Coordinate Regulation of TET2 and
EBNA2 Controls the DNA Methylation
State of Latent Epstein-Barr Virus
Fang Lu, Andreas Wiedmer, Kayla A. Martin, Priyankara J. M. S. Wickramasinghe, Andrew V. Kossenkov, Paul M. Lieberman
The Wistar Institute, Philadelphia, Pennsylvania, USA
ABSTRACT Epstein-Barr virus (EBV) latency and its associated carcinogenesis are regulated by dynamic changes in DNA methylation of both virus and host genomes. We show here that the ten-eleven translocation 2 (TET2) gene, implicated in hy-droxymethylation and active DNA demethylation, is a key regulator of EBV latency type DNA methylation patterning. EBV latency types are defined by DNA methyl-ation patterns that restrict expression of viral latency genes. We show that TET2 mRNA and protein expression correlate with the highly demethylated EBV type III la-tency program permissive for expression of EBNA2, EBNA3s, and LMP transcripts. We show that short hairpin RNA (shRNA) depletion of TET2 results in a decrease in la-tency gene expression but can also trigger a switch to lytic gene expression. TET2 depletion results in the loss of hydroxymethylated cytosine and a corresponding in-crease in cytosine methylation at key regulatory regions on the viral and host
ge-nomes. This also corresponded to a loss of RBP-jbinding and decreased histone
H3K4 trimethylation at these sites. Furthermore, we show that the TET2 gene itself is regulated in a fashion similar to that of the EBV genome. Chromatin immunoprecipi-tation high-throughput sequencing (ChIP-seq) revealed that the TET2 gene contains
EBNA2-dependent RBP-jand EBF1 binding sites and is subject to DNA
methylation-associated transcriptional silencing similar to what is seen in EBV latency type III
ge-nomes. Finally, we provide evidence that TET2 colocalizes with EBNA2-EBF1-RBP-j
binding sites and can interact with EBNA2 by coimmunoprecipitation. Taken to-gether, these findings indicate that TET2 gene transcripts are regulated similarly to EBV type III latency genes and that TET2 protein is a cofactor of EBNA2 and coregu-lator of the EBV type III latency program and DNA methylation state.
IMPORTANCE Epstein-Barr virus (EBV) latency and carcinogenesis involve the selec-tive epigenetic modification of viral and cellular genes. Here, we show that TET2, a cellular tumor suppressor involved in active DNA demethylation, plays a central role in regulating the DNA methylation state during EBV latency. TET2 is coordinately regulated and functionally interacts with the viral oncogene EBNA2. TET2 and EBNA2 function cooperatively to demethylate genes important for EBV-driven B-cell growth transformation.
KEYWORDS Epstein-Barr virus, EBV, TET2, DNA methylation, EBNA2, epigenetics, epigenome, hydroxymethylation
E
pstein-Barr virus (EBV) is a human gammaherpesvirus that is associated withseveral human cancers, including Burkitt’s lymphoma (BL), Hodgkin’s lym-phoma, B-lymphoblastic lymlym-phoma, nasopharyngeal carcinoma, and lymphoepithe-lial subtypes of gastric carcinomas (1, 2). During primary infection of naive resting B cells, EBV drives a proliferation program resembling an antigen-driven B-cell germinal center reaction that ultimately differentiates into a pool of latently infected, long-lived
Received15 May 2017Accepted24 July
2017
Accepted manuscript posted online9
August 2017
CitationLu F, Wiedmer A, Martin KA,
Wickramasinghe PJMS, Kossenkov AV, Lieberman PM. 2017. Coordinate regulation of TET2 and EBNA2 controls the DNA methylation state of latent Epstein-Barr virus. J Virol 91:e00804-17.https://doi.org/10.1128/JVI .00804-17.
EditorJae U. Jung, University of Southern
California
Copyright© 2017 American Society for
Microbiology.All Rights Reserved.
Address correspondence to Paul M. Lieberman, Lieberman@wistar.org.
GENOME REPLICATION AND REGULATION OF VIRAL GENE EXPRESSION
crossm
on November 7, 2019 by guest
http://jvi.asm.org/
memory B cells (3). The virus encodes a number of proteins and noncoding RNAs that control and coordinate the viral and host gene expression programs during this process of latency establishment (3). This process can also increase the risk of lymphoid and epithelial malignancies, especially in the context of immunosuppression.
The establishment of EBV latent infection involves progressive epigenetic modifi-cations of both virus and host genomes (4, 5). DNA methylation, in particular, plays a central regulatory role in establishing and regulating EBV latency (4, 6, 7). Viral genomes arrive as unmethylated DNA during primary infection and remain unmethylated at active promoter regions throughout the growth and proliferation stages. This form of latency, referred to as latency type III, involves the expression of at least 9 viral proteins (EBNAs, LMPs), several noncoding RNAs (EBV-encoded small RNAs [EBERs], BamHI right transcripts [BARTs]), and numerous microRNAs (miRNAs). EBV lymphoblasts are selec-tively eliminated by a strong immunological response to many of these viral antigens, and epigenetic silencing is required for viral genome persistence in long-lived differ-entiated memory B cells. This form of latency, referred to as type I, involves the restricted expression of only EBNA1 and noncoding RNAs. The remaining viral latency genes are silenced, in part, by DNA methylation. In addition, high levels of DNA methylation are required for the efficient lytic cycle reactivation of the viral genome, since the immediate early protein BZLF1 (also referred to as Z, Zebra, or Zta) recognizes only the methylated DNA of several regulatory sites in the viral genome (8–10). In some
latency types, the DNA-demethylating agent 5=azacytidine can induce lytic cycle gene
transcription or a transition from type I to type III latency (11–13). In addition, high levels of host genome DNA methylation are observed in EBV-positive gastric carcinoma and nasopharyngeal carcinoma (14, 15). These studies underscore the importance of DNA methylation in regulating viral and host gene expression during EBV latency.
EBV encodes several factors that can modulate the host and viral epigenetic state (16). EBNA2 is among the first viral genes expressed upon primary infection of B lymphocytes. EBNA2 is a transcriptional coactivator that mimics intracellular Notch
binding to the sequence-specific transcription factor RBP-j(also called RBPJ, CBF1, and
CSL) at promoters and enhancers of genes important for B-cell proliferation (17–21; reviewed in references 16 and 22). EBNA2 colocalizes on chromatin with several other cellular transcription factors, including Pu.1 and EBF1, that have key regulatory roles in lymphoid development (23, 24). Recent genome-wide chromatin immunoprecipitation for high-throughput sequencing (ChIP-seq) studies reveal that EBNA2 stabilizes the
formation of new RBP-jand EBF1 binding sites, suggesting that it reprograms
tran-scription in latently infected B lymphocytes (25). EBNA2 is expressed during the proliferative phase of EBV latency III establishment, but its expression is epigenetically silenced during the resting memory B-cell phase of EBV latency I. This silencing is typically associated with DNA methylation of the transcriptional regulatory elements in the EBNA2 promoters, termed Cp and Wp. Methylation of these sites blocks DNA
binding by sequence-specific factors like RBP-j, which results in transcriptional silence
at Cp and Wp.
The ten-eleven translocation (TET) genes have been implicated in the active
dem-ethylation of 5= methylcytosine DNA (26, 27). TET family members share a core
enzymatic domain that is capable of oxidizing methyl cytosine to hydroxymethylcyto-sine in a process that facilitates the activate demethylation of CpG DNA (28, 29). The TET2 gene is among the most frequently mutated genes in hematological malignancies (26). In mice, the combined loss of TET2 and TET1 results in an increase in B-cell malignancies (30). In one clinical study, a lymphoma patient with a TET2 mutation was found to respond to azacytidine, suggesting that TET2-associated DNA demethylation is an important driver of lymphomagenesis (31). TET2 is subject to complex transcrip-tional and posttranscriptranscrip-tional regulation. Coexpression of CXXC4/IDAX, an evolutionary partner protein of TET2, has been linked to the posttranslation degradation of TET2 (32). TET2 is also subject to posttranslational suppression by numerous miRNAs (33–36). Here, we present evidence that TET2 is also regulated at the level of DNA methylation silencing and that its expression is coordinately regulated with EBV latency type. We
on November 7, 2019 by guest
http://jvi.asm.org/
also show that TET2 works cooperatively with EBNA2 and RBP-jto regulate the DNA methylation state and transcription programs of EBV.
RESULTS
EBV genome DNA methylation correlates inversely with TET2 expression. To investigate the relationship between EBV DNA methylation status and TET2 gene expression, we first validated that DNA methylation was elevated at key viral regulatory regions in a cell line with type I latency, but not in two related cells with type III latency (Fig. 1A). We compared the isogenic Burkitt lymphoma cell lines Mutu I and Mutu III with either type I or type III latency gene expression patterns, respectively. We also assayed an LCL cell line generated with the same EBV genome as Mutu I cells. All LCLs have type III latency. The DNA methylation state was analyzed by a methylcytosine-DNA immunoprecipitation (MeDIP) assay using real-time PCR amplimers at viral regu-latory regions for EBER noncoding RNAs, the family of repeat (FR) region of OriP, and the promoter regions for LMP2A, LMP1, Cp, Qp, Zp, or Rp (Fig. 1A). As expected, we found that LMP2A, LMP1, and Cp were highly methylated in type I but not in type III latency cells. We also observed high levels of Rp methylation in type I but not type III latency. No methylation was observed at EBER, Qp, or Zp in either type I or type III. These findings confirm that key viral regulatory regions have elevated DNA methylation in type I compared to type III latency (37, 38).
We next assayed whether TET2 expression correlated with EBV latency type (Fig. 1B and C). Reverse transcription-quantitative PCR (RT-qPCR) assays revealed that in Mutu III and LCL cells, TET2 mRNA was readily detected, while this was undetectable in Mutu I cells (Fig. 1B). Western blotting revealed similar patterns for TET2 protein expression in Mutu III and LCL but not in Mutu I (Fig. 1C). We also observed that IDAX, a protein
FIG 1DNA methylation status and TET2 expression in EBV latency types. (A) MeDIP assay on Mutu I (blue), Mutu III (yellow), or LCL (red) was performed by qPCR for EBV regulatory regions for EBERs, family of repeats (FR), LMP2A promoter, LMP1 promoter, Cp, Qp, Zp, and Rp, and the percentage of input DNA was quantified. (B) RT-qPCR for TET2 mRNA analysis in Mutu I, Mutu III, or LCL, as indicated, relative to GAPDH. (C) Western blot with antibodies to TET2, IDAX, EBNA2, or GAPDH in cell extracts from Mutu I, Mutu III, or LCL.
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.86.326.70.360.2]reported to bind and destabilize TET2, was expressed at similar levels in all three cell types (Fig. 1C). EBNA2 protein expression was also tested to confirm the latency type of each cell (Fig. 1C). These findings suggest that TET2 expression is coordinately regulated with EBV latency type.
TET2 mRNA and protein expression correlates with EBV type III latency pat-terns in BL and LCL cells.To determine if the expression of TET2 correlated with EBV latency type more generally, we expanded our survey of Burkitt’s lymphoma (BL) cells that have well-characterized type I or type III latency gene expression patterns (Fig. 2A and B). RT-qPCR revealed high levels of TET2 mRNA in Kem III and two different LCLs but not in Kem I or Akata cells, which have type I latency (Fig. 2A). Western blot analysis showed that the TET2 protein is expressed at measurable levels in type III latency cell lines but is essentially undetectable in type I latency cell lines, including EBV-negative Akata and DG75 BL cell lines (Fig. 2B). Raji, a BL-derived cell line with atypical type III latency expression features, had low TET2 mRNA but expressed an aberrantly high-molecular-weight form of TET2 (Fig. 2A and B), potentially consistent with its unusual gene expression pattern.
To determine if additional regulators of DNA methylation had a similar pattern of gene expression, we assayed other TET family members, TET1 and TET3, as well as DNA methyltransferase DNMT1, DNMT3a, and DNMT3b genes (Fig. 2C). We found that TET3, DNMT1, and DNMT3a genes were slightly elevated in LCLs compared to Mutu I, but none of these candidate genes were as enriched in LCLs relative to Mutu I as was TET2. The TET3 protein level was then examined in all the cell lines as illustrated in Fig. 2B. The Western blot analysis showed that most of the BL and LCL cells expressed similar levels of the TET3 protein, except DG75, which had a slightly higher level, and Raji, which had a slightly lower level of TET3 (Fig. 2D). These findings suggest that TET2 expression correlates better than several other DNA methylation regulators with EBV methylation status and latency type.
TET2 depletion deregulates the EBV latency gene expression program.To determine if TET2 expression is important for EBV latency, we depleted TET2 using lentivirus short hairpin RNA (shRNA) transduction in LCLs (Fig. 3). We tested five different shRNAs and found only one (shTET2-1) that could deplete TET2 consis-tently. As shown in Fig. 3A, shTET2-1 had approximately 50% depletion efficiency,
and a less efficient shTET2-2 had⬃38% depletion efficiency. We found that partial
depletion of TET2 led to a partial reduction of latency proteins EBNA2, LMP1, and EBNA3C and a corresponding increase in lytic proteins Zta and EA-D (Fig. 3A). Similar changes in EBV gene expression were observed by RT-qPCR (Fig. 3B). TET2 depletion by shTET2-1 led to a reduction of EBNA2, EBNA3s, LMP1, and LMP2A mRNA, with a corresponding increase in Zta and Rta. Lytic cycle activation was further corroborated by qPCR of viral DNA (Fig. 3C). These findings indicate that TET2 depletion in type III latency leads to the loss of latency gene expression and an increase in lytic gene transcription and viral DNA replication.
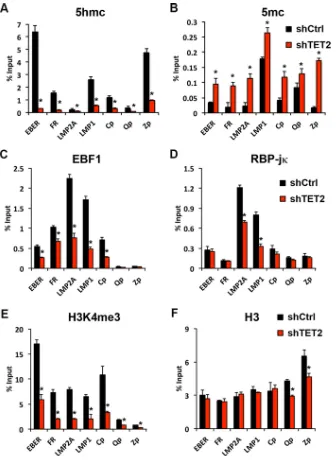
TET2 depletion alters the EBV epigenome.To investigate the mechanism of how TET2 depletion may alter the EBV epigenome, we next assayed cytosine hydroxy-methylation (5hmc) and hydroxy-methylation (5mC) by DIP assay in LCLs (Fig. 4A and B). We found that EBV regulatory regions, including the EBERs, FR, Cp, LMP1p, and Zp, had elevated levels of 5hmc, which were extensively lost after shRNA depletion of TET2 (Fig. 4A). In contrast, 5mC increased after shRNA depletion of TET2 at all EBV regulatory sites, especially FR, LMP2Ap, Cp, and Zp. Qp remained mostly resistant to these DNA methylation-associated changes. Our previous study has shown that EBF1 bound to
EBER and FR and that both EBF1 and RBP-jbound to LMP2Ap, LMP1p, and Cp in LCLs
(25). Many of these EBF1 and RBP-jK binding sites were dependent on EBNA2 expres-sion and showed epigenetic variance between type I and type III cell lines (25). We now
show that TET2 depletion in an LCL led to a reduced association of EBF1 and RBP-jto their
binding sites at LMP2Ap, LMP1p, and Cp (Fig. 4C and D). Neither EBF1 nor RBP-jbound
significantly to the Qp or Zp region, indicating that their ChIP binding was specific.
on November 7, 2019 by guest
http://jvi.asm.org/
Interestingly, TET2 depletion also led to a loss of H3K4me3 at most of these viral regulatory elements (Fig. 4E). Histone H3 was not changed significantly, except for a loss at Qp and Zp (Fig. 4F). The effect of TET2 depletion on cellular genes is shown in Fig. 5.
TET2 depletion alters the cellular epigenome. We have previously found that EBNA2-responsive promoters are regulated similarly on the viral and host genomes
FIG 2TET2 expression in B lymphocytes with EBV type III latency. (A) RT-qPCR for TET2 mRNA assayed in EBV-negative DG75 and Akata, in EBV type I latency Akata EBV⫹, Mutu I, Kem 1, and in EBV type III latency Kem III, Raji, LCL187, and LCL352 cells. (B) Western blot with antibodies to TET2 or GAPDH for cells as described for panel A. (C) RT-qPCR for TET2, TET1, or TET3 (left panel) or DNMT1, DNMT3a, or DNMT3b (right panel) in Mutu I (blue) or LCL (red). (D) Western blot with antibodies to TET3 or GAPDH for cells as described for panels A and B.
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.70.337.68.599.2](25). Therefore, we next asked whether TET2 depletion had a similar effect on cellular regulatory elements that are known to be responsive to EBNA2. We found that cellular genes for interleukin-7 (IL-7), HES1, and FCER2, which were previously found to be responsive to EBNA2, were regulated in a fashion similar to that of EBV genes after shTET2 depletion in LCLs (Fig. 5). Like EBV regulatory elements (Fig. 4), IL-7, HES1, and FCER2 showed high levels of 5hmc that could be depleted by shRNA targeting of TET2 (Fig. 5A). Depletion of TET2 also led to an increase in DNA methylation at IL-7 and HES1
(Fig. 5B), partial loss of EBF1 at FCER2, and partial loss of RBP-jat IL-7 and FCER2 (Fig.
5C and D). A more significant loss of H3K4me3 was observed, while total H3 did not change significantly (Fig. 5E and F). The mRNA level of IL-7, HES1, or FCER2 was examined in LCL with or without shTET2 depletion (Fig. 5G). The IL-7 transcription level was slightly increased, while HES1 and FCER2 transcription levels were modestly decreased when TET2 was knocked down. Taken together, these findings indicate that TET2 is important for maintaining hydroxymethylation at viral and cellular regulatory elements and the consequent epigenetic programming important for maintaining type III latency.
Identification of EBNA2, EBF1, and RBP-jbinding sites on the TET2 gene.To understand how the TET2 gene may be coregulated with EBV type III latency, we analyzed existing ChIP-seq data sets from LCL (type III) and Mutu I (type I) for EBNA2
(LCL only), EBF1, RBP-j, H3K4me3, and H3K36me3 around the TET2 gene locus (Fig. 6).
Significant H3K4me3 was observed at the TET2 transcription start site (TSS), and significant H3K36me3 was observed through the TET2 gene body in LCL relative to Mutu I, consistent with TET2 transcriptional activation in LCL relative to Mutu I. A
significant EBNA2 peak (EBNA2_A) was identified at a position⬃350 kb upstream of the
TET2 TSS (Fig. 6A and B). This position is also upstream of the transcription start site for IDAX, which is a divergently transcribed neighboring gene of the TET2 gene. This EBNA2 peak (EBNA2_A) overlapped a significant EBF1 peak common to both Mutu I and
LCL and a smaller RBP-jpeak that was enriched in LCL relative to Mutu I. Another
significant EBNA2 peak (EBNA2_B), which is 80 kb away from the TET2 TSS, overlapped
a smaller RBP-jpeak. The two major RBP-jbinding sites, upstream of the TSS (RBP_L)
and within the TET gene body (RBP_R), also overlapped relatively small EBNA2 peaks.
The two RBP-jbinding sites were confirmed by conventional ChIP-qPCR and shown to
be highly enriched in LCLs but were not detected in Mutu I cells (Fig. 6C). Taken
together, these findings indicate that EBNA2 can colocalize with EBF1 or RBP-j at
multiple sites throughout the TET2 gene locus and that RBP-j binding to TET2 is
regulated in a latency type-dependent manner.
FIG 3shRNA depletion of TET2 alters EBV gene expression. (A) Western blot analysis of LCLs transduced with either shTET2-1, shTET2-2, or control lentivirus and assayed with antibody to TET2, EBNA2, LMP1, EBNA3C, ZTA, EA-D, TET3, or actin. The depletion percentage of each shTET2 was determined by the signal intensity of TET2 relative to that of actin and then compared to that of shCtrl. (B) RT-qPCR for shTET2-1 (red), shTET2-2 (blue), or control (black) lentivirus-transduced LCLs assayed for RNA expression of EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, LMP1, LMP2A, ZTA, or RTA relative to GAPDH, as indicated. (C) qPCR of intracellular DNA for EBV using primers for either Cp relative to cellular actin.*,P⬍0.05.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.44.427.69.222.2]Epigenetic regulation of the TET2 gene locus is similar to that of EBV epi-genotypes.We next investigated the DNA methylation status of the TET2 gene locus in cell lines with either type I or type III EBV latency (Fig. 6D). We found that the regions overlapping the major transcription start site for TET2 (TSS_1 and TSS_2) were highly methylated in Mutu I (type I) cells but not detectably methylated in Mutu III or LCL (type
III cells). We did not detect high levels of methylation at the RBP-jsites (RBP_L or
RBP_R) in either type I or type III cells. We next tested whether DNA methylation at the TSS was associated with transcriptional silencing in type I latency (Fig. 6E). Mutu I cells
treated with 5=azacytidine showed a large (⬎10-fold) increase in TET2 mRNA relative
to control-treated samples by RT-PCR (Fig. 6E). This increase in transcription correlated with an increase in histone H3K4me3 at the TET2 TSS (Fig. 6F). The levels of TET2 mRNA expression and H3K4me3 did not reach those of LCLs, suggesting that additional
FIG 4TET2 depletion alters the EBV epigenome. (A to F) LCLs transduced with shTET2-1 (red) or control lentivirus (black) were assayed by DIP for 5hmC (A) or 5mC (B) or by ChIP for EBF1 (C), RBP-j(D), H3K4me3 (E), or total H3 (F) at EBV regions for EBER, FR, LMP2Ap, LMP1p, Cp, Qp, or Zp.*,P⬍0.05.
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.41.377.69.531.2]factors other than DNA demethylation drive TET2 transcription. Nevertheless, 5= aza-cytidine had no effect on TET2 mRNA or H3K4me3 methylation in LCLs, further confirming the hypomethylation status of TET2 TSS in LCLs (Fig. 6E, right panel, and F).
EBNA2 is required for TET2 expression.The contribution of EBNA2 in regulating TET2 expression was tested using the EREB2.5 LCLs with conditional expression of EBNA2 (Fig. 7A and B). In the absence of estradiol (E2), EBNA2 is sequestered in the cytoplasm and fails to bind and activate target genes in the nucleus (39). Western blot
FIG 5TET2 depletion alters EBNA2-responsive genes in the cellular genome. The process was as described for Fig. 4, except at cellular genes. (A to F) LCLs transduced with shTET2-1 (red) or control lentivirus (black) were assayed by DIP for 5hmC (A) or 5mC (B) or by ChIP for EBF1 (C), RBP-j(D), H3K4me3 (E), or total H3 (F) cellular genes for IL-7, HES1, FCER2, or actin. (G) RT-qPCR for shTET2-1 (red) or control (black) lentivirus-transduced LCLs assayed for RNA expression of IL-7, HES1, or FCER2 relative to GAPDH, as indicated.*,P⬍0.05.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.69.341.68.576.2]analysis revealed that the removal of E2 led to a rapid loss of the TET2 protein (Fig. 7A). EBNA2 protein levels do not decrease as rapidly, but previous studies indicate that its nuclear localization and transcription function is rapidly curtailed. Inactivation of EBNA2 led to a corresponding decrease in TET2 mRNA to virtually undetectable levels by 72 h (Fig. 7B). These changes corresponded to a complete loss of EBNA2 binding to the TET2
gene loci by ChIP assay (Fig. 7C) and an ⬃2-fold decrease of RBP-jbinding to its
FIG 6Epigenetic features of TET2 gene locus in type I and III cell lines. (A) ChIP-seq tracks for EBNA2, EBF1, RPB-j, H3K4me3, or H3K36me3 in LCL (red) or Mutu I (blue) cells over the regions spanning TET2 and CXXC4/IDAX gene loci. Binding sites for EBNA2 are designated EBNA2_A and EBNA2_B and indicated by asterisks. (B) Zoom-in of ChIP-seq track for RBP-jfor EBNA2 and RBP-jin Mutu I (blue) or LCL (red) cells over the TET2 promoter locus. Binding sites for RBP-j are designated RBP_L and RBP_R and indicated by asterisks. (C) ChIP-qPCR for validation of RBP-jbinding at sites RBP_L and RBP_R. (D) MeDIP for 5mC in Mutu I (blue), Mutu III (yellow), or LCL (red) cells at TET2 loci for TSS_1, TSS_2, RBP_L, RBP_R, or at actin. (E) RT-qPCR for TET2 mRNA in Mutu I or LCL cells treated with 10M 5=azacytidine (orange) or control (black) for 72 h. (F) ChIP-qPCR for H3K4me3 with primers for the TET2 TSS_1/2 in Mutu I (left) or LCL (right) cells treated with 5=azacytidine (orange) or control (black) for 72 h.
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.43.543.77.560.2]binding sites (Fig. 7D). These findings indicate that EBNA2 is required for TET2 mRNA expression, and this function likely depends on EBNA2 binding to the TET2 gene loci.
RBP-jis required for TET2 expression in type III latently infected B lympho-cytes.To better understand how EBNA2 may regulate TET2 in type III latency, we asked
whether TET2 expression was dependent on the EBNA2-interacting partner RBP-j.
RBP-jbinding sites were identified in the TET2 gene locus by ChIP-seq (Fig. 6A and B)
and validated by ChIP-qPCR (Fig. 6C). To determine whether RBP-jwas important for
TET2 expression in LCLs, we depleted RBP-j using three different shRNA-targeting
sequences (Fig. 7E and F). In all three samples, TET2 protein levels were reduced,
correlating with the depletion of RBP-j(Fig. 7E). In addition, the depletion of RBP-j
correlated with the loss of TET2 mRNA expression (Fig. 7F). These findings indicate that
RBP-jis functionally important for TET2 mRNA expression in LCLs.
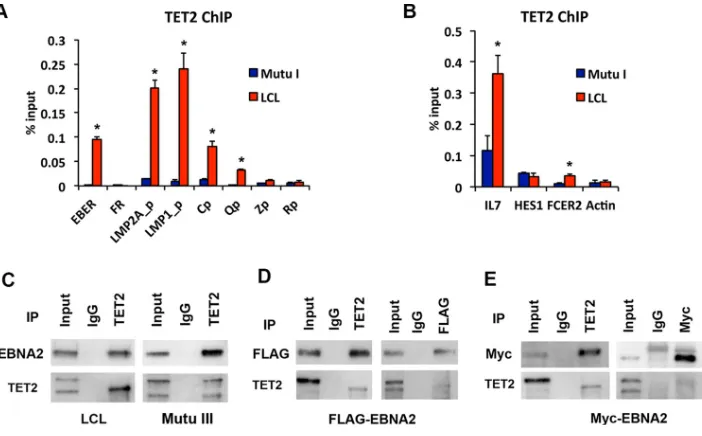
TET2 colocalizes and coimmunoprecipitates with EBNA2.To better understand how TET2 may coregulate EBNA2-dependent genes, we tested its ability to associate
FIG 7TET2 expression depends on EBNA2 and RBP-jin type III latency. (A) EREB cells withdrawn from estradiol (E2) for 24, 48, or 72 h were assayed by Western blotting for TET2, EBNA2, or actin. (B) RT-qPCR results for TET2 mRNA for EREB cells treated as described for panel A. (C) ChIP-qPCR results for EBNA2 at EBNA2_A, EBNA2_B, RBP_L, or RBP_R sites at TET2 loci or at control actin in EREB cells in the presence (⫹E2) or absence (⫺E2) of estradiol for 48 h. (D) ChIP-qPCR results for RBP-jat RBP_L or RBP_R sites or at control actin in EREB cells in the presence (⫹E2) or absence (⫺E2) of estradiol for 48 h. (E) LCLs transduced with three different shRNAs for RBP-jor control (shCtrl) and assayed by Western blotting with antibody to TET2, RBP-j, or actin. (F) Cells were treated as described for panel E and then assayed by RT-qPCR for TET2 mRNA relative to actin.*,P⬍0.05.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.50.362.69.469.2]with viral and cellular chromatin sites that are also known to be bound and regulated by EBNA2 (Fig. 8A and B). ChIP-qPCR with TET2 antibody revealed that TET2 is enriched in LCLs at EBER, LMP2Ap, LMP1p, and Qp relative to Mutu I (Fig. 8A). Similarly, TET2 was enriched at cellular IL-7 and FCER2 genes, especially in LCLs (Fig. 8B). These findings indicate that TET2 co-occupies sites bound and regulated by EBNA2.
We next tested whether the EBNA2 protein interacts with the TET2 protein by coimmunoprecipitation (co-IP) (Fig. 8C to E). Immunoprecipitation of TET2 in LCL or Mutu III (Fig. 8C) or 293T cells transfected with either FLAG-tagged EBNA2 (Fig. 8D) or Myc-tagged EBNA2 (Fig. 8E) revealed a strong co-IP of EBNA2 with antibody to EBNA2 (Fig. 8C), FLAG epitope (Fig. 8D), or Myc epitope (Fig. 8E). We were unable to copre-cipitate TET2 with EBNA2 antibody IP in LCL or Mutu III cells. However, we observed a weak co-IP of TET2 in 293T cells with FLAG-EBNA2 and Myc-EBNA2, suggesting that this interaction may be sensitive to EBNA2 antibodies. Taken together, these studies
suggest that an interaction between EBNA2 and TET2 may direct TET2 to RBP-jtarget
sites on the EBV and cellular genomes (Fig. 9).
DISCUSSION
Control of EBV latency involves dynamic interactions between virus and host epigenetic factors. Here, we have found a coordinate regulation of EBV latency type with the expression of the cellular TET2 gene. Specifically, we find that TET2 expression correlates with EBV type III latency program (Fig. 1 and 2) and is required to maintain EBV latency (Fig. 3). TET2 depletion led to a loss of cytosine hydroxymethylation and an
increase in cytosine methylation at viral and cellular sites regulated by EBNA2-RBP-j
(Fig. 4 and 5). We found that the TET2 gene is subject to regulation by EBNA2-RBP-j
and can be silenced by DNA methylation in the absence of these factors (Fig. 6). Loss
of EBNA2 or depletion of RBP-jled to a loss of TET2 expression, explaining how it is
coregulated with EBV type III latency genes (Fig. 7). Finally, we show by ChIP that TET2
can colocalize with EBNA2 and RBP-jat several regulatory regions of the genome and
by co-IP that TET2 can associate with EBNA2 (Fig. 8). We suggest that TET2 cooperates
FIG 8TET2 colocalization and coimmunoprecipitation with EBNA2. (A) ChIP-qPCR for TET2 in Mutu I (blue) or LCL (red) at EBV genome sites for EBER, FR, LMP2Ap, LMP1p, Cp, Qp, Zp, or Rp. (B) Same as described for panel A, except assayed as cellular RBP-jbinding sites at IL-7, HES1, FCER2, or actin. (C) LCL or Mutu III cell lysate was subjected to IP with TET2 or IgG control and assayed by Western blotting with EBNA2 or TET2. Input is 5% of total. (D) 293T cells transfected with FLAG-EBNA2 were subjected to IP with TET2 or IgG (left panel) or with FLAG or IgG (right panel) followed by Western blotting with FLAG or TET2 antibody. (E) 293T cells transfected with Myc-EBNA2 were subjected to IP with TET2 or IgG (left panel) or with Myc or IgG (right panel) and then assayed by Western blotting with Myc or TET2 antibody.*,P⬍0.05.
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.44.395.70.288.2]with EBNA2 to reverse DNA methylation, as well as maintain high levels of cytosine
hydroxymethylation and H3K4me3 at RBP-jtranscriptional regulatory targets.
TET2 has been shown to play an important role in regulating DNA methylation, and in particular in promoting active demethylation through successive hydroxylation reactions followed by TDG-associated base excision repair (40). TET2 may also function in maintaining an activated chromatin state through generation of hydroxymethylated cytosine, which may function as an independent epigenetic mark (41). TET2 has also
been shown to directly interact with O-GlcNAc transferase (OGT), facilitate histone
O-GlcNAcylation, and promote histone H3K4me3 methylation to regulate gene tran-scription (42). Our findings suggest that TET2-dependent hydroxymethylation provides an important signal for EBV gene regulation. We found that shRNA depletion of TET2 leads to a rapid loss of both cytosine hydroxymethylation as well as H3K4me3 and transcription from EBV regulated promoters. The loss of TET2-dependent hydroxy-methylation appears to be more rapid than the more gradual loss of DNA hydroxy-methylation. These findings support the model that TET2-dependent hydroxymethylation is an independent epigenetic mark regulating promoter H3K4me3 and transcriptional activ-ity of EBV latency type III genes.
Cytosine methylation and hydroxymethylation can have profound effects on site recognition by many sequence-specific DNA binding proteins. It is well established that cytosine methylation promotes ZTA binding to regulatory elements in Rp and facilitates EBV reactivation from latency (8–10, 43). CTCF binding is also sensitive to site-specific DNA methylation, and TET2 has also been implicated in regulating CTCF binding, especially at sites that may control differential mRNA splicing (44). TET2-mediated hydroxymethylation can also create bivalency at CpG sites that are important for rapid response genes and stem cell fate decisions (45). Whether TET2 also regulates EBV type III promoters through CTCF binding, chromatin bivalency, or mRNA splicing will be important to investigate in the future.
TET2 is an important regulator of hematopoietic cell differentiation and suppressor of tumorigenesis. TET2 mutations are found frequently in hematological malignancies (46). TET2 is highly expressed in hematopoietic stem cells and in progenitor cells and is downregulated during differentiation (47). TET2 expression levels may also help to stratify the two major forms of diffuse large B-cell lymphomas (DLBCLs), with the high levels of TET2 and hypomethylation found in the activated B-cell (ABC) type and low levels of TET2 and hypermethylation in the germinal center (GC) type (48, 49). A recent
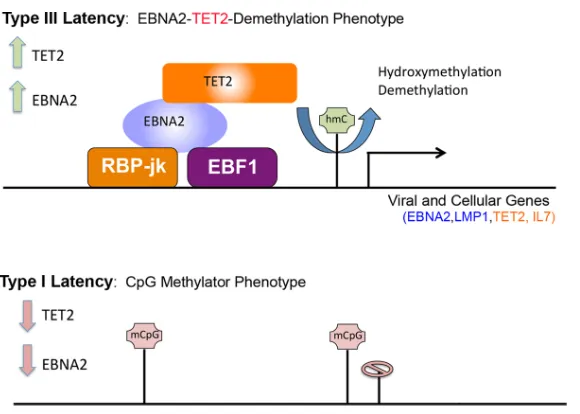
FIG 9Coordinate regulation of the EBV epigenome and TET2 gene transcript by TET2 and EBNA2. Model depicting TET2 interacting with EBNA2 at RBP-j sites to regulate the demethylation of the EBV epigenome and the TET2 gene locus in type III latency.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.61.351.66.273.2]study from the Kenney lab has shown that EBV type I latency has GC-like features while type III latency has ABC-like features (48). This study also showed that TET2 cooper-ates with EBNA2 to activate methylated promoters. Previous studies from this group indicated that TET2-associated hydroxymethylation attenuates EBV lytic reactiva-tion through preventing methylcytosine recognireactiva-tion by Zta (50). Thus, TET2 regu-lates multiple aspects of the EBV epigenome during latency.
TET2 has also been implicated in the epigenetic control of other viruses. Hepatitis B virus (HBV) X protein forms a complex with TET2, EZH2, and DNMT3L to demethylate RelA sites involved in the activation of viral target genes, such as EpCAM (51). Human herpesvirus-6B (HHV6B) induces hypomethylation on chromosome 17p13.3 to promote its own site-specific integration through a mechanism thought to involve TET2 (52). TET2 may also function as a tumor suppressor and restriction factor for virus-associated hypermethylation (53). EBV is known to induce hypermethylation in EBV-associated gastric carcinomas, and this was shown to correspond to a loss of TET2 expression and function (54). How EBV may downregulate TET2 in epithelial cancers is not yet known, but the process may be similar to how it is downregulated in EBV latency type I B cells.
In conclusion, our findings and those of others implicate TET2 as an important regulator of host and viral epigenomes during EBV latent infection. Moreover, we find
that TET2 cooperates with EBNA2 to target RBP-j-responsive genes, including the type
III transcripts of the EBV genome and the TET2 gene itself. This feed-forward, integrated regulatory mechanism may be an essential feature of viral latency control, as well as critical for the establishment of stable gene expression programs.
MATERIALS AND METHODS
Cells.EBV-positive BL cell lines Mutu I, Mutu III, Kem I, and Kem III were obtained from Jeffrey Sample, Penn State University Hershey Medical School, Hershey, PA. DG75 and Raji were obtained from ATCC. Akata cell lines with or without EBV were obtained from Elena Mattia, Sapienza University of Rome, Rome, Italy. LCL cell lines were generated from human B cells (provided by the Wistar Institute Phlebotomy Core) transformed with Mutu I virus. LCLs, DG75, Raji, Akata, Mutu I, Mutu III, Kem I, and Kem III cells were maintained in RPMI containing 12% fetal bovine serum (FBS) and antibiotics (penicillin and streptomy-cin). EREB 2.5, a lymphoblastoid cell line expressing the estrogen-inducible EBNA2-estrogen receptor (ER) fusion protein (55), was maintained in RPMI containing 12% FBS, antibiotics (penicillin and streptomycin), and 1M estradiol. 293T cells were obtained from ATCC and grown in Dulbecco’s modified Eagle medium (DMEM) with 10% FBS and antibiotics.
Lentiviral transduction.pLKO.1 vector-based shRNA constructs for TET2 (shTET2-1, TRCN0000144344; shTET2-2, TRCN0000122172) or RBPj (sh RBPj-1, TRCN0000016203; shRBPj-2, TRCN0000016204; shRBPj-3, TRCN0000016205) were obtained from Open Biosystems. Control shRNAs (shControl or shCtrl) were generated in the pLKO.1 vector with the target sequence 5=-TTATCGCGCATATCACGCG-3=. Lenti-viruses were produced by the use of envelope and packaging vectors pMD2.G and pSPAX2 as described previously (56). Mutu I or LCL cells were infected with lentiviruses carrying pLKO.1-puro vectors by spin infection at 450⫻gfor 90 min at room temperature. The cell pellets were resuspended and incubated in fresh RPMI medium and then treated with 2.5g/ml puromycin at 48 h after the infection. The RPMI medium with 2.5g/ml puromycin was replaced every 2 to 3 days. The cells were collected after 8 days of puromycin selection and then subjected to further analyses, as indicated.
MeDIP and hMeDIP assays.Genomic DNA was purified using the Wizard Genomic DNA purifi-cation kit (A1120; Promega) and then subjected to methylcytosine-DNA immunoprecipitation (MeDIP) or hydroxymethylcytosine-DNA-IP (hMeDIP) assays. The MeDIP or hMeDIP assays were performed using the MagMeDIP kit (C02010021; Diagenode) or the hMeDIP kit (C02010031; Diag-enode). qPCR assays were then performed as described previously (57). Quantification of precipi-tated DNA was determined using real-time PCR and the deltaCt(cycle threshold) method for relative quantitation (ABI 7900HT Fast real-time PCR system). Primers for DIP assays are listed in Table S2 in the supplemental material.
RNA extraction and quantitative RT-PCR.RNA was isolated from 2⫻106cells using the RNeasy kit
(Qiagen) and then further treated with DNase I by using the DNase treatment and removal kit (Ambion). RT-PCR was performed as previously described (58). Real-time PCR was performed with a SYBR green probe in an ABI Prism 7900, and the deltaCTor delta deltaCTmethod was used for relative quantitation.
Primer sequences for RT-PCR are listed in Table S1 in the supplemental material.
Coimmunoprecipitation.Co-IP was performed as described previously (59). Rabbit IgG (Santa Cruz Biotechnology sc-2027), rabbit anti-TET2 (EMD Millipore ABE364), Myc (Cell Signaling 2278), mouse IgG (Santa Cruz Biotechnology sc-2025), or mouse anti-FLAG M2 (Sigma F1804) was used in co-IP.
Antibodies used for Western blotting. Antibodies used for Western blotting were rabbit polyclonal RBP-j (Abcam AB25949), TET2 (Proteintech 21207-1-AP), TET3 (GeneTex GTX121453), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Cell Signaling catalog number 2118), Myc (Cell
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
Signaling catalog number 2278); rabbit polyclonal anti-Zta was generated from full-length Zta at Pocono Rabbit Farms; also used were rat monoclonal anti-EBNA2 (EMD Millipore MABE8), mouse monoclonal anti-TET2 (EMD Millipore MABE462), LMP1 (Dako M0897), EA-D (Abcam ab49668), sheep polyclonal anti-EBNA3C (Exalpha F125P), actin-peroxidase antibody (Sigma A3854), FLAG-peroxidase antibody (Sigma A8592).
ChIP-qPCR assays.ChIP-qPCR assays were performed as described previously (57). Quantification of precipitated DNA was determined using real-time PCR and the delta CTmethod for relative
quantitation (ABI 7900HT Fast real-time PCR system). Rabbit IgG (Santa Cruz Biotechnology sc-2027), rabbit anti-EBF1 (EMD Millipore AB10523), RBP-j(Abcam AB25949), histone H3K4me3 (EMD Milli-pore 07-473), histone H3 (EMD MilliMilli-pore 07-690), and TET2 (Proteintech 21207-1-AP) were used in ChIP assays. Mouse IgG (Santa Cruz Biotechnology sc-2025) and mouse anti-EBNA2 (gift from Paul Farrell, UK) antibodies were also used in ChIP assays. Primers for ChIP assays are listed in Table S2 in the supplemental material.
ChIP-seq and genomic data processing.ChIP-seq and genome data processing of histone H3K36me3 were performed as described previously (20). Rabbit anti-histone H3K36me3 (Abcam ab9050) was used in ChIP-seq. The EBNA2, RBP-j, EBF1, and H3K4me3 genome browser tracks were generated previously (20) and are available at NCBI GEO data sets.
SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at https://doi.org/10.1128/JVI
.00804-17.
SUPPLEMENTAL FILE 1,PDF file, 0.1 MB.
ACKNOWLEDGMENTS
We thank the Wistar Institute Cancer Center Core Facilities for Genomics, Bioinfor-matics, and Flow Cytometry. We thank Betina Kempkes (HelmholtzZentrum München) for providing EREB2.5 cells, Elena Mattia (La Sapienza University, Rome) for Akata cells, and Paul Farrell (IC, London) for the antibody to EBNA2.
This work was funded by grants from the NIH (R01 CA093606, P30 CA010815-48, and R01 DE017336) to P.M.L.
REFERENCES
1. Longnecker R, Kieff E, Cohen JI. 2013. Epstein-Barr virus, p 1898 –1959.In
Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 1. Lipincott, Philadelphia, PA.
2. Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768.https://doi.org/10.1038/nrc1452.
3. Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1:75– 82.https://doi.org/10.1038/35095584. 4. Hammerschmidt W. 2015. The epigenetic life cycle of Epstein-Barr virus.
Curr Top Microbiol Immunol 390:103–117.https://doi.org/10.1007/978 -3-319-22822-8_6.
5. Tempera I, Lieberman PM. 2014. Epigenetic regulation of EBV persis-tence and oncogenesis. Semin Cancer Biol 26:22–29.https://doi.org/10 .1016/j.semcancer.2014.01.003.
6. Woellmer A, Hammerschmidt W. 2013. Epstein-Barr virus and host cell methylation: regulation of latency, replication and virus reactivation. Curr Opin Virol 3:260 –265.https://doi.org/10.1016/j.coviro.2013.03.005. 7. Niller HH, Tarnai Z, Decsi G, Zsedenyi A, Banati F, Minarovits J. 2014. Role of epigenetics in EBV regulation and pathogenesis. Future Microbiol 9:747–756.https://doi.org/10.2217/fmb.14.41.
8. Woellmer A, Arteaga-Salas JM, Hammerschmidt W. 2012. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog 8:e1002902.https://doi.org/10.1371/journal .ppat.1002902.
9. Dickerson SJ, Xing Y, Robinson AR, Seaman WT, Gruffat H, Kenney SC. 2009. Methylation-dependent binding of the Epstein-Barr virus BZLF1 protein to viral promoters. PLoS Pathog 5:e1000356.https://doi.org/10 .1371/journal.ppat.1000356.
10. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. 2004. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat Genet 36:1099 –1104.https://doi.org/10.1038/ng1424. 11. Li L, Su X, Choi GC, Cao Y, Ambinder RF, Tao Q. 2012. Methylation profiling of Epstein-Barr virus immediate-early gene promoters, BZLF1 and BRLF1 in tumors of epithelial, NK- and B-cell origins. BMC Cancer 12:125.https://doi.org/10.1186/1471-2407-12-125.
12. Robertson KD, Hayward SD, Ling PD, Samid D, Ambinder RF. 1995.
Transcriptional activation of the Epstein-Barr virus latency C promoter after 5-azacytidine treatment: evidence that demethylation at a single CpG site is crucial. Mol Cell Biol 15:6150 – 6159.https://doi.org/10.1128/ MCB.15.11.6150.
13. Robertson KD, Barletta J, Samid D, Ambinder RF. 1995. Pharmacologic activation of expression of immunodominant viral antigens: a new strategy for the treatment of Epstein-Barr-virus-associated malignancies. Curr Top Microbiol Immunol 194:145–154.
14. Dai W, Zheng H, Cheung AK, Lung ML. 2016. Genetic and epigenetic landscape of nasopharyngeal carcinoma. Chin Clin Oncol 5:16.https:// doi.org/10.21037/cco.2016.03.06.
15. Abe H, Kaneda A, Fukayama M. 2015. Epstein-Barr virus-associated gastric carcinoma: use of host cell machineries and somatic gene mu-tations. Pathobiology 82:212–223.https://doi.org/10.1159/000434683. 16. Kang MS, Kieff E. 2015. Epstein-Barr virus latent genes. Exp Mol Med
47:e131.https://doi.org/10.1038/emm.2014.84.
17. Zimber-Strobl U, Strobl LJ, Meitinger C, Hinrichs R, Sakai T, Furukawa T, Honjo T, Bornkamm GW. 1994. Epstein-Barr virus nuclear antigen 2 exerts its transactivating function through interaction with recombina-tion signal binding protein RBP-J kappa, the homologue of Drosophila Suppressor of Hairless. EMBO J 13:4973– 4982.
18. Laux G, Adam B, Strobl LJ, Moreau-Gachelin F. 1994. The Spi-1/PU.1 and Spi-B ets family transcription factors and the recombination signal bind-ing protein RBP-J kappa interact with an Epstein-Barr virus nuclear antigen 2 responsive cis-element. EMBO J 13:5624 –5632.
19. Waltzer L, Logeat F, Brou C, Israel A, Sergeant A, Manet E. 1994. The human J kappa recombination signal sequence binding protein (RBP-J kappa) targets the Epstein-Barr virus EBNA2 protein to its DNA respon-sive elements. EMBO J 13:5633–5638.
20. Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to re-sponse elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A 91:7568 –7572.https://doi.org/10.1073/pnas.91 .16.7568.
21. Henkel T, Ling PD, Hayward SD, Peterson MG. 1994. Mediation of
on November 7, 2019 by guest
http://jvi.asm.org/
Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92–95.https://doi.org/10.1126/ science.8016657.
22. Kempkes B, Ling PD. 2015. EBNA2 and its coactivator EBNA-LP. Curr Top Microbiol Immunol 391:35–59.https://doi.org/10.1007/978-3-319-22834 -1_2.
23. Johannsen E, Koh E, Mosialos G, Tong X, Kieff E, Grossman SR. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent mem-brane protein 1 promoter is mediated by J kappa and PU.1. J Virol 69:253–262.
24. Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E. 2011. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A 108:14902–14907.https://doi.org/10.1073/pnas.1108892108.
25. Lu F, Chen HS, Kossenkov AV, DeWispeleare K, Won KJ, Lieberman PM. 2016. EBNA2 drives formation of new chromosome binding sites and target genes for B-cell master regulatory transcription factors RBP-jkappa and EBF1. PLoS Pathog 12:e1005339.https://doi.org/10.1371/ journal.ppat.1005339.
26. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. 2015. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev 263:6 –21.https://doi.org/10.1111/imr.12239.
27. Rasmussen KD, Helin K. 2016. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 30:733–750. https://doi.org/10 .1101/gad.276568.115.
28. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, Xu GL. 2011. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307.https://doi.org/10.1126/science.1210944. 29. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. 2010. Role
of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466:1129 –1133.https://doi.org/10 .1038/nature09303.
30. Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, Cao Z, Shi H, Wang J, Lin L, Chen S, Yuan W, Qin Z, Ni H, Nimer SD, Yang FC, Jaenisch R, Jin P, Xu M. 2015. Combined loss of Tet1 and Tet2 promotes B cell, but not myeloid malignancies, in mice. Cell Rep 13:1692–1704.https://doi .org/10.1016/j.celrep.2015.10.037.
31. Saillard C, Guermouche H, Derrieux C, Bruneau J, Frenzel L, Couronne L, Asnafi V, Macintyre E, Trinquand A, Lhermitte L, Molina T, Suarez F, Lemonnier F, Kosmider O, Delarue R, Hermine O, Cheminant M. 2016. Response to 5-azacytidine in a patient with TET2-mutated angioimmu-noblastic T-cell lymphoma and chronic myelomonocytic leukaemia pre-ceded by an EBV-positive large B-cell lymphoma. Hematol Oncolhttps:// doi.org/10.1002/hon.2319.
32. Ko M, An J, Bandukwala HS, Chavez L, Aijo T, Pastor WA, Segal MF, Li H, Koh KP, Lahdesmaki H, Hogan PG, Aravind L, Rao A. 2013. Modulation of TET2 expression and 5-methylcytosine oxidation by the CXXC domain protein IDAX. Nature 497:122–126.https://doi.org/10.1038/nature12052. 33. Cheng J, Guo S, Chen S, Mastriano SJ, Liu C, D’Alessio AC, Hysolli E, Guo Y, Yao H, Megyola CM, Li D, Liu J, Pan W, Roden CA, Zhou XL, Heydari K, Chen J, Park IH, Ding Y, Zhang Y, Lu J. 2013. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep 5:471– 481.https://doi.org/10.1016/j.celrep.2013.08.050.
34. Fu X, Jin L, Wang X, Luo A, Hu J, Zheng X, Tsark WM, Riggs AD, Ku HT, Huang W. 2013. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A 110:17892–17897.https://doi.org/10.1073/pnas.1317397110. 35. Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M,
Manova-Todorova K, Teruya-Feldstein J, Avigan DE, Delwel R, Pandolfi PP. 2013. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and trans-formation. Cell Stem Cell 13:87–101.https://doi.org/10.1016/j.stem.2013 .06.003.
36. Dalerba P, Clarke MF. 2013. Oncogenic miRNAs and the perils of losing control of a stem cell’s epigenetic identity. Cell Stem Cell 13:5– 6.https:// doi.org/10.1016/j.stem.2013.06.012.
37. Ambinder RF, Robertson KD, Tao Q. 1999. DNA methylation and the Epstein-Barr virus. Semin Cancer Biol 9:369 –375.https://doi.org/10.1006/ scbi.1999.0137.
38. Robertson KD, Manns A, Swinnen LJ, Zong JC, Gulley ML, Ambinder RF. 1996. CpG methylation of the major Epstein-Barr virus latency promoter in Burkitt’s lymphoma and Hodgkin’s disease. Blood 88:3129 –3136.
39. Kempkes B, Zimber-Strobl U, Eissner G, Pawlita M, Falk M, Hammer-schmidt W, Bornkamm GW. 1996. Epstein-Barr virus nuclear antigen 2 (EBNA2)-oestrogen receptor fusion proteins complement the EBNA2-deficient Epstein-Barr virus strain P3HR1 in transformation of primary B cells but suppress growth of human B cell lymphoma lines. J Gen Virol 77(Part 2):227–237.https://doi.org/10.1099/0022-1317-77-2-227. 40. Schuermann D, Weber AR, Schar P. 2016. Active DNA demethylation by
DNA repair: facts and uncertainties. DNA Repair (Amst) 44:92–102.
https://doi.org/10.1016/j.dnarep.2016.05.013.
41. Ko M, An J, Rao A. 2015. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr Opin Cell Biol 37: 91–101.https://doi.org/10.1016/j.ceb.2015.10.009.
42. Deplus R, Delatte B, Schwinn MK, Defrance M, Mendez J, Murphy N, Dawson MA, Volkmar M, Putmans P, Calonne E, Shih AH, Levine RL, Bernard O, Mercher T, Solary E, Urh M, Daniels DL, Fuks F. 2013. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32:645– 655. https://doi.org/10.1038/ emboj.2012.357.
43. Bergbauer M, Kalla M, Schmeinck A, Gobel C, Rothbauer U, Eck S, Benet-Pages A, Strom TM, Hammerschmidt W. 2010. CpG-methylation regulates a class of Epstein-Barr virus promoters. PLoS Pathog 6:e1001114.https://doi.org/10.1371/journal.ppat.1001114.
44. Marina RJ, Sturgill D, Bailly MA, Thenoz M, Varma G, Prigge MF, Nanan KK, Shukla S, Haque N, Oberdoerffer S. 2016. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splic-ing. EMBO J 35:335–355.https://doi.org/10.15252/embj.201593235. 45. Kong L, Tan L, Lv R, Shi Z, Xiong L, Wu F, Rabidou K, Smith M, He C,
Zhang L, Qian Y, Ma D, Lan F, Shi Y, Shi YG. 2016. A primary role of TET proteins in establishment and maintenance of De Novo bivalency at CpG islands. Nucleic Acids Res 44:8682– 8692.https://doi.org/10 .1093/nar/gkw529.
46. Chiba S. 2016. Dysregulation of TET2 in hematologic malignancies. Int J Hematol 105:17–22.https://doi.org/10.1007/s12185-016-2122-z. 47. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry
C, Figueroa ME, Vasanthakumar A, Patel J, Zhao X, Perna F, Pandey S, Madzo J, Song C, Dai Q, He C, Ibrahim S, Beran M, Zavadil J, Nimer SD, Melnick A, Godley LA, Aifantis I, Levine RL. 2011. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transforma-tion. Cancer Cell 20:11–24.https://doi.org/10.1016/j.ccr.2011.06.001. 48. Wille CK, Li Y, Rui L, Johannsen EC, Kenney SC. 2017. Restricted TET2
expression in germinal center type B cells promotes stringent Epstein-Barr virus latency. J Virol 91:e01987-16. https://doi.org/10 .1128/JVI.01987-16.
49. Hardee J, Ouyang Z, Zhang Y, Kundaje A, Lacroute P, Snyder M. 2013. STAT3 targets suggest mechanisms of aggressive tumorigenesis in dif-fuse large B-cell lymphoma. G3 (Bethesda) 3:2173–2185.https://doi.org/ 10.1534/g3.113.007674.
50. Wille CK, Nawandar DM, Henning AN, Ma S, Oetting KM, Lee D, Lambert P, Johannsen EC, Kenney SC. 2015. 5-hydroxymethylation of the EBV genome regulates the latent to lytic switch. Proc Natl Acad Sci U S A 112:E7257–7265.https://doi.org/10.1073/pnas.1513432112.
51. Fan H, Zhang H, Pascuzzi PE, Andrisani O. 2016. Hepatitis B virus X protein induces EpCAM expression via active DNA demethylation di-rected by RelA in complex with EZH2 and TET2. Oncogene 35:715–726.
https://doi.org/10.1038/onc.2015.122.
52. Engdahl E, Dunn N, Fogdell-Hahn A. 2016. Investigation of reference gene expression during human herpesvirus 6B infection indicates pep-tidylprolyl isomerase A as a stable reference gene and TATA box binding protein as a gene up-regulated by this virus. J Virol Methods 227:47– 49.
https://doi.org/10.1016/j.jviromet.2015.10.011.
53. Namba-Fukuyo H, Funata S, Matsusaka K, Fukuyo M, Rahmutulla B, Mano Y, Fukayama M, Aburatani H, Kaneda A. 2016. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget 7:81512– 81526. https://doi.org/10 .18632/oncotarget.13130.
54. Kaneda A, Matsusaka K, Aburatani H, Fukayama M. 2012. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res 72:3445–3450.https://doi.org/10.1158/0008-5472.CAN-11-3919. 55. Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart JW, Kremmer E,
Dele-cluse HJ, Rottenberger C, Bornkamm GW, Hammerschmidt W. 1995. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J 14:88 –96. 56. Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S,
Wikramasinghe P, Davuluri RV, Bartolomei MS, Riethman H, Lieberman
TET2 Regulation of EBV Epigenotypes Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
PM. 2012. A role for CTCF and cohesin in subtelomere chromatin orga-nization, TERRA transcription, and telomere end protection. EMBO J 31:4165– 4178.https://doi.org/10.1038/emboj.2012.266.
57. Chau CM, Lieberman PM. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J Virol 78:12308 –12319.https:// doi.org/10.1128/JVI.78.22.12308-12319.2004.
58. Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the
latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J Virol 80:5273–5282.https:// doi.org/10.1128/JVI.02541-05.
59. Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molec-ular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9:e1003672.https://doi.org/10.1371/journal .ppat.1003672.