infected cells were determined at different times postinfection. THP-1 cells were infected with VSV at different MOIs for 16 h (D) or various stimuli as indicated (E and F), and the expression of miR-526a was measured as described for panel A. (G) THP-1 cells were transfected with Flag-tagged IRF3, IRF7, or p65, and miR-526a expression was measured 24 h later as described for panel A. (H and I) THP-1 cells were transfected with the indicated siRNA oligonucleotides (10 nM) for 48 h and then infected with VSV at an MOI of 0.1 for 16 h. MiR-526a expression was measured as described for panel A. Cell-based studies were performed at least three independent times with comparable results. Data are means⫾standard deviations (SD). Student’sttest was used for statistical analysis. *,P⬍0.05, #,P⬍0.01, and **,P⬍0.01 compared to time zero samples or matched controls.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.75.510.63.622.2]quickly from 2 to 36 h and peaked at 16 h after VSV challenge,
concomitant with the kinetics of VSV replication in the culture
supernatants of infected cells (
Fig. 1B
). However, miR-526b was
not induced (
Fig. 1C
). Furthermore, VSV induced miR-526a
ex-pression in a dose-dependent manner (
Fig. 1D
). MiR-526a was
also upregulated in THP-1 transfected with poly(I·C) (MDA-5
agonist), poly(dA·dT), or cyclic bis(3
=
5
=
)-diguanylic acid
(c-di-GMP) or infected with NDV (
Fig. 1E
). The expression of
miR-526a could be induced by LPS (a TLR4 agonist) or HSV infection
to a much lesser extent than VSV infection (
Fig. 1E
), indicating
that miR-526a might participate in the cytosolic innate immune
response triggered by RNA virus.
The C-terminal regulatory domain of RIG-I recognizes RNA
carrying 5
=
-triphosphates (3pRNA) (
31
). To selectively assess the
role of miR-526a in cytosolic signaling pathway, THP-1 cells were
treated with the RIG-I agonist 3pRNA or poly(I·C). As shown in
Fig. 1F
, miR-526a was upregulated by poly(I·C) to a lesser extent
than 3pRNA. Therefore, miR-526a may participate mainly in the
RIG-I signaling pathway.
We then assessed the contribution of the IRF pathway and the
NF-
B pathway to the regulation of miR-526a expression.
VSV-induced upregulation of miR-526a expression was significantly
increased in cells transfected with the IRF3 or IRF7 expression
plasmid but was unchanged in p65-overexpressing cells (
Fig. 1G
).
Consistent with the regulatory role of IRF3/7 in miR-526a
expres-sion, VSV-induced upregulation of miR-526a expression was
at-tenuated in siIRF7- and siIRF3-treated macrophages (
Fig. 1H
).
MDA-5, IFNAR, MAVS, and RIG-I knockdown cells also showed
defective miR-526a induction in response to VSV infection,
whereas VSV-induced miR-526a upregulation was intact in p65
knockdown cells (
Fig. 1I
), further demonstrating the involvement
of the IRF3/7 pathway in the regulation of miR-526a expression.
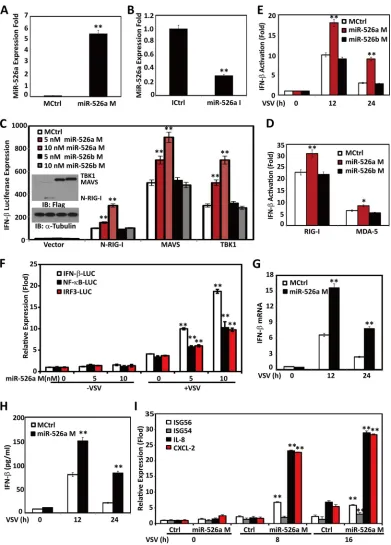
MiR-526a positively regulates VSV-triggered IFN-I
produc-tion.
To further determine whether virus-induced miR-526a
expres-sion could affect the innate immune response, we investigated the
role of miR-526a in IFN-I production using miR-526a mimics and
miR-526a inhibitors. MiRNA mimics are double-stranded RNAs
synthesized to simulate naturally occurring mature microRNAs,
while miRNA inhibitors are chemically modified antisense
single-stranded RNAs that are widely employed in miRNA loss-of-function
studies. As shown in
Fig. 2A
and
B
, transfection of miR-526a mimics
(miR-526a M) increased miR-526a expression about 6-fold in
macrophages, whereas miR-526a inhibitors (miR-526a I)
de-creased its expression level over 70%. Increasing amounts of
miR-526a mimics were cotransfected with the expression construct for
RIG-I 2CARD (N-RIG-I), MAVS, TBK1, or MDA-5 into 293T
cells together with an IFN-

luciferase reporter (IFN-

-luc);
miR-526b mimics were used as negative miRNA controls. Thirty-six
hours after transfection, the luciferase activity was measured and
normalized based on pRL luciferase activity. As shown in
Fig. 2C
and
D
, overexpression of miR-526a, but not miR-526b, potently
activated the IFN-

promoter mediated by RIG-I, MDA-5, and its
downstream molecules. We next investigated the function of
miR-526a in VSV-induced IFN-

production. Consistent with the
stimulating effect of miR-526a on the IFN-promoter, miR-526a
enhanced IFN-promoter activity substantially in response to VSV
infection (
Fig. 2E
and
F
). Similar enhancement of VSV-induced
activation of NF-
B and IRF3 luciferase reporters by miR-526a
was also observed (
Fig. 2F
). Congruence results showed that
miR-526a overexpression increases VSV-triggered IFN-

production
at both the mRNA and protein levels (
Fig. 2G
and
H
). We went
further to investigate the effect of miR-526a on VSV-induced
pro-duction of proinflammatory cytokines and chemokines and
found that miR-526a significantly enhanced VSV-induced
pro-duction of IL-8 (interleukin 8), ISG56 (IFN-stimulated gene 56),
ISG54 (IFN-stimulated gene 54), and CXCL-2 (chemokine ligand
2) in macrophages (
Fig. 2I
). Taken together, these results
demon-strate that miR-526a positively regulates VSV-triggered IFN-I
production.
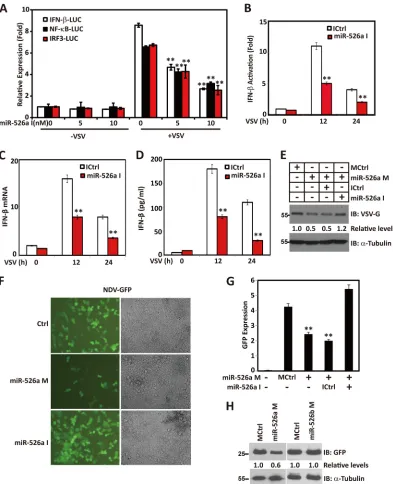
MiR-526a suppresses viral replication.
Consistent with the
positive effect of miR-526a on IFN-I production, overexpression
of miR-526a inhibitors attenuated IFN-promoter activity
sub-stantially in response to VSV infection (
Fig. 3A
and
B
). Similar
reduction of VSV-induced activation of NF-
B and IRF3
lucifer-ase reporters by miR-526a inhibitors was also observed (
Fig. 3A
).
Congruence results showed that miR-526a inhibits VSV-triggered
IFN-

production at both the mRNA and protein levels (
Fig. 3C
and
D
).
We then investigated the biological significance of virally
in-duced-miR-526a upregulation by measuring VSV G protein levels
in the whole-cell lysates of the infected macrophages and found
that 526a mimics inhibits VSV replication, whereas
miR-526a inhibitors rescue the suppressed VSV replication (
Fig. 3E
).
Furthermore, another RNA virus, NDV-GFP, was also tested in
this study. By quantification of NDV-GFP with
fluorescence-ac-tivated cell sorting (FACS) and immunofluorescent analysis, we
found that miR-526a mimics inhibit NDV-GFP replication by
approximately 50%, whereas miR-526a inhibitors reverse the
sup-pression of NDV replication (
Fig. 3F
and
G
). While a similar
re-duction of NDV-GFP protein by miR-526a mimics was also
ob-served, miR-526b mimics failed to exert any effect on NDV
replication (
Fig. 3H
). These results provide biological evidence
that miR-526a acts as a positive regulator of RIG-I-mediated type
I IFN signaling and thereby modulates the innate antiviral cellular
response.
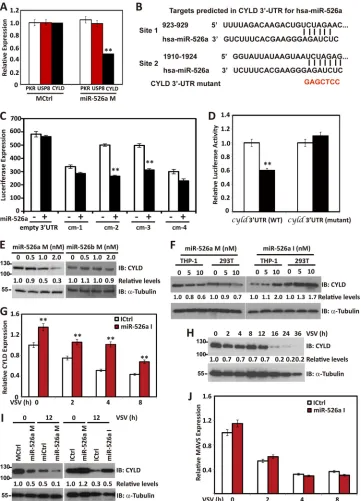
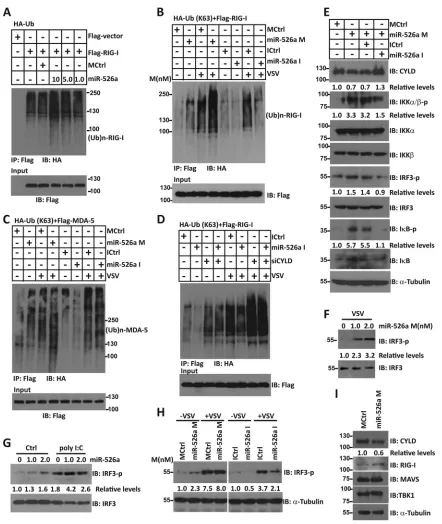
MiR-526a targets CYLD.
To characterize the mechanism
associ-ated with miR-526a function, we used a web-based miRNA target
prediction program TargetScanHuman (
http://www.targetscan.org
)
to search for potential miR-526 targets. Among the possible targets
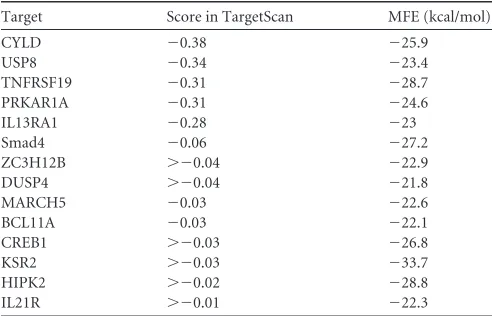
of miR-526a (some of the predicative targets are listed in
Table 4
),
we focused on CYLD, USP8 (ubiquitin-specific protease 8), and
PKR (protein kinase R), which could contribute to the antiviral
phenotype observed. Result showed that only CYLD mRNA
ex-pression was significantly downregulated by miR-526a
overex-pression (
Fig. 4A
). CYLD mRNA has 4 putative miR-526a target
sites. To certify the possibility that CYLD is regulated by
miR-526a, we constructed reporter plasmids by cloning 4 fragments of
the human CYLD 3
=
UTR containing prediction sites to the 3
=
UTR region of the firefly luciferase gene in the pGL3-cm vector
and named them cm-1 to cm-4. By cotransfection of the reporter
plasmids and internal control pRL plasmids into 293T cells, we
observed that miR-526a downregulated luciferase gene expression
in cm-2 and cm-3 plasmids (representing site 1 and site 2 in
Fig.
4B
;
Fig. 4C
). Mutations in the two sites of cm-2 and cm-3
com-pletely abolished the ability of miR-526a to inhibit luciferase
ac-tivity (
Fig. 4D
). Furthermore, transfection of miR-526a mimics
decreased CYLD protein expression (
Fig. 4E
), whereas miR-526a
inhibitors rescue the suppressed CYLD expression (
Fig. 4F
). To
investigate whether the suppression of CYLD by miR-526a is
functionally relevant, we analyzed the CYLD expression in
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 2MiR-526a positively regulates VSV-triggered type I IFN production. (A and B) THP-1 cells were transfected with miR-526a mimics (miR-526a M) or control mimics (MCtrl) (A) or with miR-526a inhibitors (miR-526a I) or control inhibitors (ICtrl) (B). The final RNA concentration was 5 nM, and the miR-526a expression was measured by RT-PCR. (C and D) The indicated amounts of miR-526a or miR-526b mimics were cotransfected with the indicated plasmid together with IFN--luc, as well as pRL as an internal control. The luciferase activity was measured 36 h later and normalized for transfection efficiency. (E) 293T cells were transfected with control mimics, miR-526a mimics, or miR-526b mimics. After 12 h, cells were transfected with IFN--luc and infected with VSV at an MOI of 1 for the indicated times. The luciferase activity was measured and normalized for transfection efficiency. (F) Transfection of 293T cells with increasing amounts of miR-526a mimics or control mimics. After 12 h, the cells were transfected with IFN--luc, IRF3-luc, or NF-B-luc; 24 h later, the cells were infected with VSV at an MOI of 1 for another 16 h. The luciferase activity was then measured and normalized for transfection efficiency. (G and H) THP-1 cells were transfected with miR-526a mimics or control mimics and infected with VSV at an MOI of 1 for 16 h. IFN-mRNA (G) and protein (H) levels were analyzed by RT-PCR and AlphaLISA, respectively. (I) THP-1 cells were transfected with miR-526a mimics (5 nM) and infected with VSV at an MOI of 1 for 16 h. RT-PCR was performed using the primers specific for IFN-responsive genes, including ISG56, ISG54, IL-8, and CXCL-2. Cell-based studies were performed at least three independent times, with comparable results. Data are means⫾SD. Student’s ttest was used for statistical analysis. **,P⬍0.01 compared to the corresponding control.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.97.488.63.608.2]FIG 3MiR-526a suppresses viral replication. (A) 293T cells were transfected with increasing amount of miR-526a inhibitors. After 12 h, cells were transfected with IFN--luc, IRF3-luc, or NF-B-luc for 24 h and infected with VSV at an MOI of 1 for 16 h. The luciferase activity was measured and normalized for transfection efficiency. (B) 293T cells were transfected with control inhibitors or miR-526a inhibitors. After 12 h, cells were transfected IFN--luc for 24 h and infected with VSV at an MOI of 1 for the indicated times. The luciferase activity was measured and normalized for transfection efficiency. (C and D) THP-1 cells were transfected with control inhibitors or miR-526a inhibitors for 48 h and infected with VSV at an MOI of 1 for the indicated times. IFN-mRNA (C) and protein (D) levels were analyzed by RT-PCR or AlphaLISA. (E) THP-1 cells were transfected with control mimics or miR-526a mimics together with control inhibitors or miR-526a inhibitors, respectively, for 48 h followed by VSV infection. After 16 h, the level of VSV G protein was monitored by immunoblotting using anti-VSV G antibody.␣-Tubulin was used as an equal loading control. (F) 293T cells were transfected with miR-526a mimics or miR-526a inhibitors. After 48 h, cells were infected with NDV-GFP, and images were taken at 16 h postinfection. (G) As described for panel F, NDV-GFP was quantified by FACS. NDV-GFP expression was defined as the product of the percent GFP-positive cells and the geometric mean of the fluorescence index (MFI). (H) 293T cells were transfected with miR-526a mimics or miR-526b mimics. After 48 h, cells were infected with NDV-GFP. The level of NDV replication was monitored by immunoblotting using anti-GFP antibody.␣-Tubulin was used as an equal loading control. Numbers below certain Western blots indicate relative levels determined by software-based quantification of the representative results shown. Cell-based studies were performed at least three independent times with comparable results. Data are means⫾SD. Student’sttest was used for statistical analysis. **,P⬍0.01 compared to the corresponding control.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.95.488.65.549.2]sponse to VSV infection. Concomitant with enhanced miR-526a
expression induced by VSV challenge, CYLD expression was
re-duced at both the mRNA (
Fig. 4G
) and protein (
Fig. 4H
) levels in
the presence of viral infection. Moreover, virus-induced
down-regulation of CYLD was inhibited by the expression of miR-526a
inhibitors (
Fig. 4G
and
I
); however, virus-induced
downregula-tion of MAVS was unchanged by miR-526a inhibitors (
Fig. 4J
).
Together, these findings suggest that endogenous human CYLD is
a novel target of miR-526a, and miR-526a acts as a positive
regu-lator of innate antiviral response by suppressing CYLD
expres-sion.
MiR-526a regulates antiviral response through
enhance-ment of K63-linked RIG-I ubiquitination.
The removal of
Lys-63-linked polyubiquitination chains from RIG-I by CYLD has
been shown to inactivate the RIG-I-mediated antiviral response.
As miR-526a targeted CYLD for degradation, we tested whether
RIG-I is a target for miR-526a by transfecting 293T cells with
full-length RIG-I in the presence of miR-526a mimics. As shown
in
Fig. 5A
, RIG-I underwent increased polyubiquitination in a
miR-526a mimic dose-dependent manner. Using a ubiquitin
mu-tant with only one lysine at position 63 available for conjugation
(HA-Ub K63), we further revealed that VSV infection leads to
increased K63-linked polyubiquitination of RIG-I, which is
en-hanced by miR-526a mimics but abrogated by miR-526a
inhibi-tors (
Fig. 5B
). In particular, VSV-induced MDA-5 ubiquitination
levels were nearly unchanged by miR-526a mimics or miR-526a
inhibitors (
Fig. 5C
). Furthermore, miR-526a inhibitors failed to
exert its suppressive effect on RIG-I ubiquitination in siCYLD
cells (
Fig. 5D
). These data indicated that miR-526a specifically
targets CYLD expression and the subsequent RIG-I
ubiquitina-tion.
To better understand how miR-526a activates RIG-I signaling,
IRF3 phosphorylation level was analyzed with miR-526a
overex-pression. As illustrated in
Fig. 5E
, miR-526a strongly stimulated
IRF3 phosphorylation, I
B phosphorylation, and IKK
ε
phos-phorylation induced by VSV. However, addition of miR-526a
in-hibitors drastically reduced the signals for IRF3 activation and
NF-
B activation. Similarly, IRF3 phosphorylation induced by
VSV infection (
Fig. 5F
and
H
) or poly(I·C) transfection (
Fig. 5G
)
was significantly enhanced by miR-526a mimics. Conversely,
miR-526a inhibitors blocked the activation of IRF3 induced by
VSV infection (
Fig. 5H
). Meanwhile, miR-526a mimics reduced
the amount of CYLD protein, whereas the levels of RIG-I and
TBK1 proteins remained unchanged (
Fig. 5I
). These observations
suggest that enhancement of K63-linked RIG-I ubiquitination by
miR-526a may be a mechanism by which miR-526a activates the
IFN-I innate immune response.
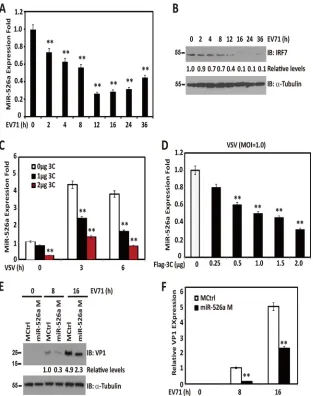
Downregulation of miR-526a by EV71 3C protease impairs
the innate immune response.
Since miR-526a upregulation
in-duced by viral infection was partially mediated by IRF7, which can
be disrupted by EV71 3C protease (
8
), we investigated the
expres-sion kinetics of miR-526a in response to EV71 infection.
Interest-ingly, EV71 infection reduced miR-526a expression, concomitant
with the declined IRF7 expression (
Fig. 6A
and
B
). Since EV71 3C
protease induced IRF7 cleavage, we evaluated whether EV71 3C
affects the expression of miR-526a. As shown in
Fig. 6C
and
D
,
ectopically expressed EV71 3C blocked the upregulation of
miR-526a induced by VSV infection in a dose-dependent manner.
Fi-nally, we assessed whether miR-526a is functionally linked to
EV71 infection. Specifically, miR-526a mimic-treated 293T cells
were infected with EV71. As shown in
Fig. 6E
, miR-526a mimics
reduced the production of VP1 over 90% at 8 h postinfection. VP1
mRNA expression was also significantly reduced by miR-526a
overexpression (
Fig. 6F
). These results suggest that control of
IRF7-regulated miR-526a expression by the 3C protein may
rep-resent a novel viral mechanism to escape cellular responses.
DISCUSSION
Various molecules have been shown to regulate the induction of
IFN-I by targeting distinct components of the virus-triggered
sig-naling pathways (
32–34
). Here, we report a novel positive
feed-back regulator in RIG-I signaling at the level of miRNAs. First, we
found that RNA viral infection upregulates the expression of
miR-526a in macrophages through IRF-dependent pathways. Second,
we showed that miR-526a positively regulates viral-triggered
IFN-I production and that the inducible miR-526a expression
suppresses virus replication. Third, we also demonstrated that
miR-526a targets human CYLD, which regulates IFN signaling by
removing K63-linked RIG-I ubiquitination. Fourth, the present
study showed that the miR-526a level is reduced by EV71 infection
or 3C expression, which correlates with a decreased IRF7 level in
host cells. Finally, cells with overexpressed miR-526a were highly
resistant to EV71 infection. These findings not only indicate that
miR-526a is a positive regulator of the RIG-I-mediated innate
antiviral response but also suggest a novel mechanism for the
eva-sion of innate immune control by EV71.
CYLD is a known deubiquitinase, originally identified as a
ge-netic defect in familial cylindromatosis (
20
). Previous reports
in-dicated that CYLD inhibits IRF3 signaling and subsequent IFN-

production by removing Lys-63-linked polyubiquitin chains of
RIG-I (
21
,
35
). Here we report that miR-526a triggers stronger
K63-linked RIG-I ubiquitination, coincident with enhanced IRF3
signaling and NF-
B signaling. Our findings and those in a
previ-ous report (
35
) suggest that CYLD is significantly downregulated
by VSV. While the molecular mechanism regarding virus-induced
CYLD downregulation remains elusive, our results indicate that
miR-526a modulates CYLD protein availability at both the mRNA
and protein levels, revealing the possible mechanism of
infection-triggered CYLD downregulation.
[image:8.585.40.286.86.244.2]Besides its important role in RIG-I signaling, CYLD also acts as
a negative regulator of NF-
B, Wnt/

-catenin signaling, and Jun
TABLE 4miR-526a targets associated with immunity in humans, aspredicted by TargetScan
Target Score in TargetScan MFE (kcal/mol)
CYLD ⫺0.38 ⫺25.9
USP8 ⫺0.34 ⫺23.4
TNFRSF19 ⫺0.31 ⫺28.7
PRKAR1A ⫺0.31 ⫺24.6
IL13RA1 ⫺0.28 ⫺23
Smad4 ⫺0.06 ⫺27.2
ZC3H12B ⬎⫺0.04 ⫺22.9
DUSP4 ⬎⫺0.04 ⫺21.8
MARCH5 ⫺0.03 ⫺22.6
BCL11A ⫺0.03 ⫺22.1
CREB1 ⬎⫺0.03 ⫺26.8
KSR2 ⬎⫺0.03 ⫺33.7
HIPK2 ⬎⫺0.02 ⫺28.8
IL21R ⬎⫺0.01 ⫺22.3
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 4MiR-526a targets CYLD. (A) 293T cells were transfected with miR-526a mimics or control mimics. After 48 h, PKR, USP8, and CYLD expression levels were analyzed by RT-PCR. (B) Human CYLD might be the molecular target of miR-526a. Shown is a sequence alignment of miR-526a and its target sites in the 3=UTR of CYLD, which was downloaded from TargetScan (http://www.targetscan.org). Red letters are mutated sequences. (C) 293T cells were cotransfected with luciferase reporter plasmids cm-1 to cm-4 containing predicted sites of the human CYLD 3=UTR. After 36 h, the luciferase activity was measured and normalized for transfection efficiency. (D) 293T cells were cotransfected with the WT or CYLD 3=UTR luciferase reporter plasmid containing mutations in luciferase reporter plasmids cm-2 and cm-3. After 36 h, the luciferase activity was measured and normalized for transfection efficiency. (E) 293T cells were transfected with increasing amounts of miR-526a or miR-526b mimics for 48 h, and the CYLD level was detected by immunoblotting using anti-CYLD antibody.
␣-Tubulin was used as equal loading control. (F) 293T and THP-1 cells were transfected with miR-526a mimics or miR-526a inhibitors, and CYLD level was detected by immunoblotting using anti-CYLD antibody.␣-Tubulin was used as equal loading control. (G and J) THP-1 cells were transfected with control inhibitors or miR-526a inhibitors. After 48 h, CYLD (G) and MAVS (J) expression levels were detected by RT-PCR. (H) THP-1 cells were infected with VSV at an MOI of 1 for the indicated times. The expression of CYLD was measured by immunoblotting using anti-CYLD antibody.␣-Tubulin was used as an equal loading control. (I) THP-1 cells were transfected with control mimics or miR-526a mimics together with control inhibitors or miR-526a inhibitors following VSV infection at an MOI of 1 for 12 h. CYLD expression was detected by immunoblotting using anti-CYLD antibody.␣-Tubulin was used as an equal loading control. Numbers below certain Western blots indicate relative levels determined by software-based quantification of the representative results shown. Cell-based studies were performed at least three independent times, with comparable results. Data are means⫾SD. Student’sttest was used for statistical analysis. **,P⬍0.01 compared to the corresponding control.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.112.473.66.567.2]FIG 5MiR-526a regulates the IFN-I innate immune response through enhancement of K63-linked RIG-I ubiquitination. (A) Cells expressing Flag-RIG-I, HA-Ub, and increasing doses of miR-526a mimics were immunoprecipitated with an anti-Flag antibody and analyzed by immunoblotting with an anti-Flag antibody. (B and C) Flag-RIG-I (B) or Flag-MDA-5 (C) was cotransfected with HA-Ub K63; miR-526a mimics together with control inhibitors or miR-526a inhibitors were infected with VSV at an MOI of 1 for 16 h. Samples were subjected to immunoprecipitation with an anti-Flag antibody and analyzed by immunoblotting with an anti-Flag antibody. (D) Flag-RIG-I was cotransfected with HA-Ub K63. miR-526a inhibitors together with siCYLD oligonucleotides (10 nM) were infected with VSV at an MOI of 1 for 16 h. Samples were subjected to immunoprecipitation with an anti-Flag antibody and analyzed by immuno-blotting with an anti-Flag antibody. (E) 293T cells were transfected with miR-526a together with control inhibitors or miR-526a inhibitors for 48 h and infected with VSV (MOI⫽1) for 16 h. The levels of CYLD, IKK␣, IKK, IKK␣/-p, IRF3, IRF3-p, IB, and IB-p were monitored by immunoblotting.␣-Tubulin was used as equal loading control. (F and G) 293T cells were transfected with increasing amount of miR-526a mimics for 24 h and infected with VSV (MOI⫽1) for 16 h (F) or transfected with poly(I·C) for 24 h (G). IRF3 and IRF3 phosphorylation was analyzed by immunoblotting with anti-IRF3 and IRF3-p antibodies. (H) THP-1 cells were transfected with miR-526a mimics or miR-526a inhibitors followed by VSV infection for 12 h. The levels of IRF3 or IRF3-p at the indicated times were monitored by immunoblotting.␣-Tubulin was used as an equal loading control. (I) THP-1 cells were transfected with miR-526a mimics or control mimics, and the levels of CYLD, RIG-I, MAVS, and TBK1 were monitored by immunoblotting.␣-Tubulin was used as an equal loading control. Numbers below certain Western blots indicate relative levels determined by software-based quantification of the representative results shown. Similar results were obtained in three independent experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.73.515.61.585.2]N-terminal kinase signaling pathways. Our findings may have
im-plications for the involvement of miR-526a in the development
and progression of other human disorders, such as multiple
my-eloma and colon and hepatocellular carcinomas, linked to loss of
CYLD (
36–38
). MiR-526a may thus play an even more profound
role in host antiviral response and cancer. Since viruses also play a
role in the pathogenesis of several tumor types, miRNAs
influenc-ing viral-host interactions have important implications not only
for infectious diseases but also for carcinogenesis. Therefore,
miR-526a may have the potential to become a novel drug target in
virally induced infectious or malignant diseases.
TargetScan was applied in this study to identify potential
miR-526a targets in the human genome. The minimum free energy
(MFE) of the miRNA-target duplex was determined to predict the
miRNA target sites. Low-MFE values between the miRNAs and
the target sites reveal energetically more probable hybridizations
between the miRNAs and the target genes. The possible targets of
miR-526a in the human genome that may contribute to the
anti-viral effect are listed in
Table 4
. Vita software was applied to
iden-tify the potential miR-526a targets in the EV71 genome. A
previ-ous report (
41
) showed two potential miR-296-5p targets (nt 2115
to 2135 and nt 2896 to 2920) in the EV71 genome which were
validated by luciferase reporter assays and Western blotting;
therefore, miR-296 and miR526b were also analyzed in parallel.
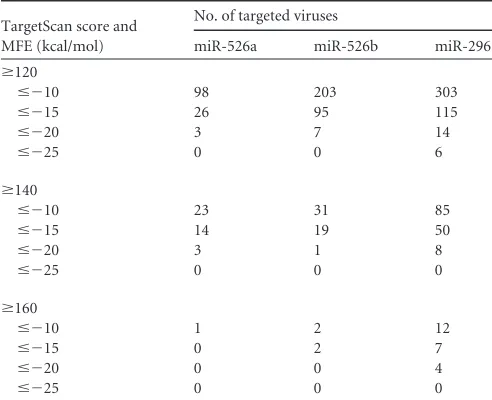
Table 5
gives the statistics of miRNA targets on viruses with
dif-ferent predictive parameters. For instance, if the MFE cutoff is set
at
⬍⫺
20 kcal/mol, and the score cutoff is set at 120, the numbers
of targeted viruses for miR-526a, miR-526b, and miR-296 are 3, 7,
and 14, respectively. Although our data showed a role for the
miR-526a-CYLD-RIG-I axis in regulating antiviral responses, we
can-FIG 6Downregulation of miR-526a by EV71 3C impairs IFN-I production. (A and B) 293T cells were infected with EV71 at an MOI of 1 for the indicated times. MiR-526a expression was detected by RT-PCR (A), and IRF7 was detected by immunoblotting (B). (C) 293T cells were transfected with different amounts of Flag-3C followed by VSV infection at an MOI of 1. Cells were harvested at the indicated times. MiR-526a expression was monitored by RT-PCR. (D) 293T cells were transfected with increasing amounts of Flag-3C followed by infection with VSV at an MOI of 1. MiR-526a expression was monitored by RT-PCR. (E and F) 293T cells were transfected with control mimics or miR-526a mimics followed by infection with EV71 at an MOI of 1 for the indicated times. The level of VP1 was detected by immunoblotting (E) or RT-PCR (F). Numbers below certain Western blots indicate relative levels determined by software-based quantification of the representative results shown. Cell-based studies were performed at least three independent times, with comparable results. Data are means⫾SD. Student’s ttest was used for statistical analysis. **,P⬍0.01 compared to the corresponding control.on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.137.450.65.461.2]not exclude the possibility that miR-526a may have other possible
targets in humans or viruses that could contribute to the antiviral
phenotype observed. Besides that, miR-526a is a member of the
miR-515 family, which includes a number of members that have
high degree of homology. In fact, stem-loop RT analyses that are
specific for mature miRNAs and can discriminate among related
miRNAs that differ by as little as one nucleotide were used for
better specificity and efficiency in this study (
39
). However, as we
did not assess the expression levels of pri- and pre-miR-526a,
which are dissimilar to other family members, we still cannot
ex-clude the possibility of other miRNAs from the -515 family
regu-lating the innate immune signaling pathway. Such a possibility
needs to be addressed in our future studies. In addition, detection
of the miR-526a precursors in the future will also clarify if IRF3/7
regulates miR-526a expression directly on the miRNA gene
pro-moter or just as an indirect effect mediated by the products of
other IRF3/7-regulated genes that might influence miRNA.
Taken together, the results of present study identified a novel
positive regulator of the RIG-I signaling pathway and proposed a
new mechanism for the evasion of innate immune control by
EV71. The viral infection was first sensed by RIG-I, which in turn
initiated IFN-I production against the infection; the
simultane-ously activated IRF3 and IRF7 upregulated the expression of
miR-526a, which then enhanced the innate antiviral immune response
by suppressing CYLD expression and subsequent RIG-I
activa-tion. EV71-encoded 3C protein targeted miR-526a
downregula-tion to suppress the IFN signaling pathway by IRF7 cleavage,
which finally led to the extensive replication of and persistent
in-fection with the invading virus.
ACKNOWLEDGMENTS
This work was supported in part by the Basic Research Program of China (2012CB518900), the National Natural Science Foundation of China (31170029, 31270911, 31300637, 31100960, and 31270800). The funders had no role in study design, data collection and analysis, decision to pub-lish, or preparation of the manuscript.
REFERENCES
1.Palacios G, Oberste MS.2005. Enteroviruses as agents of emerging in-fectious diseases. J. Neurovirol.11:424 – 433.http://dx.doi.org/10.1080 /13550280591002531.
2.McMinn PC.2002. An overview of the evolution of enterovirus 71 and its clinical and public health significance. FEMS Microbiol. Rev.26:91–107. http://dx.doi.org/10.1111/j.1574-6976.2002.tb00601.x.
3.Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, Racaniello VR.2007. MDA-5 is cleaved in poliovirus-infected cells. J. Virol.81:3677–3684.http://dx.doi.org/10.1128/JVI.01360-06.
4.Barral PM, Sarkar D, Fisher PB, Racaniello VR.2009. RIG-I is cleaved during picornavirus infection. Virology391:171–176. http://dx.doi.org /10.1016/j.virol.2009.06.045.
5.Harris KG, Coyne CB.2013. Enter at your own risk: how enteroviruses navigate the dangerous world of pattern recognition receptor signaling. Cytokine63:230 –236.http://dx.doi.org/10.1016/j.cyto.2013.05.007. 6.Arita M, Wakita T, Shimizu H.2012. Valosin-containing protein (VCP/
p97) is required for poliovirus replication and is involved in cellular pro-tein secretion pathway in poliovirus infection. J. Virol.86:5541–5553. http://dx.doi.org/10.1128/JVI.00114-12.
7.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J.2011. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol.85:8811– 8818.http://dx.doi.org/10.1128 /JVI.00447-11.
8.Lei X, Xiao X, Xue Q, Jin Q, He B, Wang J.2013. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol.87:1690 –1698.http://dx.doi.org/10.1128/JVI.01855-12.
9.Lu J, Yi L, Zhao J, Yu J, Chen Y, Lin MC, Kung HF, He ML.2012. Enterovirus 71 disrupts interferon signaling by reducing the level of inter-feron receptor 1. J. Virol.86:3767–3776.http://dx.doi.org/10.1128/JVI .06687-11.
10. Akira S, Takeda K.2004. Toll-like receptor signalling. Nat. Rev. Immu-nol.4:499 –511.http://dx.doi.org/10.1038/nri1391.
11. Meylan E, Tschopp J.2006. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol. Cell22:561–569.http://dx .doi.org/10.1016/j.molcel.2006.05.012.
12. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE.2006. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol.7:1250 –1257. http://dx.doi.org/10.1038/ni1412.
13. Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S.2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol.6:981–988. http://dx.doi.org/10.1038/ni1243.
14. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced in-nate antiviral responses. Nat. Immunol.5:730 –737.http://dx.doi.org /10.1038/ni1087.
15. Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB.2005. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19:727–740.http://dx.doi.org/10.1016/j.molcel.2005.08.014.
16. Qi B, Huang Y, Rowe D, Halliday G.2007. VISA—a pass to innate immunity. Int. J. Biochem. Cell Biol.39:287–291.http://dx.doi.org/10 .1016/j.biocel.2006.08.016.
17. Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ.2006. The specific and essential role of MAVS in antiviral innate immune responses. Immunity24:633– 642.http://dx.doi.org/10.1016/j.immuni.2006.04.004. 18. Hiscott J, Lin R, Nakhaei P, Paz S.2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med.12:53–56.http://dx.doi.org/10.1016 /j.molmed.2005.12.003.
19. Seth RB, Sun L, Ea CK, Chen ZJ.2005. Identification and characteriza-tion of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell122:669 – 682.http://dx.doi.org/10.1016/j.cell .2005.08.012.
20. Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, Jones C, Hansen J, Blair E, Hofmann B, Siebert R, Turner G, Evans DG, Schrander-Stumpel C, Beemer FA, van Den Ouweland A, Halley D, Delpech B, Cleveland MG, Leigh I, Leisti J, Rasmussen S.2000. Identification of the familial cylin-dromatosis tumour-suppressor gene. Nat. Genet.25:160 –165.http://dx .doi.org/10.1038/76006.
[image:12.585.40.286.77.277.2]21. Zhang M, Wu X, Lee AJ, Jin W, Chang M, Wright A, Imaizumi T, Sun TABLE 5miR-526a targets on EV71 predicted by Vita
TargetScan score and MFE (kcal/mol)
No. of targeted viruses
miR-526a miR-526b miR-296
ⱖ120
ⱕ⫺10 98 203 303
ⱕ⫺15 26 95 115
ⱕ⫺20 3 7 14
ⱕ⫺25 0 0 6
ⱖ140
ⱕ⫺10 23 31 85
ⱕ⫺15 14 19 50
ⱕ⫺20 3 1 8
ⱕ⫺25 0 0 0
ⱖ160
ⱕ⫺10 1 2 12
ⱕ⫺15 0 2 7
ⱕ⫺20 0 0 4
ⱕ⫺25 0 0 0
on November 7, 2019 by guest
http://jvi.asm.org/
SC.2008. Regulation of IkappaB kinase-related kinases and antiviral re-sponses by tumor suppressor CYLD. J. Biol. Chem.283:18621–18626. http://dx.doi.org/10.1074/jbc.M801451200.
22. Brummelkamp TR, Nijman SM, Dirac AM, Bernards R.2003. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature424:797– 801.http://dx.doi.org/10.1038/nature01811. 23. Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D,
Courtois G.2003. The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature424:801– 805.http: //dx.doi.org/10.1038/nature01802.
24. Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G.2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature424: 793–796.http://dx.doi.org/10.1038/nature01803.
25. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R.2006. Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell125:665– 677.http://dx.doi.org/10.1016/j.cell.2006 .03.041.
26. Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X.2009. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN produc-tion in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 183:2150 –2158.http://dx.doi.org/10.4049/jimmunol.0900707. 27. Nahid MA, Satoh M, Chan EK.2011. MicroRNA in TLR signaling and
endotoxin tolerance. Cell. Mol. Immunol.8:388 – 403.http://dx.doi.org /10.1038/cmi.2011.26.
28. Park MS, Shaw ML, Munoz-Jordan J, Cros JF, Nakaya T, Bouvier N, Palese P, Garcia-Sastre A, Basler CF. 2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.77: 1501–1511.http://dx.doi.org/10.1128/JVI.77.2.1501-1511.2003. 29. Wei C, Ni C, Song T, Liu Y, Yang X, Zheng Z, Jia Y, Yuan Y, Guan K,
Xu Y, Cheng X, Zhang Y, Yang X, Wang Y, Wen C, Wu Q, Shi W, Zhong H.2010. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 185:1158 –1168.http://dx.doi.org/10.4049/jimmunol.0903874. 30. Livak KJ, Schmittgen TD.2001. Analysis of relative gene expression data
using real-time quantitative PCR and the 2(⫺⌬⌬CT) method. Methods
25:402– 408.http://dx.doi.org/10.1006/meth.2001.1262.
31. Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, Hopfner KP.2008. The C-ter-minal regulatory domain is the RNA 5=-triphosphate sensor of RIG-I. Mol. Cell29:169 –179.http://dx.doi.org/10.1016/j.molcel.2007.10.032.
32. Komuro A, Bamming D, Horvath CM.2008. Negative regulation of cytoplasmic RNA-mediated antiviral signaling. Cytokine 43:350 –358. http://dx.doi.org/10.1016/j.cyto.2008.07.011.
33. Zhang M, Tian Y, Wang RP, Gao D, Zhang Y, Diao FC, Chen DY, Zhai ZH, Shu HB.2008. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res.18:1096 – 1104.http://dx.doi.org/10.1038/cr.2008.277.
34. Takeuchi O, Akira S.2009. Innate immunity to virus infection. Immunol. Rev.227:75– 86.http://dx.doi.org/10.1111/j.1600-065X.2008.00737.x. 35. Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cardenas
WB, Yount JS, Moran TM, Basler CF, Komuro A, Horvath CM, Xavier R, Ting AT.2008. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep.9:930 –936.http://dx.doi .org/10.1038/embor.2008.136.
36. Hashimoto K, Mori N, Tamesa T, Okada T, Kawauchi S, Oga A, Furuya T, Tangoku A, Oka M, Sasaki K.2004. Analysis of DNA copy number aberrations in hepatitis C virus-associated hepatocellular carcinomas by conventional CGH and array CGH. Modern Pathol.17:617– 622.http: //dx.doi.org/10.1038/modpathol.3800107.
37. Hellerbrand C, Bumes E, Bataille F, Dietmaier W, Massoumi R, Bosserhoff AK.2007. Reduced expression of CYLD in human colon and hepatocellular carcinomas. Carcinogenesis 28:21–27. http://dx .doi.org/10.1093/carcin/bgl081.
38. Jenner MW, Leone PE, Walker BA, Ross FM, Johnson DC, Gonzalez D, Chiecchio L, Dachs Cabanas E, Dagrada GP, Nightingale M, Protheroe RK, Stockley D, Else M, Dickens NJ, Cross NC, Davies FE, Morgan GJ. 2007. Gene mapping and expression analysis of 16q loss of heterozygosity identifies WWOX and CYLD as being important in determining clinical outcome in multiple myeloma. Blood110:3291–3300.http://dx.doi.org /10.1182/blood-2007-02-075069.
39. Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ.2005. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 33:e179. http://dx.doi.org/10.1093/nar /gni178.
40. Tsitsiou E, Lindsay MA.2009. MicroRNAs and the immune response. Curr. Opin. Pharmacol. 9:514 –520. http://dx.doi.org/10.1016/j.coph .2009.05.003.
41. Zheng Z, Ke X, Wang M, He S, Li Q, Zheng C, Zhang Z, Liu Y, Wang H.2013. Human microRNA hsa-miR-296-5p suppresses enterovirus 71 replication by targeting the viral genome. J. Virol.87:5645–5656.http://dx .doi.org/10.1128/JVI.02655-12.
on November 7, 2019 by guest
http://jvi.asm.org/


![FIG 1 Viral infection upregulates miR-526a expression through the IRF pathway. THP-1 cells were infected with VSV (multiplicity of infection [MOI]LPS (0.1in each sample](https://thumb-us.123doks.com/thumbv2/123dok_us/144867.22011/4.585.75.510.63.622/infection-upregulates-expression-pathway-infected-multiplicity-infection-sample.webp)