0022-538X/91/020913-09$02.00/0
CopyrightC) 1991, AmericanSociety forMicrobiology
An RNA Hairpin
at
the
Extreme
5'
End of the Poliovirus RNA
Genome
Modulates Viral Translation in Human Cells
ERIC A. F. SIMOES"2 ANDPETERSARNOW13*
Departmentof Biochemistry, Biophysics and
Genetics,'
Departmentof Microbiology andImmunology,3 andDepartment
of
Pediatrics,2
University
of
Colorado Health SciencesCenter,
Denver,
Colorado 80262Received 3 August 1990/Accepted 26 October 1990
Several mutations were introduced into an infectious poliovirus cDNA clone by inserting different oligodeoxynucleotide linkers into preexisting DNA restriction endonuclease sites in the viral cDNA. Ten mutated DNAs were constructed whose lesions mapped in the5' noncoding region or in thecapsid coding region oftheviralgenome.EightofthesemutatedcDNAsdidnotgive risetoinfectious virusupontransfection into human cells, one yielded viruswith a wild-type phenotype, andone gaveriseto a viralmutant with a small-plaque phenotype. Thislastmutant,designated 1-5NC-S21, bearsa6-nucleotide insertion in theloopof a stable RNAhairpinatthevery5'end of theviralgenome. Detailedanalysis ofthebiological properties of 1-5NC-S21 showed that the primarydefectinmutant-infected cellsisafivefolddecrease in translationrelative towild-type-infected cells. Transfection intoHeLa cellsofinvitro-synthesizedRNA molecules bearingeither the 5' noncoding region of1-5NC-S21orwild-type poliovirusupstream ofaluciferasereportergeneshowed that themutatedRNAhairpinwasresponsible fortheobserved decreaseinviral translationinmutant-infected cells andconferred this defecttoheterologousRNAs. Thesefindings indicatethatanRNAhairpinlocated at theextreme5' end of the viral RNA andhighlyconservedamongenterovirusesand rhinoviruses profoundly affects thetranslation efficiencyofpoliovirusRNA ininfectedcells.
The discovery that cDNA copies of eucaryotic RNA viruses and RNA transcripts produced from those cDNAs canyield infectiousvirusupontransfection intoappropriate
hostcells (20, 27, 43) has made these viruses amenable to
genetic manipulations (see reference 35 for a review). For example, poliovirus, a member ofthe picornavirus family (31), is currently under intense scrutiny. The genome of poliovirusconsists ofapositive-stranded RNAmolecule of approximately 7,500 nucleotides with a virally encoded protein, VPg, covalently attachedtoits 5' end andapoly(A) homopolymertract atits3' end (31). Incontrast to cellular mRNA molecules, which contain a m7G(5')ppp(5')N cap structure at their 5' ends, polysome-associated poliovirus RNAterminates with 5'-terminalpUpU. Alongopen
read-ing frame starting atposition 743 is translated into a viral polyproteinof200,000Da.Precedingtheopenreadingframe is a 743-nucleotide-long 5' noncoding (5NC) region that containssignals importantfor(i)theinitiation oftranslation, (ii) the initiation ofreplicationofpositive-strandRNA mol-ecules,and (iii)thedetermination ofthe viral neurovirulent phenotype. Sequence analysis has revealed several interest-ing features of this region (14, 26). First, the primary nucleotide sequence is highly conserved among the three poliovirus serotypes (40). Second, eight unused AUG codonsprecedethe AUGcodonthat is used for theinitiation of translation of the viral polyprotein (14, 26). Third, as
many as23 RNA hairpin structures have beenpredicted in thisregion bycomputer-assisted analysis (1, 25, 29, 36); the presenceof several of them has beenconfirmedby biochem-icaltests (18, 21, 36).
One of theseconfirmed RNAhairpins islocated 9 nucle-otides downstream of the 5' end of the viral RNA (18). Its secondary structure was determined by using nucleases as structural probes (18). The first genetic evidence for the
*Correspondingauthor.
importance ofthis RNA hairpin in the viral life cycle was provided by Racaniello and Meriam (28), who isolated a mutant(R1-5NC-1)in whichtherewas adeletion of nucleo-tide10,whichwaspredictedtobe involved inbasepairingat
the bottom ofthehairpinstructure.Thismutantdisplayeda temperature-sensitive phenotype for RNA synthesis and a slight defect, which was not temperature dependent, in protein synthesis (28). Revertants ofR1-5NC-1 restoredbase pairingatthe bottom of thehairpin. This finding suggested that this hairpin structure is involved in translation and amplification of the viral RNA. However, it has not been clear whether itsprimaryrole is inmodulating viral transla-tion, replicatransla-tion,orboth. Wereporthere theconstruction of a mutant(1-5NC-S21) that bearsa6-nucleotide insertion in thesingle-stranded loop ofthishairpinstructure.Evidence is presentedthat themutant is defective intranslation and not in replication of the viral RNA. This mayindicate that the stem of the hairpin is important for efficient viral RNA replicationand thatthe entirehairpinisinvolvedindirecting efficient translationof the viral RNA in human cells.
MATERIALS ANDMETHODS
Cells and viruses. HeLa cells were grown in Dulbecco's modified Eagle'smedium(DMEM) supplementedwith 10% bovine serum(Irvine Scientific). Wild-type poliovirustype1
(PV1) and viable mutant viruses were isolated from single plaques derivedfrom HeLa cells that had been transfected withinvitro-synthesized full-length RNAmolecules as
pre-viously described (33).
Bacterial strains andplasmids.Allplasmidsweregrownin Escherichia coliHB101, obtainedoriginallyfrom C. Cepko (Harvard Medical School). The construction of plasmids p5'polioandT7-polio has been described(33).
Enzymes. Restrictionenzymeswerepurchasedfrom New
EnglandBioLabsandPromega. T4 DNAligase,T4
polynu-913
on November 10, 2019 by guest
http://jvi.asm.org/
cleotide kinase, T7 RNA polymerase, and RNasin were obtained from Promega.
Construction of linker-insertion mutations. Plasmid p5'polio, containing the first 1,809 nucleotides of a full-length cDNA clone of PV1 (T7-polio) (33) was partially digested with RsaI in the presence of 20
Vxg
of ethidium bromide per ml. Full-length, 4,629-bp linear fragments were separatedfrom smaller fragments on a1%low-melting-point agarose gel. The gel segment containing the full-length fragment was excised, and the DNA was isolated (34). Phosphorylated HpaI orSmaI oligodeoxynucleotidelinkers (Collaborative Research Inc., Waltham, Mass.), each 6bpin length, were ligated overnight at 14°C to full-length linear molecules (34). Circularized molecules were cut to comple-tionwith HpaI or SmaI,respectively; linearmoleculeswere thenisolated bylow-melting-point agarose gel electrophore-sis and ligated. Ligated molecules were transformed into HB101, and recombinants were selected that had lost an appropriate RsaI restriction site and had acquired aunique HpaI or SmaI site.Mutantp5NC-SS21 wasconstructedbyintroducinga6-bp Sall linkerinto the SmaI site (at position21) of p5NC-S21 (Table 1).
Mutantswith mutationsmapping in the first 502 bpofthe viral cDNA were reconstructed into afull-length infectious cDNA, T7-polio (33), by using a three-fragment ligation strategy. Briefly, mutated plasmid p5'polio was digested withPvuII andPflmI, yielding a 536-bp fragment. Plasmid T7-polio wascutwitheitherPflmI orwith acombination of PflmI and PvuII, yieldingfragments of3,131 and 6,162bp, respectively. After purification bylow-melting-pointagarose electrophoresis, the three fragments were ligated together, and full-length cDNAs bearing the desired mutations were identified. Plasmids bearing mutations betweenpositions456 and 1513 weredigested withBsmI, whichcutsonlyatthese twopositionsinp5'polioorT7-polio. The1,057-bpfragment harboring the mutation was isolated and ligatedto thelarge fragmentobtained fromdigestion ofT7-polio with BsmI.
T7-polio plasmids were linearized by digestion with EcoRI, and full-length viral transcripts were produced in vitro by the addition of T7 RNA polymerase (33). Subse-quently, RNAs were transfected into HeLa cells as de-scribedpreviously(33).Plaqueswereusuallyobserved 36to 48 h after transfection. Multiple virus isolates were then recovered from the agar overlay, and viral stocks were prepared and stored at -20°C.
To confirm that thephenotype of themutantswas dueto the introduced mutation, infectious cDNAs expressing mu-tant viruses were reconstructed by mix and match experi-ments(4)with each mutationfollowed bysequencingof the fragmentspanning the introduced mutation.
One-stepgrowth curve.Themeasurementofvirus growth andrelease was performed in HeLa cells on 60-mm dishesas described previously (3).
Measurement of viral RNA synthesis. HeLa cell monolay-ers were infected with 1-5NC-S21 or wild-type virus at a multiplicity of infection (MOI) of 20. After adsorption at 37°C for 30 min, medium was added and the plates were incubatedat37°C. At various times after infection the dishes were placed on ice. The cells were washed with ice-cold phosphate-buffered saline (PBS) and scraped into 500 ,ul of PBS.Aftercentrifugation at 800 xgfor 1 min andaspiration of the PBS, the cells were lysed by suspension in a cold solution of 10 mM Tris hydrochloride (pH
7.4)-i
mM EDTA-0.5% Nonidet P-40 (Shell Chemicals) at 0°C for 5 min. Nuclei and debris were pelleted by centrifugation.Cytoplasmic RNAs were denatured by the addition of 6x SSC(lxSSCis 0.15 M NaCl
plus
0.015 M trisodiumcitrate)
and 7.4% (wt/vol) formaldehyde at 60°C for 15 min. The RNA was bound to nitrocellulose by aspiration with a slot-blot apparatus (Schleicher & Schuell). Filters were
bakedat80°Cfor 90 min andprehybridized in sealedbagsin asolution containing 50% formamide, 5x SSC, 8x Denhardt solution, 50 mM Na2PO4 (pH 6.5), 0.1% sodium dodecyl sulfate, and 25 ,ug ofsalmon sperm DNA per mlat65°C for 1 h. Hybridization was continued overnight at65°C in the same solution containing a
32P-labeled
in vitro-made tran-script ofpoliovirus minus-stranded RNA, spanning nucleo-tides 670 to 220 of PV1. Afterhybridization, the filterswere washed threetofive times in asolutioncontaining 0.1x SSC and 0.1% sodium dodecyl sulfate at 65°C, air dried, and exposedtoX-rayfilmat -70°C. Quantitation of theamount of32P hybridizedtoRNAateach time pointwasperformed by densitometric analysis of the bands seen on the X-ray film with a computing densitometer (Molecular Dynamics, Sunnyvale, Calif.). Comparisons were made in the linear range of exposure of the X-ray film, as determined by analysis of several different exposures.Measurement of RNA stability. HeLa cell monolayer cul-tures in 60-mm dishes were infected with poliovirus at an MOI of 20 as described above. At 4 h after infection the medium was removed, and DMEM containing 2 mM guani-dine hydrochloride was added to the monolayer to stop viral RNA synthesis (see reference 2 for a review). Cells were harvested at various times after guanidine hydrochloride addition as described above. RNA wasdenatured, bound to filters, and hybridized to [32P]RNA probes as described above. To detect glucose-regulated protein 78-immunoglob-ulinbinding protein (BiP), mRNA a probe containing nucle-otides 1610 through 1496 of a hamster Bip cDNA was used
(32).
Measurement of viral protein synthesis. Infection proce-dures were as described above. At various times after infection, cells were incubated for 30 min in methionine-depleted DMEM containing 100 ,uCi of [35S]methionine per ml. The monolayers were washed with cold PBS, and the cells were lysed onice in a solutioncontaining 50 mM Tris hydrochloride (pH 8.0), 150 mM NaCl, 5 mM EDTA, and 0.5% Nonidet P-40. Nuclei were pelleted by centrifugation, and the labeledproteins in the supernatant wereanalyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The gels were treated with Fluoro-Hance (Research Prod-ucts International Corp. Prospect, Ill.), dried, and exposed to X-ray film at -70°C.
Transfection of in vitro-synthesized hybrid RNA molecules into HeLa cells. Plasmids containing the promoter for T7 RNA polymerase and the 5NC regions of luciferase or wild-type or mutant (S21, SS21) poliovirus immediately upstream of the coding sequence for luciferase were con-structed as described previously (19). RNAs synthesized in vitro from theseplasmids were analyzed by electrophoresis in 1% agarose gels containing ethidium bromide and photo-graphed. The relative amounts of RNAs were then deter-mined by densitometry of the Polaroid picture. Transfection of in vitro-synthesized RNA molecules was performed by a DEAE-dextran transfection method (33). Briefly 60-mm dishesof HeLa cells were infected with wild-type PV1at an MOI of 20, or mockinfected, and incubatedfor 3 h at37°C. Equal quantities of the different RNA molecules were then transfected, in parallel, into the PV1-infected or mock-infected cells (19). One hour later (4 h after infection), the cells were harvested, washed in ice-coldPBS, and lysed by
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
SS21 S140
IS52
v
VP4
S702(5NC-S702)
S21(5NC-S21)
S1265 H1265 S1278
S1238 S1329
At
VP2
FIG. 1. Mutagenesis of the 5NC region and the capsid coding region of the poliovirus typeIgenome. Numbersindicate the positions of theintroduced mutations. Designations of viablemutantsareshownwithin parentheses.Nucleotide insertionsare asfollows:S, CCCGGG;
H,GUUAAC; SS,CCCGUCGACGGG.
three freeze-thaw cycles. Luciferaseactivities in theextracts
weremeasuredasdescribedpreviously(9)withaMonolight
2001 Luminometer (Analytical Luminescence Laboratory Inc., San Diego, Calif.).
Measurement oftransfection efficiencies ofin vitro-synthe-sized RNA molecules into HeLacells. Radioactively labeled hybridRNAmoleculeswereproduced in vitro with T7 RNA polymerase (see above) by adding [32P]CTP to the incuba-tion mix. Labeled RNA molecules were transfected into HeLacellsandgrownin60-mm dishes by using the DEAE-dextrantransfection procedure (see above). Cellswere har-vested after incubation for 10 min, and cytoplasmic RNAs were prepared as described above. Input RNAs and
cyto-plasmic RNAs were analyzed on a denaturing agarose gel containing 6% formaldehyde. After the gel run, the RNAs were transferred to a nitrocellulose filter. The filter was dried,exposedtoanX-ray film, and autoradiographed.
RESULTS
Mutagenesis of RsaI restriction endonuclease sites in the 5NC- and capsid-coding region of the viralgenome. Defined mutations wereintroduced into preexisting RsaI sites inan infectious poliovirus cDNA. Mutations at eight RsaI sites
were obtained, mapping both in the 5NC region and in the capsid-coding regionoftheviralgenome(Fig. 1). Synthetic
oligodeoxynucleotidelinkerscontaining6bp encoding HpaI
or SmaI sites were inserted into RsaI sites present in the 5'-proximal 1,329-bp cDNA of an infectious poliovirus cDNAclone. Mutantswereobtained with insertions at8of 14 possible RsaI sites. Five mutations mapped in the viral 5NCregion(S21, SS21, S52, S140, S702), and five mapped in the region encoding capsid protein VP2 (S1238, S1265, H1265, S1278, S1329). Positions of introduced mutations
wereverified byrestrictionenzymedigests; full-length
tran-scripts were made from these plasmids in vitro and
trans-fected intohumanHeLa cells.Cellswereoverlaidwithagar
and incubated at 32.5, 37, or 39.5°C. Full-length cDNAs containing insertions of 6 bpat positions 140, 1238, 1265, 1278, and 1329 did not give rise to viable virus underany
conditions. However, two mutated plasmids gave rise to
viable viruses displaying wild-type-sized or small-sized plaque phenotypes. These plasmids contained 6-nucleotide insertions at positions 702 (1-5NC-S702, wild-type-sized plaques) and 21 (1-5NC-S21, small plaques) in the viral
genome(Table 1).Avirus withawild-typeplaque phenotype
(1-5NC-S702) could be recovered bearing a 6-nucleotide insertionatposition702. Thiswasnotsurprising,because it hasbeen shown thatmany mutationscanbetoleratedinthe 5NC region between nucleotide positions 600 and 743 (10, 17, 41).
A 6-bp insertion (5' CCCGGG 3') at position 21 in the
full-length cDNA gave rise to a viable virus (1-5NC-S21) with a small-plaque phenotype (Fig. 2), whereas a 12-bp insertion (5' CCCGTCGACGGG 3', SS21) at the same positionin the cDNAcould not berecoveredasvirus after transfection (Table 1). No temperature or cold sensitivity
was observed in the plaque phenotype of 1-5NC-S21. Be-cause the mutant contained an insertion in a loop of a predicted hairpin structure that has been shown to be important in the amplification of the viral RNA (28), we further investigated the molecular events leading to the small-plaque phenotypeof1-5NC-S21.
One-stepgrowthcurveof1-5NC-S21 andwild-typeviruses. As afirstapproachtocharacterizethe defect in1-5NC-S21,
one-step growth curves of mutant and wild-type viruses were obtained.The mutantdisplayed afinal viralyield that wasfivefold less than that of the wildtype(Fig. 3).
Further-TABLE 1. Summary of constructed poliovirusmutants
Clone Wild-typesequence Mutant sequence Phenotypea
1 p5NC-S21 GTTGT21ACCCA GTTGTCfCGGGACCCA M(1-5NC-S21)
2 p5NC-SS21 GTTGT21ACCCA GTTGTCCCGTCGACGGGACCCA L
3 p5NC-S52 CTAGT52ACTCC CTAGTCCCGGGACTCC L
4 p5NC-S140 GGGGT140ACAAA GGGGTCCCGGGACAAA L
5 p5NC-S702 TAAGT702ACAAT TAAGTCCCGGGACAAT WT(1-5NC-S702)
6 pVP2-S1238 TATGT1238ACTAC TATGTCCCGGGACTAC L
7 pVP2-S1265 CGGGT1265ACACC CGGGTCCCGGGACACC L
8 pVP2-H1265 CGGGT1265ACACC CGGGTGTuAACACACC L
9 pVP2-S1278 CATGT1278ACAGT CATGTCCCGGGACAGT L
10pVP2-S1329 GCCGT1329ACCAG GCCGTCCQGGGACCAG L
a L,Lethalmutation;WT,wild type;M,mutant.Thevirusesobtainedareindicatedwithinparentheses.
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.87.521.77.180.2] [image:3.612.55.559.605.715.2]A.
I
[image:4.612.83.294.66.532.2]B
FIG. 2. Plaque phenotype of wild-type (A) and 1-5NC-S21 (B) viruses. HeLa cellswereinoculated with wild-typeormutantvirus, overlaid withagar,and incubatedat37°C. Plateswerestained with crystal violet 48 h later.
more, accumulation of virions was delayed in
mutant-in-fectedcellsrelative tothat inwild-type-infected cells. Viral RNA synthesis in 1-5NC-S21- and wild-type-infected cells. The relative amounts ofpositive-strand RNA mole-culesproduced in mutant- and wild-type-infected cells was measured todeterminewhether there was adefect inRNA replication in 1-5NC-S21-infected cells. Synthesis of polio-viral RNA in infected cells is usually monitored by the incorporation ofradioactive uridine in thepresenceof dacti-nomycin. This drugblocks the enzymatic activities of cellu-larRNApolymerases butnotof poliovirus RNA polymerase (2). However, mutant 1-5NC-S21was observedto be sensi-tive to dactinomycin (data not shown). Therefore, total
10000-phtucell 100
10
0 2 4 6 8 10
HOURS AFTER INFECTION
FIG. 3. One-step growth curves of wild-type and 1-5NC-S21 viruses.HeLacells wereinfectedwithwild-typeor1-5NC-S21 virus at an MOI of 20, and the total virus yield was determined. The numbers of PFU per cell at different times after infection are indicated: O,wild-type virus;*, 1-5NC-S21 virus.
cytoplasmic RNA wasisolatedfrom mutant- and wild-type-infected cells at several times after infection and immobi-lizedonnitrocellulose membranes. Theamountof positive-stranded viral RNA was detected afterhybridizationwith a radioactively labeledminus-stranded viral RNA and visual-ized by autoradiography (Fig. 4A). Figure 4B displays the quantitation of the data showninFig. 4A. Mutant1-5NC-S21 produced 85to90%asmuch RNAasdidwild-type virus in similarlyinfected cells(Fig. 4). Despite a smalldelayinthe synthesis of1-5NC-S21 RNA, the amount of mutant RNA produced isnearlyidentical to that ofwild-type RNA(Fig. 4B).
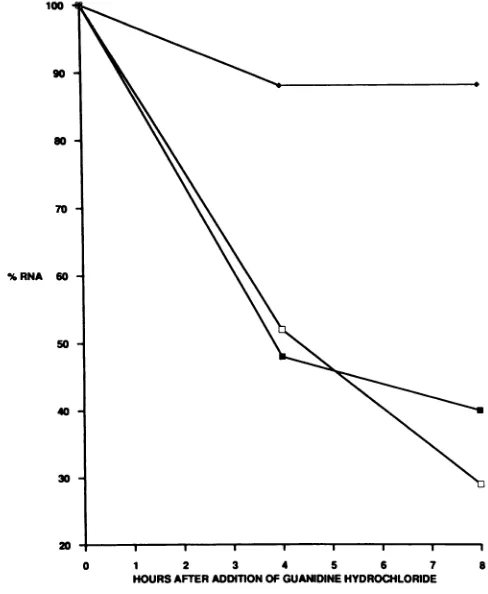
Stability of mutant (1-5NC-S21) and wild-type RNAs in infected cells. Although similaramounts of viral RNAwere detected in mutant- and wild-type-infected cells, it was possible that mutant and wild-type RNAs have different half-lives and thatonly asubpopulationof the mutantRNA wasactually available forpackaginginto virions.Therefore, we tested the half-lives ofmutant and wild-type RNAs in infectedcells, afternew viral RNAsynthesis wasinhibited by the addition ofguanidine hydrochloride (see Materials and Methods). The mutant and wild-type RNAs have the samestabilityininfected cells(half-life,4 to 5h)(Fig. 5). In addition, the decay of the mRNA encoding BiP was mea-sured in mutant- andwild-type-infected cells. We chose to monitor BiP mRNAbecause its level is thesameininfected and uninfected cells (32). The half-life of BiP was greater than 8 h in mutant-infected cells (Fig. 5) and wild-type-infected cells(datanotshown).Thus, these results showthat
--- ---
---
-
--- ...
-
---F- I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.327.558.69.368.2]A
S21
WT
B
18000
16000
14000
12000
OD UNITS 10000
8000
4000
2000
100
0-
2-
4-6-
_
8-
w-10-a-
0-
2-4- va
6-
_
8-
_
10-
-0 2 4 6 8 10
[image:5.612.60.295.70.533.2]HOURSAFTER INFECTION
FIG. 4. Synthesis of viral RNA in HeLa cells infected with wild-type (WT) and 1-5NC-S21 (S21) polioviruses. Viral RNA
synthesiswasmonitoredasdescribedinMaterialsandMethods.(A) Autoradiograph of the slot blot hybridization. (B) Densitometric
scan(optical density [OD]at633nm) oftheautoradiograph shown
inpanel A: r1, wild-type poliovirus; *, 1-5NC-S21 virus.
thefivefoldreductionof viralgrowth in1-5NC-S21-infected cells isnotlikelytobe duetoadecreasedstabilityofmutant RNAs.
Proteinsynthesisin1-5NC-S21- andwild-type-infectedcells. To test whether the small-plaque phenotype of1-5NC-S21 was due to a decrease in viralprotein synthesis, we moni-tored viral translation in cells infected with mutant and wild-type viruses at MOIs of 10 and 1 (Fig. 6A and B, respectively). Fewer viral proteins were made in mutant-infected cells than in the wild-type infection at 3 h after infectionwith an MOI of 10 (Fig. 6A) orat6 and9 h after infectionatanMOIof1(Fig. 6B). Furthermore,inhibitionof
90
70
% RNA 60
50
40
50
20 I
0 1 2 3 4 5 6 7 8
HOURSAFTER ADDMON OF GUANIDINEHYDROCHLORIDE
FIG. 5. Stability ofwild-typeand 1-5NC-S21 RNAs in infected HeLacells.Analysiswasperformedasdescribedin Materialsand Methodsand the legend toFig. 4B. The amount of RNA at the time ofaddition of 2 mMguanidinehydrochloridewasdesignated100%. As acontrol,thedegradation ofthecellularmRNAencoding BiPis included.Symbols: O,wild-typeRNA; *,1-5NC-S21RNA;*,BiP mRNA.
host-cell translation by the mutant virus was significantly delayedcomparedwith thatinwild-type-infected cells (Fig. 6B). Thisdefect couldbe overcomebyahigher MOI (Fig. 6). Late in infection the same amounts of viral proteins were made in mutant- and wild-type-infected cells (Fig. 6A). Immunoprecipitation of the viral proteins revealed that polyprotein processingwas notdisturbed in mutant-infected cells(datanotshown).Thus, theseresults show that 1-5NC-S21 has a defect in viral translation, resulting in a low production of viral proteins and a delayed inhibition of cellular translation.
Transfectionof in vitro-synthesizedhybridRNA molecules into HeLa cells.Anearly defect in translation could be dueto aprimary defect in viral translationor to asecondary effect ofdelayed uncoating ofthe viral RNA. In addition, it was possible that long-range
pleiotropic
effectsperturbing
the overallstructureof the viralRNAgenomewereresponsible forthedecreased translation of 1-5NC-S21 RNA.To test whether the 5NC region alone of 1-5NC-S21 generated a defect in translation, we constructed
hybrid
RNAmoleculescontainingthe5NCregionsofwild-typeand mutantpoliovirus (S21, SS21) linkedtothecoding
region
of thefirefly luciferase gene(19).First, we tested whether all hybrid RNAscould be intro-duced with similar efficiency into HeLa cells.
Therefore,
radioactively labeled hybrid RNAs were produced in vitro
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.312.556.77.374.2]A S21 WT B S21 WT
M 3 5 7 3 5 7 M 6 9 6 9
P1- -F -,
-
3CD-2C- < _
4
FIG. 6. Protein synthesis in wild-type (WT)- and 1-5NC-S21-infected cells. Mock-1-5NC-S21-infected (M) and virus-infected HeLa cells
werepulse-labeledwith
[35S]methionine
for 30min atthe indicated hoursafter infection. Solubleextracts werepreparedand analyzed by sodiumdodecyl sulfate-polyacrylamidegel electrophoresis. An autoradiograph is shown displaying extracts obtained from cells infected withaMOI of 10(A)or1(B).PolioviralproteinsP1,3CD, and2Careindicated.by
T7 RNApolymerase,
andsimilaramounts of RNAsweretransfected into HeLa cells (see Materials and Methods).
Cytoplasmic
extracts wereprepared
10 min aftertransfec-tion,
andtransfected RNAswereanalyzed
informaldehyde-containing
agarosegels
and thenautoradiographed.
Bothuncapped
wild-type
and mutant (S21, SS21) andcapped
luciferase
hybrid
RNAs were introduced with a similarefficiency
into HeLa cells(Fig.
7). A small percentage of bothuncapped
andcapped
RNAswasdegraded
atthattime.Usually,
the half-life of the bulk of transfected RNAs in thecytoplasm
is less than 20 min. However, the fraction oftransfected RNAs that becomes associated with
polysomes
has a half-life of greater than 2 h (12a). Densitometric
analysis
of the bands inFig.
7showed that0.01% of theinput
RNAwas taken upinto the
cytoplasm
of the cell.Next, different amounts of
hybrid
RNA molecules weretransfected into uninfected and
wild-type-infected
HeLa cells, and the translational efficiencies of the RNAs weredetermined
by
monitoring
theproduction
of luciferase mol-ecules.Capped
RNAscontaining
the 5NCregion
of lu-ciferase linkedtoluciferase could be translated in uninfected HeLacells butnotinpoliovirus-infected
HeLa cells(Fig.
8). Thisindicates thatcapped
RNAmolecules could besuccess-fully
introduced into mammalian cells (12a) and that these RNA molecules could be translated in, uninfected cells;furthermore,
the amount of translated product was propor-tional to the amount of RNA introduced. The lack ofexpression
in infected cells wasexpected,
sincepoliovirus
infection inhibits
cap-dependent
translation (11, 38).Uncapped
RNAscontaining
the 5NC region ofwild-type
poliovirus
linked to luciferase were translatedefficiently
in-WT S21 SS21 LUC
c c
FIG. 7. Transfection efficiencies ofin vitro-synthesized hybrid RNA molecules. HeLa cells (2 x 106) were transfected with radioactively labeledRNAs asdescribed in Materials and Methods. Cytoplasmic RNAs were prepared 10 min after transfection and analyzed in aformaldehyde-containing agarose gel. An autoradio-graph of thegel is shown. Input (i) and cytoplasmic (c) species of uncapped wild-type (WT), uncapped mutant (S21 and SS21), and capped luciferase(LUC) hybridRNAs areshown.
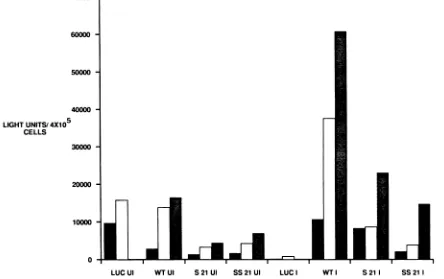
uninfected HeLa cells as well as in infected HeLa cells. Furthermore, translation of 5NC poliovirus-luciferase RNAs wasfivefold more efficient in infected cells than in uninfected cells. This demonstrates that we could mimic the cap-independent translation of genes containing the 5NC region ofpoliovirus (22, 42) by introducing RNA molecules directly into mammalian cells. This method circumvents possible splicing and transport problemsthatcertainRNA molecules may encounterwhenexpressedfromDNAplasmids.
Uncapped hybridRNAmoleculescontainingthe 6-nucle-otide insertion of 1-5NC-S21 or a 12-nucleotide insertion at the same position (Materials and Methods) translated five-fold less well than the unmutagenized viral 5NC region in both uninfected and infected cells (Fig. 8). Thus, the trans-lational defect of 1-5NC-S21 can be mimicked in hybrid RNA molecules. Theadditional mutationatthe same posi-tion(SS21),whichunfortunatelycouldnotbe recoveredas a virus, displayed a similar translational defect, further sub-stantiating the importance of the RNA hairpin structure in viraltranslationalregulation.
DISCUSSION
Tenmutationswereintroduced intoaninfectious poliovi-ruscDNAclone.Onlytwoof the mutatedplasmidsgave rise to viable virus upon transfection into human HeLa cells. One mutant, 1-5NC-S21, containinga6-nucleotide insertion at position 21 in the viral 5NC region, displayed a small-plaque phenotype. 1-5NC-S21 producedfivefoldfewer viri-onsthandidwild-typevirus butsynthesized nearlywild-type amounts ofRNA during a single infectious cycle. In con-trast,viralproteinsynthesiswasdiminishedearlyin mutant-infected cells and the inhibitionofhost cell translationwas impaired. Therefore,it islikelythat thedecreasedviralyield resultedfroma decrease in viralcapsid proteinproduction, since sufficientquantitiesofmutantviral proteintosupport viral RNA synthesis were apparently present. It has been
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.323.557.73.261.2] [image:6.612.83.279.75.315.2]70000
-60000
-50000
-
40000-LIGHTUNITS/
4X105
CELLS30000
-20000
-10000
0
LUCUl WTUl S21 Ul SS21Ul LUCI WTI S21 1 SS21 1
FIG. 8. Translation ofinvitro-synthesizedhybridRNA molecules in HeLa cells. HeLa cells(2 x 106)wereeitherleft uninfected(UI)or
infected withwild-typepoliovirusat anMOIof 20(I).Three hourslater, cellsweretransfectedwithdifferentamounts(1jig[_],2 ,ug[ELi],
or 4
Vig
[El])ofhybridRNAmoleculesbearingdifferent5NCregions(capped luciferase [LUC], uncapped wildtype[WT],uncappedS21, uncappedSS21). Soluble extracts wereprepared1hlater,and thetranslational efficiencies ofthe RNAsweredeterminedbymonitoringthe productionofluciferaseprotein. Luciferase light unitsper 4x 105cellsareshown. Thisexperimentwasrepeatedthree times.Dependingonthedensityof the cellsinthedish,thetransfection efficienciesvaried somewhat. However, thelinearity ofRNAuptakeremainedsimilar.
observedthat verylittle viralproteinis needed for viral RNA synthesisto occur(4, 10, 41). Thedelayed inhibition of host cell translation inmutant-infected cellsis mostlikely alsoa resultof the low levels ofprotein 2A, known to be involved in theshut-off of cellular translation (4, 16), early in mutant-infected cells.
The1-5NC-S21mutation is containedwithin theloopofa stableRNAhairpin structure located ninenucleotides from the 5' end of the viral genome (18). The phenotype of 1-5NC-S21 indicates that this RNA hairpin modulates the translationalefficiency ofthe viral genome, atleastat early times after viral infection. This is somewhat surprising, because sequences sufficient for viral RNA translation by internal ribosomebindinghave beenmappedbetween posi-tions 200 and 600 (5, 22, 24). Ofcourse,we cannotruleout theunlikely possibilitythatboth the S21 and SS21 mutations resulted in again of function phenotype. In any case, the RNA hairpin could be especially important during viral translation immediately after infection, before host cell translation has been inhibited. Certain RNA-RNA or RNA-protein interactions mayplay arole in modulating efficient translationof the viral genomeby internal ribosomebinding, particularlyat atime when competitionwith cap-dependent hostmRNAtranslation is severe.
Itisnoteworthy that 1-5NC-S21 is sensitiveto dactinomy-cin,adrugthat inhibitsDNA-dependentRNApolymerases. Interestingly, mostpoliovirusmutantsbearing mutations in the5NCregionaredactinomycin sensitive(28,41). Because dactinomycinbinds witha1,000-foldloweraffinityto
single-or double-stranded RNA than to DNA (12, 37), it seems likely thatalabile cellularfactoris neededforthegrowthof 1-5NC-S21 and certain other mutantsbearing lesions in the 5NCregion (28, 41) butnotfor growth ofwild-type poliovi-rus.
The presenceof the RNAhairpin in thewild-typegenome has been confirmed by nucleolyticprobing(18). Computer-assistedpredictions ofthewild-type, S21, and SS21 hairpin structuressuggest thatS21 and SS21containlongerstemand smallerloopstructures than does the wild type with calcu-lated freeenergies (7)of-19.9, -22.1, and -34.7 kcal/mol forwild-type, S21, and SS21hairpins,
respectively
(Fig.
9). If the mutated RNA moleculesactuallydisplaythepredicted 5'-terminal structures in infected cells, potential RNA-pro-tein or RNA-RNA interactions could be perturbed. How-ever,thesestructures werepredicted by experimentalvalues calculatedundernonphysiological
conditions (7), and addi-tional experimental proof is clearly needed to verify the existence of the predicted mutated hairpins in mammalian cells.RNAhairpinstructureshavebeenimplicatedinthe trans-lational control in several procaryotic and
eucaryotic
sys-tems. Forexample, Tang and Draper showedthat an RNA hairpin is responsible for translationalrepression
of the a operon ofE. coliby one of its encodedproteins,
S4(39).
Bacteriophage R17 regulates the translationof its
replicase
genebybindingofthephagecoatproteinto anRNA
hairpin
thatincludes thereplicaseAUGstartcodon
(8).
The work of Kozak (15) and Pelletier and Sonenberg (23) demonstratedon November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.84.528.80.359.2]0G'V
C ^> GC Go C U C U-A G-C G-C G-C G-C U-A C-G U-AG-C-A C G-C-A C C A 6
[image:8.612.71.302.80.239.2]88 21
FIG. 9. Predicted RNA hairpinstructuresinwild-type (WT) and
mutant (S21, SS21) poliovirus genomes. The free energies of the
structurescalculated frompublished tables (7)were -19.9, -22.1,
and -34.7 kcal/mol for the wildtype,S21, andSS21, respectively.
that the introduction of RNA hairpins into 5NC regions of cellular mRNAscan inhibit translation in mammalian cells. Subsequently, it was reported that an RNA hairpin in the 5NC region offerritin mRNAbindsacytosolic protein in the absence of iron; binding of this proteinrepressestranslation of the ferritin mRNA (13, 30, 44). RNAhairpins have also been implicated in translational regulation by ribosomal frameshifting (6).
The 5'-proximal RNA hairpin mutated in this study is highly conserved between enteroviruses and rhinoviruses (29). Both the conservation and the phenotypes ofmutants seem to suggestafunctionalsignificance of this structurein the viral life cycle. However, the mechanism by which the RNA hairpin regulates poliovirus translation remains to be elucidated. Additional mutagenesis of the hairpin is required
to reveal the structural andprimary sequencerequirements
involved in translational regulation. Determination of how this 5'-proximal hairpin interacts with downstream se-quencesknown to conferinternal ribosomebinding and the identification of proteins that bindtothe5'-proximal hairpin will help elucidate the apparently complicated interaction betweenpoliovirus translation and the inhibition of host cell translation, both early and late in viralinfection.
ACKNOWLEDGMENTS
We thank Karla Kirkegaard for many helpful discussions and critical reading of the manuscript. We appreciate themany invalu-able suggestionsby Simon Hambidge and Dennis Macejak through-outthecourseof this work.
This study was supported by Public Health Service grants Al-30345, AI-25105, and AG-07347 from the National Institutes of Health.
REFERENCES
1. Agol, V.I.1990. Current approachestotheproblem of
poliovi-rusattenuation, p. 311-318. In M. A.Brinton and F. X. Heinz
(ed.), Newaspects ofpositive-strand RNA viruses. American Society forMicrobiology, Washington, D.C.
2. Baltimore, D. 1969. Thereplication of picornaviruses, p. 101-176. In H. Levy (ed.), The biochemistry of picornaviruses. MarcelDekker, Inc., New York.
3. Bernstein, H. D., P. Sarnow, and D. Baltimore. 1986. Genetic complementation among poliovirus mutants derived from an
infectious cDNAclone. J. Virol. 60:1040-1049.
4. Bernstein,H.D., N.Sonenberg,and D. Baltimore.1985. Polio-virus mutant thatdoes not selectively inhibit host cellprotein synthesis. Mol. Cell. Biol. 5:2913-2923.
5. Bienkowska-Szewczyk, K., and E. Ehrenfeld. 1988.Aninternal 5'-noncodingsequencein poliovirus RNA that mediates
trans-lational initiation.J. Virol. 62:3068-3072.
6. Brierley, I., P.Digard, and S. C. Inglis. 1989. Characterization ofan efficient coronavirus ribosomalframeshifting signal:
re-quirementforanRNApseudoknot. Cell57:537-547.
7. Cantor,C.R., and P. R. Schimmel. 1980. Statisticalmechanics andkinetics of nucleic acidinteractions,p. 1183-1264.InC.R. Cantorand P. R. Schimmel(ed.),Biophysicalchemistry, part III: thebehavior ofbiologicalmacromolecules. W. H. Freeman andCo., San Francisco.
8. Carey,J., V.Cameron,P. L.deHaseth,and0. C. Uhlenbeck. 1983.Sequence-specific interaction ofR17coatprotein with its ribonucleic acidbinding site. Biochemistry22:2601-2610. 9. DeWet, J. R., K. V.Wood,M.DeLuca,D. R.Helinski,andS.
Subramani. 1987. Fireflyluciferasegene:structureand expres-sion in mammaliancells. Mol. Cell. Biol. 7:725-737.
10. Dildine, S. L., and B. L. Semler. 1989. The deletion of41 proximal nucleotides reverts apoliovirusmutantcontaininga
temperature-sensitive lesion in the 5' noncoding regionof ge-nomicRNA. J.Virol. 63:847-862.
11. Etchison, D., S. C. Milburn, I. Edery, N. Sonenberg, and J. W. B.Hershey. 1982.Inhibition ofHeLacellprotein synthe-sisfollowing poliovirus infection correlates withtheproteolysis of a 220,000-dalton polypeptide associated with eukaryotic initiation factor 3 and cap binding protein complex. J. Biol. Chem.257:14806-14810.
12. Gellert, M., C. E. Smith, D. Neville,and G. Felsenfeld. 1965. Actinomycin binding to DNA: mechanism and specificity. J. Mol.Biol. 11:445-457.
12a.Hambidge, S.,and P.Sarnow.Submittedforpublication.
13. Hentze, M. W., T. A. Rouault, S. W. Caughman, A. Dancis, J.B. Harford, and R. D.Klausner. 1987. Acis-actingelement necessary and sufficient for translational regulation ofhuman ferritinexpression inresponse toiron. Proc. Natl. Acad. Sci. USA84:6730-6734.
14. Kitamura, N., B. L.Semler,P.G.Rothberg,G. R.Larsen,C.J. Adler, A.J. Dorner, E. A.Emini,R.Hanecak, J. J.Lee,S.van der Werf, C. W. Anderson, and E. Wimmer. 1981. Primary
structure, gene organization and polypeptide expression of poliovirusRNA. Nature(London)291:547-553.
15. Kozak, M. 1986. Influences ofmRNA secondary structure on
initiationbyeukaryoticribosomes. Proc. Natl.Acad.Sci. USA 83:2850-2854.
16. Krausslich,H.G.,M.J.H.Nicklin,H.Toyoda,D.Etchison,and E. Wimmer.1987.Poliovirusproteinase2Ainducescleavageof eucaryotic initiation factor 4F polypeptide p220. J. Virol. 61: 2711-2718.
17. Kuge,S.,andA. Nomoto. 1987.Construction of viable deletion andinsertion mutants of the Sabin strain oftype 1 poliovirus: function ofthe 5' noncoding sequence in viral replication. J. Virol. 61:1478-1487.
18. Larsen, G.,B. L.Semler,and E. Wimmer.1981. Stablehairpin
structure within the 5'-terminal 85 nucleotides of poliovirus
RNA. J. Virol. 37:328-335.
19. Macejak,D.G.,S.J.Hambidge,L.Najita,and P. Sarnow.1990. EIF-4F-independent translation ofpoliovirusRNAand cellular mRNA encoding glucose-regulated protein78/immunoglobulin heavy-chain bindingprotein,p. 152-157. In M. A. Brintonand F. X.Heinz(ed.),Newaspectsofpositive-strandRNAviruses. AmericanSociety forMicrobiology, Washington, D.C. 20. Mizutani,S., andR. J. Colonno. 1985. In vitro synthesisofan
infectious RNA from cDNA clones ofhuman rhinovirus type 14. J. Virol. 15:8783-8798.
21. Najita,L., and P. Sarnow. 1990. Oxidation-reduction sensitive interactionofacellular 50-kDaproteinwithanRNAhairpinin the 5' noncoding regionofthe poliovirus genome. Proc. Natl. Acad. Sci. USA87:5846-5850.
22. Pelletier, J., G. Kaplan, V. R. Racaniello, and N. Sonenberg.
1988. Cap-independent translation ofpoliovirusmRNA is
con-A U C G C U C U-A G-C a-C G-C G-C U-A C-G U-A G-C'
A C A C C A 6
0 0 G
C
,co
GC U C U-A G-C G-C G-C G-C U-A C-G U-A G-C'A C A C C A
WT 821
on November 10, 2019 by guest
http://jvi.asm.org/
ferred by sequence elements within the 5' noncoding region. Mol. Cell. Biol. 8:1103-1112.
23. Pelletier, J., and N. Sonenberg. 1985. Insertion mutagenesis to increasesecondary structure within the 5'noncodingregion of a eukaryoticmRNAreducestranslational efficiency. Cell 40:515-526.
24. Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryoticmRNAdirected by a sequence derived frompoliovirus RNA. Nature(London)334:320-325.
25. Pilipenko,E. V., V. M. Blinov, L. I. Ramanova, A. N.Sinyakov, S. V. Maslova, and V. I. Agol. 1989. Conserved structural domains inthe5'-untranslated region of picornaviralgenomes: ananalysis of thesegmentcontrollingtranslation and neurovir-ulence. Virology 168:201-209.
26. Racaniello,V R.,and D. Baltimore. 1981. Molecularcloning of polioviruscDNAand determination of the complete nucleotide sequence of the viral genome. Proc. Natl. Acad. Sci. USA 78:4887-4891.
27. Racaniello, V. R., and D. Baltimore. 1981. Cloned poliovirus complementaryDNAis infectious in mammalian cells. Science 214:916-919.
28. Racanielo,V. R.,and C. Meriam. 1986. Poliovirus temperature-sensitive mutantcontainingasinglenucleotidedeletion inthe 5'-noncodingregion of theviralRNA.Virology 155:498-507. 29. Rivera, V. M., J. D. Welsh, and J. V. Maizel, Jr. 1988.
Comparativesequenceanalysis ofthe 5'noncodingregionofthe enteroviruses and rhinoviruses. Virology 165:42-50.
30. Rouault, T. A., M. W. Hentze,S.W.Caughman, J. B. Harford, and R. D. Klausner. 1988. Bindingofacytosolicproteintothe iron-responsive element of human ferritin messenger RNA. Science 241:1207-1210.
31. Rueckert, R. R. 1990. Picornaviridae and their replication, p. 507-548. In B. N. Fields and D. M. Knipe (ed.), Virology. RavenPress, NewYork.
32. Sarnow, P. 1989. Translation of the glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is in-creased inpoliovirus-infected cells at a time when cap-depen-dent translation of cellular mRNAs is inhibited. Proc. Natl. Acad. Sci. USA 86:5795-5799.
33. Sarnow, P. 1989. Role of 3'-end sequences in infectivity of
poliovirus transcripts made in vitro. J. Virol. 63:467-470. 34. Sarnow, P., H. D. Bernstein, and D. Baltimore. 1986. A
polio-virustemperature-sensitiveRNAsynthesismutantlocated ina noncoding region ofthegenome. Proc. Natl. Acad. Sci. USA 83:571-575.
35. Sarnow, P., S. J. Jacobson, and L. Najita. 1990. Poliovirus genetics.Curr. Top. Microbiol. Immunol. 161:155-188. 36. Skinner, M. A., V. R. Racanlieio, G. Dunn,J. Cooper, P. D.
Minor, and J. W. Almond. 1989.Newmodelforthesecondary structure ofthe 5' non-codingRNAofpoliovirus issupported by biochemical and genetic data that also show that RNA secondary structure is important in neurovirulence. J. Mol. Biol. 207:379-392.
37. Sobell, H.1973. Thestereochemistry ofactinomycin bindingto DNA and itsimplications in molecular biology. Prog. Nucleic AcidsRes. 13:153-190.
38. Sonenberg, N. 1987. Regulation of translation by poliovirus. Adv. VirusRes.33:175-204.
39. Tang, C. K., and D. E. Draper. 1989. Unusual mRNA pseudoknot structure is recognized by a protein translational repressor. Cell57:531-536.
40. Toyoda, H., M. Kohara, Y. Kataoka, T.Suganuma,T. Omata, N. Imura, and A. Nomoto.1984.Completenucleotidesequences of all threepoliovirusserotype genomes:implication forgenetic relationshipgenefunction andantigenic determinants. J. Mol. Biol. 174:561-585.
41. Trono, D., R. Andino, and D. Baltimore. 1988. An RNA se-quence of hundreds of nucleotides atthe5' end ofpoliovirus RNA is involved in allowing viral protein synthesis. J. Virol. 62:2291-2299.
42. Trono, D., J. Pelletier, N. Sonenberg, andD. Baltimore. 1988. Translationinmammalian cellsofa genelinkedtothepoliovirus 5'noncodingregion. Science241:445-448.
43. van der Werf, S., J. Bradley, E. Wimmer, F. W. Studier, and J. J. Dunn. 1986. Synthesis of infectious poliovirus RNA by purified T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 83:2330-2334.
44. Walden, W. E., M. M. Patino, and L. Gaffield.1989.Purification ofaspecificrepressorof ferritinmRNAtranslationfrom rabbit liver. J.Biol. Chem. 264:13765-13769.