0022-538X/89/114875-07$02.00/0
Copyright C) 1989, American Society for Microbiology
Analysis of
rev
Gene Function on Human
Immunodeficiency
Virus
Type
1
Replication in Lymphoid Cells by Using
a
Quantitative
Polymerase Chain Reaction Method
SALVATOREJ. ARRIGO,12 STACY WEITSMAN,12 JOSEPH D. ROSENBLATT,2 ANDIRVIN S. Y.
CHEN'.2*
Department ofMicrobiology and
Immunology'
and Departmentof Medicine,2 University of California, Los Angeles, Schoolof
Medicine and JonssonComprehensive
CancerCenter,
LosAngeles, California
90024-1678Received 7 June1989/Accepted 27 July 1989
Most detailed analysesof the human immunodeficiency virus type 1 (HIV-1) rev gene product have relied on transfection of subgenomic env constructs into cells in whichamplificationof the transfected DNA occurs. This was necessitated by difficulties in quantitating low-abundance HIV-1 mRNA species and in distinguishing different RNAs of similar sizes. We have modffied the conventional polymerase chain reaction method for general use as an extremely sensitive procedure for quantitative analysisof RNA species. Using this method, we assessed the role of the HIV-1 rev gene in viralreplication following mutagenesis of an infectious molecular clone,
HIV-lJR-CSF.
Following transfection ofwild-type and mutant proviral constructs, we can specifically detectunspliced RNA and distinguish between the spliced tat-rev andnef mRNAs, which are not resolved by standard RNAanalyses. Our results show that the rev protein ofHIV-lJRCsF
simultaneously down regulates the expression of tat-rev and nef RNAs and up regulates the level of unspliced full-length HIV-1 RNA. A cis-acting element(s), located exclusively within the env sequences, is essential to exhibit this regulation. Fractionation of cells shows that the ultimate effect of Rev is to direct the appearance of unspliced or singly spliced RNAs in the cytoplasm. Models are discussed for possible mechanisms of Rev action.Inadditiontothe three genes expressed by all replication-competentretroviruses (gag,pol, and env), human immuno-deficiency virustype1(HIV-1)contains the additional genes vif,vpr, vpu, tat, rev,andnef(11). Three of these genes have been shown to affect the expression of HIV-1 RNA and protein. The tat gene product increases the level of both HIV-1-specific RNA and protein through a trans-acting responsive element located in the long terminal repeat (LTR) andatthe 5' terminiofall HIV-1 RNA species (6, 7, 17, 22, 23, 25, 26, 31, 34). The nefgene product appears todown regulate expression from the LTR through a negative regu-latory elementin the LTR (1, 19).
The rev gene product appears to be necessary for high-levelexpression ofgagand envmRNAsandprotein acting
at a posttranscriptional level. Rev function has been pro-posedas an inhibitor of splicing, as an activatorof nuclear transport,andas asourceof reliefto atranslationalblock(3, 8, 9, 12-14, 21, 27, 28, 30). Previous studies of the mecha-nism ofRevactionhaverelied onvariousreplicatingvector systemstogenerateassayablelevels ofvirus-specific RNA. A system commonly used to assay Rev function involves transfection of simian virus 40-transformed monkey kidney cell lines(COS) withreplicating vectors (13, 14, 20, 21, 27, 28).Thesestudiesgenerallyusedenv vectorsinwhichenvis expressed as an unspliced RNA. Proviral constructs have also been usedto examine the effect ofRev onthe expres-sion of all
HIV-1-specific
RNAs (3, 8, 9, 12, 28, 30). However, these studies did notdiscriminate among HIV-1 RNAs ofsimilar sizes. It has recently been demonstrated that Rev is necessaryforthe accumulation ofhighlevels of cytoplasmic unspliced RNA in transfected HeLa and COS cells; this was attributedto an effect on both transport and stability of the unspliced RNA (9, 21). Arev-responsive
element (RRE) found within the HIV-1 env gene has been
* Correspondingauthor.
shown to be involved in the regulation of HIV-1 RNA expression byRev (9, 12, 13, 21, 27).
Tomoreprecisely definetherole ofRevin theregulation of HIV-1 replication, we have constructed mutants in an
infectiousmolecularclone,pYKJRCSF, of a primary isolate of HIV-lJRcsF. This isolate replicates to high titers in primary human lymphoid cells (16). To detect RNA from proviralconstructsinlymphoid cells,wedevelopeda quan-titative assayforRNAby usingamodifiedpolymerase chain reaction(PCR)method. Becauseoftheincreased sensitivity and specificity ofthistechnique overthose of conventional
techniques,
we canindependentlyexamine theregulation of full-length, tat-rev, and nefRNAs (3, 22, 24). Our results demonstrate that inlymphoid cells,Revregulates the distri-bution ofHIV-1 RNAs withinthecell.MATERIALS ANDMETHODS
Cell lines and electroporation. 729 and Jurkat cell lines weremaintained in Iscove mediumsupplementedwith 5 and 10% fetal calfserum, respectively. Before electroporation, cells were suspended in RPMI 1640 medium supplemented with 10%fetal calfserum at aconcentration of2 x 107/ml. Cells (107) were electroporated with a total of 50 ,ug of plasmidDNAasdescribedpreviously (5).For RNAandp24 analysis,cells wereharvested at48 h
postelectroporation.
Plasmidconstructions.The
proviral
clonepYKJRCSF
wasdescribed
previously
(16). Mutant ASalwas madeby
filling
inaSalI siteatnucleotide(nt)8483 ofpYKJRCSFwiththe Klenow fragment of DNA polymerase. This resulted in a
frameshiftmutation in therevgeneand thecreationofa new
PvuI site verified by restriction enzyme
digestion.
Mutant ABX was generated by adding aBamHI linkerto anSspI site at nt 8262 andreplacing
thewild-type
BamHI(nt
7324)-to-XhoI (nt8911)
fragment
with theSspI-to-XhoI fragment.
This resulted in a deletion of nt 7328 to 8261 from the proviralconstruct. Mutant APstwas madein
proviral
clone4875
on November 10, 2019 by guest
http://jvi.asm.org/
LA45 LA41
I
II
-ISAISA2SD2 SA
ABX ASal
(RRE-) (rev-)
> F;ULL LENGTH
.... ... A_ env
....
...
.....
...-
.tat/rev
.1
,,,,,,...
,,,,,,....
,,,,... nefRNASPECIES
FULLLENGTH env
tat/rev
nef
M667/M668
(nt496-526/
656-637)
PCRPRODUCTS
LA8/LA9 LA45/LA41
(nt7l1-730/ (nt5964-5984/
805/786) 8395-8374)
161 95
161 ND
161 ND
161 ND
ND
123
133
FIG. 1. Oligonucleotide primer pairsfor PCRonHIV-1 RNAs. (A) Schematicrepresentation of the HIV-lJR-CSFgenome showingthe
locationand orientationoftheoligonucleotide primersused forPCR.Thesplicedonors(SD)andspliceacceptors(SA)forenv, tat-rev,and
nefRNAsareshown. Thesplicedformsof these RNAsaredepictedbelow the schematicdiagram.Thelocations of thegag,RRE-,andrev
mutations in thegenomeareshown(notdrawntoscale). (B)Sizesofamplified productsgenerated byRNAPCR withpairsofoligonucleotide primers specific forHIV-1 RNAs. ND,RNAspeciesfor whichspecific productswerenotdetected because of theirlargetheoreticalsizes.
pN/BJRCSF,which is identicaltopYKJRCSFexcept foran
additional 2 kilobasesof cellularflankingsequenceupstream of the 5' LTR. Mutant APst was generated by restriction enzyme digestion with PstI and religation, resulting in the deletion ofnt 1205to 1418 within thegaggene.
RNApreparation andp24 antigendetection. Total cellular RNA was prepared by lysingthe cells in 4.7 M urea-1.3%
sodium dodecyl sulfate-0.23 M NaCl-6.7 mM Tris (pH 8.0)-0.67 mM EDTA. This was followed by three phenol-chloroform (1:1) extractions, onechloroform-isoamyl alco-hol (24:1) extraction, and ethanol precipitation. The total recovered nucleicacidwastreated twiceat37°Cfor 1 h with RNase-freeDNase(Worthington Diagnostics)toremovethe
DNA. Nuclearandcytoplasmic RNAswerefractionated by
lysingthecellsin0.65% Nonidet P-40-150mMNaCl-10mM Tris (pH 7.8)-1.5 mMMgCl2 and by pelleting the nuclei by low-speed centrifugation. The nuclear and cytoplasmic frac-tions werethentreatedasdescribed above. Cell-free
super-natantsfromelectroporated cells were assayed forgagp24
antigen by using a specific enzyme-linked immunosorbent
assaykit(Abbott Laboratories).
RNA PCR analysis. RNA (c5
jig)
was subjected to aprimer extension hybridizationreaction inavolume of 10p,l containing10ngofprimer(antisense),250 mMKCI, 10mM
Tris(pH 8.3),and 1mM EDTAat65°C for1h. A portion (5
,ll)
of this reactionwasaddedtoeachoftwotubes contain-ing 10 ,ul of 1.5x reverse transcriptase buffer (1.5 mMdeoxynucleoside triphosphates, 7.5 mM dithiothreitol, 15 mM MgCl2, 0.09M KCl,0.63 mM Tris [pH 8.3]). Reaction mixtureswereincubated in thepresenceorabsence of5U of avian myeloblastosis virus reverse transcriptase (Life
Sci-ences, Inc.) at 37°C for 1 h. Portions (2
pd1
each) of these primerextension reactions were subsequently subjected toanalysis by PCR. The PCR amplifications each contained 0.25 mM deoxynucleoside triphosphates, 50 mM NaCl, 5 mMTris(pH 8.0),5 mMMgCl2, 100ngofthe 3'(antisense)
oligonucleotide, 0.67to 1.2 U ofTaq polymerase (Promega BiotecorNewEngland BioLabs, Inc.),and20to30ngof the 5' (sense) oligonucleotide end labeled with T4 DNA kinase
toaspecificactivity of 1 x 108to6 x 108 cpm/p.g,inatotal
volume of 25 ,ul. PCR was performed in a DNA thermal cycler (Cetus orEricomp) for 25 cycles of denaturation at
91°C for 1 min and polymerization at 65°C for 2 min. PCR-amplified products were separatedon a6%
polyacryl-amidegelandvisualizedby autoradiography.
The nucleotide sequences of the oligonucleotide primers used for PCR (Fig. 1) are as follows: for M667, 5'-GGC
TAACTAGGGAACCCACTG-3';forM668,5'-CGCGTCCC TGTTCGGGCGCC-3';for LA8, 5'-GCGCGCACAGCAAG
AGGCGA-3'; forLA9,
5'-GACGCTCTCGCACCCATCTC-A
M668LA9
SD1
APst (gag-)
B
LA8/LA41 [image:2.612.105.517.84.422.2](nt7 11-730/ 8395-8374) ...
...
... ......
on November 10, 2019 by guest
http://jvi.asm.org/
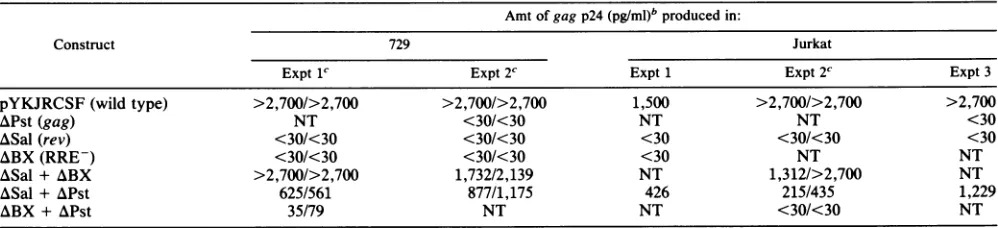
TABLE 1. Production ofgagp24from electroporated lymphoidcellsa
Amt of gag p24(pg/ml)'produced in:
Construct 729 Jurkat
Expt 11 Expt2C Expt 1 Expt2C Expt3
pYKJRCSF (wild type) >2,700/>2,700 >2,700/>2,700 1,500 >2,700/>2,700 >2,700
APst(gag) NT <30/<30 NT NT <30
ASal (rev) <30/<30 <30/<30 <30 <30/<30 <30
ABX (RRE-) <30/<30 <30/<30 <30 NT NT
ASal + ABX >2,700/>2,700 1,732/2,139 NT 1,312/>2,700 NT
ASal +APst 625/561 877/1,175 426 215/435 1,229
ABX + APst 35/9 NT NT <30/<30 NT
aCulturesupematantsfrom107cellstransfected with a total of 50
F.g
ofplasmid DNA were assayed at 48 hposttransfection.bDetected by enzyme-linked immunosorbent assay (Abbott). Values of <30 are below the sensitivity of this assay. NT, Not tested.
Results ofduplicateelectroporations are shown.
3'; for LA45, 5'-GGCTTAGGCATCTCCTATGGC-3'; and for LA41,5'-TGTCGGGTCCCCTCGTTGCTGG-3'.
RESULTS
Rev is necessary for production ofgag p24 antigen. To
determine the function of Rev in the expression of gag
proteins in lymphoid cells, mutants were generated in an
infectious clone of
HIV-lJR-CsF,
pYKJRCSF (Fig. 1A). Arevmutant(ASal) containsa4-base-pair (bp) insertionatthe Sallsite(nt 8483), resultinginaframeshiftintherevcoding
sequences;anRRE- mutant(ABX) containsadeletion ofnt
7328to8261whichencompassesthe RRE (9, 12, 21, 27) but
does not interrupt the rev coding sequences; and a gag mutant (APst) contains an out-of-frame deletion of almost
theentiregagp24 codingsequencesand is thusincapable of
expressing gag p24 antigen. This mutant was designed to
conserve expression of allthe trans-acting regulatory
pro-teins so that it could complement a defect in p24 gag
productiondueto loss ofrevfunction.
Theseconstructs wereelectroporated either separatelyor
incombination with each otherinto ahumanB-cell (729)or
T-cell (Jurkat) line. At 48 h postelectroporation, culture
supernatants were analyzed for the presence of gag p24
antigen by using a specific enzyme-linked immunosorbent
assay kit(Abbott). The results are summarized in Table 1.
The wild-type proviral construct gave rise to consistently high values ofgag p24 in both cell types tested. The gag
mutantwasusedas anegative control, since itwasincapable
of producing any gag p24 protein. The rev and
RRE-mutants were also incapable ofproducing any p24 antigen
when electroporated alone. Thus, both the rev protein and
theRREarenecessaryfor theproduction ofanydetectable
gagproteins.
Complementation experiments were performed to deter-mine whether it was possible to rescue p24 production in these mutants. High levels ofp24wereproduced when the
rev mutant was coelectroporated witheither the RRE- or
gag mutant, suggesting that the gag and RRE- mutants
produced a functional rev protein able to rescue the rev mutant in trans. In contrast, coelectroporation ofgag and RRE- mutants did not rescue p24 production orgave rise only to very low levels of measurable p24 production, indicatingthatthe RRE-mutantcontainedacis defect. This lowlevelwasmostlikelyduetorecombination betweenthe two constructs, since coelectroporation of either mutant
with avector producing only Rev did notgive rise to any
detectable p24 antigen (data not shown). The rev mutant
couldproduce wild-type levelsofp24 antigen when
coelec-troporated with the vector producing only Rev (data not shown). These results suggest that both Rev and the RRE must be present in order to obtain expression of gag p24 antigen inlymphoid cells and that both are therefore essen-tial to the replication ofHIV-1. As indicated by results in other systems, Rev functions in trans through a cis-acting element to allow expression ofgag protein.
Quantitative PCR analysis of HIV-1-specific RNAs. To detect low-abundance HIV-1 RNA transcripts in lymphoid cells, we employed the PCR method (18, 29) with RNA isolated from HIV-1-infected cells by first converting the RNA to cDNA with reverse transcriptase. This method offersalarge increaseinsensitivityoverthatavailable with conventional Northern (RNA), nuclease protection, or primer extension analyses. The oligonucleotide pairs were
designed to direct synthesis of PCR products which would spanknownsplice donorsand acceptors. Thelocation ofthe
oligonucleotide pairs
in the HIV-1 genome with respect toknown splice donors andacceptors and thepredicted sizes ofthe PCR products generated for each HIV-1 RNA with these
oligonucleotide
pairsare shown inFig. 1. Since large PCRproducts (>250 bp)are notgeneratedunderthe condi-tions usedwith theseoligonucleotide
pairs, onlythesizes of detectable products are shown.Oligonucleotide
primers M667 and M668 will direct synthesis ofa 161-bpamplified productcorrespondingtoall HIV-1RNAs;primersLA8 and LA9shouldgeneratea95-bpPCRproductspecificfor HIV-1 RNAwhich hasnotutilizedthe 5' splice donorat nt743 (full length).Primers LA45 and LA41willgeneratea123-bpPCR productspecific forHIV-1 RNA which hasjoinedthesplice donoratnt6055 with thespliceacceptorat nt8365 (tat and rev); LA8andLA41 shouldgenerate aPCR product of133 bpspecific
forHIV-1 RNAwhichhasjoined
thesplice
donor at nt 743 with thesplice
acceptor at nt 5788as well as the splice donor at nt 6055 with thesplice
acceptorat nt 8365(nef).
Total RNAprepared from
peripheral
bloodlymphocytes
infected withHIV-lJR-CsF
was treated with RNase-free DNase to remove allcontaminating
DNA and thencon-verted to cDNA with reverse transcriptase by using the appropriate3'
(antisense)
oligonucleotide
primer.
Theprod-uctsof this reactionwerethen
subjected
to25cycles
of PCR amplificationwith the addition of the 5'(sense)
oligonucle-otide primer end labeled with32p.
We have modified the standard PCRprotocol
to allowquantitative
analysis
of samples. Byusing
anend-labeledoligonucleotide primer,
we can omit thetransfer andhybridization
stepsused in other protocolsand decrease the number ofcycles
ofdenaturationon November 10, 2019 by guest
http://jvi.asm.org/
A
.iQ,t9 /L004 AXA...A. . ,.
'.rI; nf Ji fn lmrd r; nI,..
n-4~
_. __* .4- + _. +-_*_r
a
...
-.:
me
go :- 1
B
t NUCLEOTIDE
--sequential2-fold
dilutions ofRNA
LA8/LA9q 0 0 40 10 *
@*-LA45iLA4!
-AB/LA4Ti
FIG. 2. Quantitativedetection ofspecificHIV-1R] (A)Detection ofHIV-specificRNAsbyPCRonRNA
peripheral blood lymphocytes. RNA was extracted I
(inf.)and uninfected (uninf.) peripheral bloodlymph
from 105cells was subjected to primerextension rea
presence (+) orabsence(-) ofreverse transcriptase
these reactions were subjected to 25 cycles of PCR
tisense) oligonucleotidewasused forprimerextensio
(sense) oligonucleotide primerwasendlabeled with[y
included in the PCR reaction. Products were separa
polyacrylamide gelandvisualizedby autoradiography
therightindicatesizes in basepairs.Molecularweight pGEM-2 (Promega Biotec) digestedwithHpaII. (B)PC
dilutions of RNA from HIV-1-infected peripheral b]
cytes. RNA was prepared from infected cells, dilut converted to cDNAby primerextension. cDNAwas
PCRasdescribed above.
and polymerization. Decreasing the number c
PCRfrom 40 to 25 and eliminatingthe transfer
izationstepsresult inaPCR method which is bot
sensitive andquantitative.Inapplyingthis methi
critical that theannealing stepineach PCR
cyclh
and that different primer pairs be tested for al
lowbackgrounds. PCR with HIV-1-specific
olig
pairs can generateamajor specific amplified pri
predicted size from HIV-1-infected-cell RNA
Thesespecificbandsarenotpresentinuninfecte
RNA. In some cases, minor bands other than
dictedaregenerated.Nucleotidesequenceanaly
that most minor bands generated during PCI 1-specific productsbutresultfromnonspecificpr
antisense oligonucleotide primerandspecific pr
senseprimer.This nonspecific primingmostlike
the reverse transcriptase step, and these abe products are thenamplified duringPCR. Since
cleotide pairs M667-M668 and LA8-LA9 can
HIV-1-specific DNA, mock primer extension
reverse transcriptase was omitted from the r
control for any contamination of the RNA with DNA were performed; noHIV-1 DNAwasdetected in these samples.
To determine whether HIV-1-specific RNAs could be detected quantitatively by this RNA PCR procedure, se-quential twofold dilutions of total cellular RNA from HIV-1-infected cells were subjected to PCR analysis (Fig. 2B). Thesignals producedby RNA PCR were quantitativeacross a2,000-fold range of RNA concentrations with all oligonu-cleotide pairs tested. In all cases, increasing signal intensi-ties weredetected fromincreasing amountsof RNA. How-ever, for a given number of PCR amplification cycles, the magnitude of the increase in signal intensities was not linear and reached a plateau as higher signal intensities were
achieved, probablybecause concentrations of nucleotides or oligonucleotides were limiting. Thus, it is important to --F - recognizethat differences insignal strength intensities dem-onstrated by PCR may be underestimates of relative differ-ences.Absolutequantitation requires the use of appropriate -TSW RNA standards subjected to PCR inparallel. We estimate
that thesensitivityof this method ismorethan 104-fold that of standard RNA detection systems, making detection of low-levelRNA speciesand theuse of far fewer cells
possi-:,of 4;. ble.
[image:4.612.66.300.84.356.2]Effect of Rev on levels ofgag-pol, tat-rev,and
nefRNAs.
To examine the block to gag p24 production exhibited by the proviralmutants,theexpression of HIV-1-specificRNAs in electroporated lymphoid cellswasexaminedby PCR. Using specific oligonucleotide primer pairs, we can discriminate NAsbyPCR. between tat-rev and nefRNAs, unlike other researchers, from infected who have grouped these RNAs into a same-size class of from infected double-spliced RNAs.iocytes. RNA Both the rev mutant (ASal) and the RRE- mutant (ABX)
actions
in the producedRNAprofiles
verydifferent fromthatofthewildPortions of type
(Fig.
3). Both mutantsproduced
reduced levels of n,and
th(a5
unspliced full-length
RNA andaconcomitantlarge
increase-32P]ATPand in both tat-rev and nef RNAs relative to the wild type. A eted on a 6% comparison of the PCR signals produced by wild-type and
.Numbers on mutant constructs with standard curves generated by
dilu-marker(M)is tions of infected-cell DNA has allowed usto determinethe nRon twofold magnitude of these differences. The levels of tat-rev and nef lood
lympho-
RNAs in both the rev and RRE- mutants reproducibly Led, and then increase to more than 10-fold the levels in the wild type, and subjectedto the level of unspliced full-length HIV-1 RNA with these mutantsdecreasesmorethan5-fold relativetothe wild type. The levels of totalHIV-1-specific RNAwiththesemutants f cycles of were not significantly differentfrom those ofthewild type, andhybrid- suggestingthat Rev does not affect overall RNA levels of thextremely HIV-1. Coelectroporation of the rev and RRE- mutantsod,it isalso resulted in restoration ofthe high level offull-length RNA
ebeomitted seenwith the wild type. The levels ofboth tat-rev andnef ppropriately RNAs remained higher than that seen with the wild type.
;onucleotide
This canbe explained by aninability to trans-complement oduct of the thecis mutation of the RRE in the /BX mutant.(Fig. 2A). Similar results were obtained with another construct zd,total-cell which expresses Rev. The gag mutant (APst) can comple-those pre- menttherevmutant,as measuredby p24 production (Table sisindicates 1).Coelectroporationof both mutants resulted in a high level
R.
are HIV- offull-length RNA and low levels of both tat-rev and nef rimingof the RNAs(Fig. 3B), similar to those seen in the wild type. RNA iming ofthe from cells electroporated with the gag mutant exhibited lyoccurs at wild-typelevels of tat-rev and nefRNAs. However, the level rrant DNA ofunspliced full-length HIV-1 RNA was severely reduced, theoligonu- withanoverall reduction in the level of total HIV-1-specific also detect RNA. This could be due to a decrease in RNA stability,Is in which similar to that seen in gag mutants of Rous sarcoma virus reactions to (2).
on November 10, 2019 by guest
http://jvi.asm.org/
A
OLIGONUCLEOTIDE PRIMERS
C:,
LL-cn
C-1
I- 1 ><
:b L.J cr
c
Ct::]
-
+~
0 0
c, cn cn
-C <] <
RNA
SPEC ES
OLjGONUCLEOT DE
PR;MERS
M667/M668 -_ _
LA8ILA9 - *
LA45/LA4! -- *a
LAP -41i
-LA8/LA9
IJELLLENGTHll
FULL LENGTH
roef
_ e e NUCLEAR
CWY'.DKASY
-NUrLEAR
L^ /LA41,i
gf(.v
C1YTOPLASMiC
FIG. 4. Effect of Revonnuclear and cytoplasmic RNA distribu-tion. RNA from electroporated 729 cellswasseparated into nuclear andcytoplasmic fractions. Nuclearorcytoplasmic RNA from 8 x
105 electroporated cells was subjected to RNA PCR analysis by using oligonucleotides specific forfull-length andtat-revRNAs.
B
OLIGONUCLEOTIDE PRIMERS
CL
0:
LL UI t CL
C_:> tlt +
cr -
-_>0 4 a
cl) a- to
Y:) v< :L3 <f
M667/M668
-~
*LA8/LA9 - w
_ _._
LA45/LA41 -_
LA8/LA41 m
Iir.:
I.m
'i
aRNA
SPErIES
TOTAL
FULL LENGTH
fto/rev
nef
FIG. 3. Effect of RevonHIV-1 RNA accumulation. (A) Analysis of RNA from 729 cells electroporated with the indicated plasmid DNAs. In eachpair of oligonucleotide primers, equivalentamounts
of RNA from these cells were subjected to primer extension and
subsequent PCR. (B) Analysis of RNA from electroporated 729
cells.Experimentswereperformedasdescribed above.
levels of unspliced RNA in the cytoplasmic fraction. In
contrast,therevandRRE- mutantsexhibited higher levels
oftat-revRNA inthecytoplasmic fraction than did the wild
type.The decreased levels of cytoplasmic full-length HIV-1 RNA with therevand RRE- mutantswereinversely
corre-lated with the large increases in tat-rev RNA in the
cyto-plasms of cells electroporated with these mutants. The majority of spliced RNA of all the constructswas found in
the cytoplasmic fractions, indicating that tat-rev RNA is transported rapidly to the cytoplasm and does not
accumu-lateinthe nucleus.
Expression ofrevand thepresenceof the RREappearto
be necessary for the accumulation of unspliced full-length HIV-1 RNA inthe cytoplasm. IfRev couldexert aneffect
through a cis-acting element other than RRE, this effect
should be observed with the RRE- mutant, which can still
produce Rev. Since both the rev mutant, ASal, and the RRE- mutant, ABX, exhibit identical levels of each ofthe HIV-1 RNAs and identical distributions of the HIV-1 full-length RNA, it seems likely that Rev exerts its effect on
RNA expression solely through the RRE and not through
anyother element within the viralgenome.
These results indicate that Rev alters the distribution of HIV-1 RNAs by up regulating expression of full-length
HIV-1RNA anddownregulating expression of bothtat-rev andnef RNAs. The levels oftat-revandnef RNAsappearto
becoregulated by Rev, since equivalent increases in these RNAsareseenwith bothrevandRRE- mutants.The effect of Revontheregulation ofHIV-1 RNAdistributionrequires
acis-acting element, the RRE.
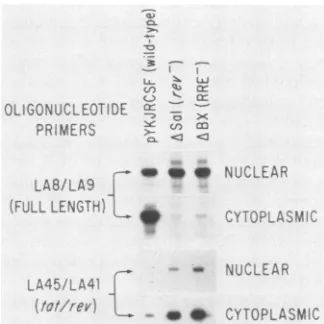
Effect of Rev on cellular distribution of HIV-1 unspliced
full-lengthRNA. To determine whether Revwasaffectingthe cellulardistribution of HIV-1 RNAs, nuclear and cytoplas-mic RNA fractions were prepared from lymphoid cells
electroporated with wild-type and mutant proviral
con-structs. Cell equivalents of RNA were then subjected to
analysis by PCR with oligonucleotide pairs specific for unspliced full-length HIV-1 RNA (LA8-LA9) and tat-rev RNA (LA45-LA41) (Fig. 4). Although nuclear levels of full-length RNA were similar to those of wild-type and
mutant constructs, only the wild type accumulated high
DISCUSSION
Using PCR, we have developed a novel method for the
quantitative detection of specific low-abundance HIV-1 RNAs. Oligonucleotide pairs which detect specific HIV-1 RNA species through in vitro synthesis of cDNA and subsequent detection by PCR amplification have been gen-erated. This method offers significant advantagesover
con-ventional methods used to measure RNA accumulation, such asNorthern, nucleaseprotection, orprimerextension analyses. By using an end-labeled oligonucleotide primer,
performingfewercyclesofPCR,andeliminatingthe transfer
and hybridization steps of the conventional PCR method, quantitative detection ofHIV-1 RNAs inlymphoid cells is
possible. Because ofthe large increase in sensitivity
pro-videdby this method, we caneasily detect low-abundance HIV-1 RNAs inelectroporated lymphoid cells andperform hundreds ofassayswith RNA from107cells. Because ofthe
specificity ofthe oligonucleotide primers, wecan
discrimi-CL
L.L- E~ L'i
t.n r
(9 __
- __
(nClC
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.355.517.85.247.2] [image:5.612.70.281.85.470.2]nate between RNAs such as tat-rev and nef, which are difficulttodistinguish by conventionaltechniques.The
non-linearnatureofthis detectionmethoddoes not affect quan-titation; over a large range of input RNAs and in a large variety of oligonucleotide pairs, we have seen a direct and reproducible correlation between the amount of specific-input RNA and the intensityof the signalproduced. Further-more,if linearity ofthedoseresponseisdesiredforagiven range of sample values, the number ofPCR cycles can be empirically determined to maintain them within the linear partofthe curve. Accuratequantitation byPCR necessitates the useofstandard curves on RNAdilutionsdone inparallel. Using this RNA PCR procedure, we can discriminate between the tat-rev and nef RNAs produced in lymphoid cells infected withHIV-1 ortransfected withHIV-1proviral
constructs. This has allowedustoanalyzetheeffect ofRev oneachof theseRNAsindependently rather thanondoubly splicedRNAs as asingle size class. Consequently,wehave determinedthat Revdown
regulates
theexpression of bothtat-rev andnefRNAs concordantly inlymphoid cells while increasingthelevelofcytoplasmic full-lengthHIV-1 RNA. Littleor no effectonthe levels of nuclearunspliced HIV-1 RNA is seen in the presence ofRev. Rev exerts its effect solely through the'RRE located in the env gene, which is presentin bothunsplicedandsingly splicedRNAs.Removal of this element results in levels and distributions of HIV-1-specific RNAs indistinguishable from those ofa rev mu-tant.These datademonstratethatthistarget
region,
whichis removed during splicing to the tat-rev andnef
RNAs, is critical forRev toexhibit
anyeffectonHIV-1 RNA expres-sion.It isinteresting thatalthoughthe rev andRRE- mutants
produce more than 10times as much tat-rev RNA as the wild-type construct does, an increase in total RNA levels was not observed. In these mutants, an increase in total RNAlevels duetoincreasedlevelsof trans-activationbyTat might be expected. It ispossible that there isnoincrease in tatproteinwith thesemutants.Alternatively, since nefRNA isalsoincreased,aconcomitantincrease innef protein might
counteract any increase in tat trans-activation. Deletions within nef have previously been shown to enhance viral replication(1, 19),and ithas been further demonstrated that the nef protein down regulates Tat-dependent transcription from theHIV-1 LTR. Thisrepression is apparentlyexerted
through
thenegativeregulatoryelement locatedin the LTR (1). Additionally, the RRE- mutant might be expected to overexpress revprotein in comparison with the wild type. Since littleor noeffectonthe leveloftotal HIV-1 RNAwas seenwiththe RRE- or rev mutants, we cannotdemonstrate anyeffect ofRev on HIV-1 RNA stability, asdescribed by Felber et al. (9), although small effects on the stability of total RNA or larger effects on the stability ofminorRNA species would not be seen.Ourparallel analysis of different RNA speciesis
consis-tent with several models for Rev action. Rev might be necessary for transport to the cytoplasm of unspliced and singly spliced RNAs containing the RRE, resulting in less substrate forsplicing to tat-rev and nef RNAs. This theory has beenpreviously proposed by otherinvestigators (9, 13, 21). Adefect in rev would lead to an increase in the nuclear pool of unspliced RNAavailablefor subsequent splicing to tat-revand nef RNAs. The fates of singly spliced RNAs are lessclear.The env RNA could potentially be further spliced totat-rev RNA, orit could be transported to the cytoplasm bytheactionof Rev.Theputative vifand vpr RNAs could be generatedbyasplice fromthesplice donor at nt 743 to splice
acceptors at nt4924 and5401,respectively (22,24).Likeenv RNA, these RNAs(which would contain theRRE)mightbe furthertransportedtothecytoplasmbytheaction ofRev,or
they could be splicedto aberrant doubly spliced products; candidatesfor suchaberrantproducts have been detected in HIV-1-infectedcells (22). Thus, the transport theorywould require that Rev act on unspliced and singly spliced RNAsto transportbeforeandaftersplicing ofthefirstintron, respec-tively. The effect of Rev on the cytoplasmic level ofsingly spliced RNAs hasbeen addressed only indirectly by
using
constructs which express env RNA as an unspliced RNA (13,21). Analternate hypothesisis that Rev might interfere with the splicing machinery by allowingthe unsplicedand singly spliced RNAs, both containing the RRE, to bypass complete splicing to doubly spliced tat-rev, nef, and other aberrant RNAs. This would result in an accumulation of unspliced andsingly spliced RNAs in thecytoplasm only in the presence of Rev. This theory would explain why the singlysplicedRNAsare notfurthersplicedtocompletion,as occurs with most cellular RNAs. Furtherregulation of the ratio of unspliced to singly spliced RNAs might be accom-plished by cis-acting sequences such as those found in Rous sarcoma virus (2). Although the effect of Rev has been demonstrated with vectors containing no known splice do-nors, implying that splicing is not involved (9, 20, 21, 27), mutations of the 5' splice donor can frequently lead to activation ofcryptic splicedonors(32, 33).Additionally, the presenceof a 5' splice donor does not appear to be necessary for spliceosome formation (4, 10, 15). Rev may prevent a permanent association of RRE-containing RNAs with the spliceosome, which might indirectly block the transport of these RNAs to the cytoplasm. Further splicing would re-move the RRE, releasing the doubly spliced RNA and allowingits transport to the cytoplasm.
ACKNOWLEDGMENTS
This work was supported by the University of California Task Force on AIDS.
We thank P. Green, W. O'Brien, J. Zack, K. Arrigo, V. Miller, and M. Yip for helpful comments and W. Aft for preparation of the
manuscript.
LITERATURE CITED
1. Ahmad, N., andS. Venkatesan. 1988. nef proteinofHIV-1isa
transcriptional repressor of HIV-1 LTR. Science 241:1481-1485.
2. Arrigo, S. J., and K. Beemon. 1988.Regulation ofRous sarcoma virus RNAsplicing and stability. Mol. Cell. Biol. 8:4858-4867. 3. Arya, S. K., C. Guo, S. F. Josephs, and F. Wong-Staal. 1985.
Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 229:69-73.
4. Bindereif, A., and M. R. Green. 1986. Ribonucleoprotein com-plexformation during pre-mRNA splicing in vitro. Mol. Cell. Biol. 6:2582-2592.
5. Cann, A. J., Y. Koyanagi, and I. S. Y. Chen. 1988. High
efficiency transfection of primary human lymphocytesand stud-ies of geneexpression. Oncogene 3:123-128.
6. Culien, B. R. 1986. trans-Activation of human
immunodefi-ciency virus occurs viaabimodalmechanism. Cell 46:973-982. 7. Dayton, A. I., J. G. Sodroski, C. A. Rosen, W. C. Goh, and W.A. Haseltine. 1986. The trans-activator gene of the human T celllymphotropic virustypeIIIisrequiredfor replication. Cell 44:941-947.
8. Feinberg, M. B., R. F. Jarrett, A. Aldovini, R. C. Gallo, and F. Wong-Staal. 1986. HTLV-IIIexpression and production involve
complex regulation at the levels ofsplicing and translation of
viralRNA. Cell 46:807-817.
9. Felber,B.K., M. Cladaras-Hadzopoulou,C. Cladaras,T.
on November 10, 2019 by guest
http://jvi.asm.org/
land, and G. N.Paviakis. 1989. rev protein ofhuman immuno-deficiency virustype 1affects the stability andtransportof the
viral mRNA. Proc. Natl. Acad. Sci. USA86:1495-1499.
10. Frendewey, D., and W. KeUler. 1985. Stepwise assembly of a
pre-mRNA splicing complex requires U-sn-RNPs and specific intronsequences. Cell 42:355-367.
11. Gallo, R., F.Wong-Staal, L. Montagnier, W. A.Haseltine,and M. Yoshida. 1988. HIV/HTLV gene nomenclature. Nature (London) 333:504.
12. Hadzopoulou-Cladaras,M., B.K. Felber, C. Cladaras, A. Atha-nassopoulos, A. Tse, and G. N.Paviakis. 1989. The rev(trslart) protein ofhuman immunodeficiency virus type 1 affectsviral mRNA andproteinexpression viaa cis-actingsequenceinthe envregion.J. Virol.63:1265-1274.
13. Hammarskjold, M.-L., J. Heimer, B. Hammarskjold, I. Sang-wan, L. Albert, and D. Rekosh. 1989. Regulation of human
immunodeficiencyvirus envexpressionby the rev gene product. J. Virol. 63:1959-1966.
14. Knight, D. M., F. A. Flomerfelt, and J. Ghrayeb.1987.
Expres-sion of the art/trs protein of HIV and study of its role in viral envelope synthesis. Science236:837-840.
15. Konarska, M. M., and P. A. Sharp. 1986. Electrophoretic separation of complexes involved in the splicing ofprecursors to
mRNAs.Cell 46:845-855.
16. Koyanagi, Y., S. Miles, R. T. Mitsuyasu, J. E. Merrill, H. V. Vinters,andI.S. Y. Chen. 1987. Dualinfectionof the central nervoussystembyAIDSviruses with distinctcellulartropisms. Science 236:819-822.
17. Le, S. V., J. H. Chen, M. J. Braun, M. A. Gonda, and J. V. Maizel. 1988. Stability ofRNAstem-loop structureand distri-bution of nonrandomstructurein thehumanimmunodeficiency virus(HIV-1). Nucleic Acids Res. 16:5153-5168.
18. Lee,H., P.Swanson, V. S.Shorty,J. A.Zack, J. D. Rosenblatt, and I. S. Y. Chen. 1989. High rate of HTLV-II infection in seropositive IV drug abusersfromNewOrleans. Science244: 471-475.
19. Luciw, P. A., C.Cheng-Mayer, and J. A. Levy. 1987. Mutational
analysisof the human immunodeficiency virus:theorf-Bregion
down-regulates virus replication. Proc. Natl. Acad. Sci. USA 84:1434-1438.
20. Malim, M. H., J.Hauber, R. Fenrick, and B. R.Cullen. 1988.
Immunodeficiency virus rev trans-activator modulates the
expression of the viral regulatory genes. Nature (London) 335:181-183.
21. Malim, M. H., J. Hauber, S.-Y. Le, J. V. Maizel, and B. R.
Cullen. 1989. The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of
un-splicedviralmRNA. Nature(London) 338:254-257.
22. Muesing,M.A.,D. H.Smith, andD.J. Capon.1987.Regulation
of mRNA accumulation by human immunodeficiency virus trans-activatorprotein. Cell 48:691-701.
23. Peterlin, M.,P.Luciw,P.Barr,andM. Walker.1986. Elevated levels of mRNA can account for the trans-activation of human
immunodeficiencyvirus. Proc. Natl. Acad. Sci. USA 83:9734-9738.
24. Rabson,A.B.,P. E.Steel,C. F.Garon,and M.A. Martin. 1983. mRNAtranscriptsrelated tofull-lengthofendogenous retrovi-ral DNA inhuman cells. Nature(London) 306:604-607.
25. Rosen,C.A., J.G.Sodroski, W. C. Goh, A. Dayton, J.Lippke,
andW. A. Haseltine. 1986. Post-transcriptional regulation
ac-counts for the trans-activation of the human T-lymphotropic virustype III.Nature(London) 319:555-559.
26. Rosen, C.A., J. G. Sodroski,and W. A. Haseltine. 1985. The
locationofcis-acting regulatorysequencesin the humanT cell
lymphotropic virus III(HTLV-III/LAV) long terminal repeat.
Cell41:813-823.
27. Rosen, C. A., E. F.Terwilliger,A.I.Dayton,J.G.Sodroski,and W.A.Haseltine. 1988.Intrageniccis-actingartgeneresponsive sequences ofthe human immunodeficiency virus. Proc. Natl.
Acad. Sci. USA 85:2071-2075.
28. Sadaie,M. R., T.Benter, and F.Wong-Staal. 1988. Site-directed mutagenesisof twotrans-regulatorygenes(tat-III, trs)of HIV-1. Science239:910-913.
29. Saiki,R.K.,D.H., Gelfand,S.Stoffel,S. J.Scharf,R.Higuchi,
G. T. Horn, K. B. Muilis, and H. A. Erlich. 1988. Primer-directedenzymatic amplificationofDNA with a thermostable DNApolymerase. Science239:487-491.
30. Sodroski,J., W.C.Goh,C.Rosen,A.Dayton, E.Terwillinger,
and W. Haseltine. 1986. A second post-transcriptional
trans-activatorgenerequiredforHTLV-III replication. Nature (Lon-don) 321:412-417.
31. Sodroski, J., C. Rosen, F. Wong-Staal, S. Z. Salahuddin, M. Popovic, S. Arya, R. C. Gallo, and W. A. Haseltine. 1985.
trans-Acting transcriptional regulation ofhuman T-cell
leuke-miavirustype IIIlong terminalrepeat. Science 227:171-173.
32. Treisman, R., S. H. Orkin, and T. Maniatis. 1983. Specific transcription and RNA splicing defects in five cloned
-thalassemiagenes. Nature(London) 302:591-596.
33. Wieringa, B., F. Meyer, J. Reiser, and C. Weissmann. 1983. Unusual splice sites revealed by mutagenic inactivation ofan
authentic splice site of the rabbit13-globingene. Nature (Lon-don)301:38-43.
34. Wright, C., B. Felber, H. Paskalis, and G. Pavlakis. 1986. Expression and characterization of the trans-activator of HTLV-III/LAV virus. Science234:988-992.

![FIG.iocytes. RNAfrom(A)peripheralactions in thePortions ofn, and th(a5-32P]ATP andeted on a 6%](https://thumb-us.123doks.com/thumbv2/123dok_us/1324018.86162/4.612.66.300.84.356/fig-iocytes-rnafrom-peripheralactions-theportions-ofn-atp-andeted.webp)