Complementation of the Function of Glycoprotein H of Human
Herpesvirus 6 Variant A by Glycoprotein H of Variant B in the Virus
Life Cycle
Hiroko Oyaizu,a,b,cHuamin Tang,a,bMegumi Ota,a,bNobuyuki Takenaka,aKeiichi Ozono,cKoichi Yamanishi,dand Yasuko Moria,b
Division of Clinical Virology, Kobe University Graduate School of medicine, Kobe, Japanb
; Laboratory of Virology and Vaccinology, Division of Biomedical Research, National Institute of Biomedical Innovation, Ibaraki, Osaka, Japana
; Department of Pediatrics, Osaka University Graduate School of Medicine, Suita, Osaka, Japanc ; and National Institute of Biomedical Innovation, Ibaraki, Osaka, Japand
Human herpesvirus 6 (HHV-6) is a T-cell-tropic betaherpesvirus. HHV-6 can be classified into two variants, HHV-6 variant A
(HHV-6A) and HHV-6B, based on genetic, antigenic, and cell tropisms, although the homology of their entire genomic
se-quences is nearly 90%. The HHV-6A glycoprotein complex gH/gL/gQ1/gQ2 is a viral ligand that binds to the cellular receptor
human CD46. Because gH has 94.3% amino acid identity between the variants, here we examined whether gH from one variant
could complement its loss in the other. Recently, we successfully reconstituted HHV-6A from its cloned genome in a bacterial
artificial chromosome (BAC) (rHHV-6ABAC). Using this system, we constructed HHV-6ABAC DNA containing the HHV-6B gH
(BgH) gene instead of the HHV-6A gH (AgH) gene in
Escherichia coli
. Recombinant HHV-6ABAC expressing BgH
(rHHV-6ABAC-BgH) was successfully reconstituted. In addition, a monoclonal antibody that blocks HHV-6B but not HHV-6A
infec-tion neutralized rHHV-6ABAC-BgH but not rHHV-6ABAC. These results indicate that HHV-6B gH can complement the
func-tion of HHV-6A gH in the viral infectious cycle.
H
uman herpesvirus 6 (HHV-6) is a T-cell-tropic
betaherpesvi-rus (
23
), which was first isolated from the peripheral blood
lymphocytes of patients with lymphoproliferative disorders and
AIDS (
24
). HHV-6 isolates can be categorized into HHV-6 variant
A (HHV-6A) and HHV-6B based on their genetic, antigenic, and
cell tropism properties (
6
,
8
,
24
,
33
). HHV-6B causes exanthem
subitum in primary infections (
34
), and HHV-6A was
predomi-nantly detected in samples from pediatric gliomas (
10
). HHV-6
infects most infants older than 6 months of age and can establish
lifelong latency; more than 90% of the general population is
sero-positive (
22
). HHV-6A was detected exclusively in 86% of the
samples from asymptomatic HHV-6-positive patients in an
Afri-can population, showing that HHV-6A is the predominant
vari-ant significvari-antly associated with viremic-infvari-ant infections in the
African population, distinct from other global cohorts (
7
).
HHV-6A can infect a broader variety of human cells than
HHV-6B (
18
), although the homology between the variants is
nearly 90% over their entire genomes (
12
–
14
). Which HHV-6
genes determine the different tropisms of the variants remain
un-known.
Herpesviruses encode several conserved envelope
glycopro-teins, one of which is glycoprotein H (gH), which is critical for
viral entry, possibly functioning in the fusion process as a complex
formed with glycoprotein L (gL) (
5
,
17
). Recently, the crystal
structure of the herpes simplex virus 2 (HSV-2) gH ectodomain
bound to gL was determined (
9
). Interestingly, gH/gL forms an
unusually tight complex with a unique architecture that does not
resemble any known viral fusogen.
Neutralizing antibodies to HHV-6 gH, established by our
lab-oratory and other laboratories (
16
,
21
,
27
), have shown that gH of
HHV-6 also plays an important role in viral entry. Anderson and
Gompels showed previously that neutralizing antifusion
antibod-ies could recognize the HHV-6 gH/gL complex in the absence of
gQ1 and gQ2 (
2
).
Unlike in HSV, in HHV-6, the gH/gL heterodimer requires
additional associated glycoproteins, gQ1 and gQ2 (
1
), for its
traf-ficking and receptor-binding functions (
30
). Interestingly,
how-ever, even though HHV-6B gH/gL also associates with a gQ1/gQ2
complex (
15
), the gH/gL/gQ1/gQ2 complex of HHV-6A binds to
its cellular receptor, human CD46, but the corresponding
com-plex in some HHV-6B strains does not (
1
,
18
). Furthermore, our
previous study showed that HHV-6A can mediate cell-cell fusion
in a variety of cells expressing human CD46 but that HHV-6B
cannot (
20
).
Since, as described above, herpesvirus-encoded gHs have
fu-sion activity, we speculated that this gH activity might differ
be-tween the variants, which could explain their different tropisms.
Given that the amino acid identity of HHV-6A gH (AgH) and
HHV-6B gH (BgH) is 94.3% (
12
–
14
), and in hopes of revealing
any difference in gH functions between the variants, we decided to
test whether gH from one variant could function to complement
the loss of endogenous gH from the other. It was shown previously
that human cytomegalovirus (HCMV) gH could substitute for
HHV-6 gH and could participate in heterologous complex
forma-tion (
3
).
Recently, we successfully reconstituted HHV-6A from its
ge-nome using the BAC (bacterial artificial chromosome) system
(
31
). Here we used the BAC system to examine whether
recombi-nant HHV-6A expressing gH of HHV-6B (BgH), but not
HHV-6A gH (AgH), would show a different or compromised
in-Received27 February 2012 Accepted16 May 2012
Published ahead of print30 May 2012
Address correspondence to Yasuko Mori, [email protected]. Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00504-12
on November 7, 2019 by guest
http://jvi.asm.org/
fectious behavior. We reconstituted infectious virus from the
HHV-6A genome encoding BgH instead of AgH and showed that
BgH can functionally replace AgH in the HHV-6A life cycle,
al-though the viral growth ability seems to be slightly decreased by
the replacement.
MATERIALS AND METHODS
Cells and viruses.The human T-lymphoblastoid cell lines JJhan and MT4 were cultured in RPMI 1640 medium supplemented with 8% fetal bovine serum (FBS). Umbilical cord blood mononuclear cells (CBMCs) were separated on a Ficoll-Conray gradient and cultured in RPMI medium containing 10% fetal bovine serum, phytohemagglutinin (5g/ml), and interleukin-2 (IL-2) (2 ng/ml) (29). HHV-6A strain U1102 or HHV-6B strain HST was propagated in stimulated CBMCs (1).
Antibodies.The polyclonal antibody for gB was described previously (19). The monoclonal antibodies (MAbs) for HHV-6A gH (gH1-1) [30], OHV-3 [21[, and U14 [28[), gQ1 (AgQ-119) (1), gQ2 (AgQ2 4-2 and AgQ2B) (15,30), and gL (AgL3) (1) were described previously.
We also raised a MAb to gH (J1) that reacts with both HHV-6A and -6B gHs, as described previously (30).
Immunoprecipitation (IP) assay and Western blotting.Antibodies were bound to protein G-Sepharose (GE Healthcare) by incubation at 4°C for 8 h. Whole-cell extracts were then immunoprecipitated with the ap-propriate protein G-Sepharose-bound antibody by incubation at 4°C for 8 h. The bound proteins were eluted with 0.1 M glycine (pH 2.8) at 4°C, collected, and neutralized with 1 M Tris-HCl (pH 9.0) to pH 7.0 to 7.4 (30). The samples were then subjected to SDS-PAGE. For Western blot analyses, the antibodies were cross-linked with protein G-Sepharose using dimethyl pimelimidate (DMP; Thermo Scientific) according to the man-ufacturer’s instructions. Western blotting was performed as described previously (1).
Immunofluorescence assay. Indirect immunofluorescence assays (IFAs) were performed as described previously (15).
DNA analysis.BAC DNAs isolated fromEscherichia coliwere digested with BamHI and separated on a 0.5% agarose gel. The DNA fragments in the gel were transferred onto a Hybond-N⫹ nylon membrane (GE Healthcare). The probe was amplified from U1102 DNA with primers
AgHF1 and BgHR3 (all primer sequences are available upon request) by PCR. Southern blot hybridization was then performed, according to the manufacturer’s instructions (GE Healthcare).
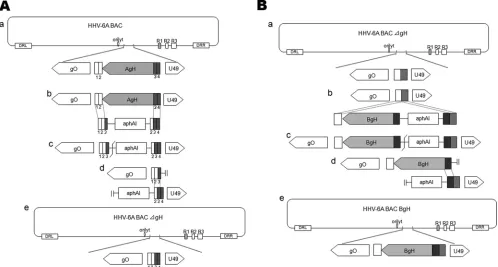
Construction of the gH-deleted and -replaced mutants in the HHV-6A BAC.The construction of the HHV-6A BAC mutants was per-formed as described previously (31,32). Schematics of the construction strategy are shown inFig. 2. The HHV-6A BAC (HHV-6ABAC) DNA fromE. coliDH10B was transformed intoE. coliGS1783 cells by using a Bio-RadE. coliPulser. We deleted the gH gene, which corresponds to bp 78034 to 80118 in the U1102 genome (GenBank accession number NC_001664) (13). Briefly, GS1783 cells containing HHV-6ABAC were cultured in LB medium containing 17g/ml chloramphenicol at 30°C overnight. The culture grown overnight was then added to warm, fresh LB medium containing 17g/ml chloramphenicol at a 1:30 ratio. The result-ing culture was incubated at 42°C for 15 min to induce Red recombina-tion. Next, the bacteria were chilled in ice water for 20 min and spun down. The pellet was washed twice with ice-cold 10% glycerol. After the last wash, the bacterial pellet was resuspended in 10% glycerol and stored at⫺80°C.
Next, the first Red recombination was performed. One hundred nano-grams of the PCR products amplified from plasmid pEP-KanS using primers AgH deletion F and AgH deletion R was transformed into pre-pared competent GS1783 cells, as described above, by electroporation. The bacteria were cultured at 30°C for 1.5 h and then plated onto LB agar plates containing 17g/ml chloramphenicol and 50g/ml kanamycin, to select forE. coliclones harboring the kanamycin resistance gene. After a 24-h incubation at 30°C, the selected clones were confirmed by PCR using the appropriate primers.
The second recombination was then performed to excise the kanamy-cin resistance gene. Briefly, 100l of a culture of GS1783 cells containing the kanamycin resistance gene grown overnight was added to 2 ml of warm medium containing 17g/ml chloramphenicol. The bacteria were grown for 2 to 4 h at 30°C, 2% arabinose was added, and the culture was incubated for another 30 to 60 min to induce the expression of the I-SceI restriction enzyme. After the incubation, the culture was transferred into a 42°C water bath for 15 to 30 min. The culture was then incubated at 30°C for 2 h before being transferred onto agar plates containing 17g/ml
FIG 1Amino acid sequence (single-letter amino acid code) alignment of HHV-6A and HHV-6B. The shaded residues are identical, and the two variants have 94.3% amino acid identity. U1102, HHV-6A; HST, HHV-6B.
on November 7, 2019 by guest
[image:2.585.76.510.70.300.2]chloramphenicol. Chloramphenicol-resistant but kanamycin-sensitive clones were selected by plating single clones onto chloramphenicol- and chloramphenicol-kanamycin-containing plates. We named the resultant BAC HHV-6ABAC⌬gH.
We next constructed a BgH-inserted mutant. The BgH sequence was amplified from HST (HHV-6B) DNA by using primers BgH F1 and BgH R1 and digested with EcoRI. We amplified the kanamycin resistance gene from pEP-KanS using primers BgH F2 and BgH R2-1 and performed a second PCR using primers BgH F2 and BgH R2-2. The resulting PCR products were digested with EcoRI to produce BgH PCR fragments. These two EcoRI-digested DNA fragments were ligated and amplified with primers BgH F1 and BgH R3, and 100 ng of the PCR product was trans-formed intoE. coliGS1783 electroporation-competent cells containing HHV-6ABAC⌬gH. The selected clones were confirmed by PCR. Next, the kanamycin resistance gene was excised by expressing the I-Sce1 restriction enzyme, followed by the induction of the Red recombination system (as described above). We named the resultant BAC HHV-6ABAC-BgH.
Viral growth assay.Recombinant-HHV-6-infected CBMCs were frozen, thawed, and centrifuged, and the supernatants were then col-lected and used as a virus stock. The titer of each virus was measured as the 50% tissue culture infectious dose (TCID50). For the measurement
of virus growth, 5⫻106CBMCs were used for incubation with each
virus at 37°C for 1 h at an MOI (multiplicity of infection) of approxi-mately 0.01. After incubation, the cells were washed and divided into 6 samples and cultured in 1 ml of culture medium (RPMI medium con-taining 10% FBS). The samples were harvested at 0 h, 8 h, 1 day, 3 days, 5 days, and 7 days postinfection (p.i.). To determine the number of
HHV-6 genome copies in each sample, the samples were treated with proteinase K (Sigma) at 55°C for 2 h. Total DNA in each sample was extracted by PCI (phenol-chloroform-isoamyl alcohol) (25:24:1), fol-lowed by ethanol precipitation. Viral genome copy numbers were de-termined by real-time PCR by using primer sequences 5=-TTTGCAGT CATCACGATCGG-3= and 5=-AGAGCGACAAATTGGAGGTTTC-3=. To measure the viral gene (gB) expression level in each sample, total RNA was extracted by using TRIzol (Invitrogen) according to the manufactur-er’s protocol. The samples were then treated with DNase I (Roche). The cDNA from each sample was used for real-time PCR using two pairs of primers, 5=-CACCAATCCGGTGACTACTG-3=and 5=-CTAGGTGTCT TGACGACAGTG-3=for the viral gene and 5=-GCACCCAGCACAATGA AGA-3= and 5=-CGATCCACACGGAGTACTTG-3= for the beta-actin gene. The viral gene expression level in each sample was normalized to the expression level of beta-actin. All real-time PCRs were conducted with a Light Cycler 480 II machine (Roche), and PCR was performed by using a SYBR green kit (Roche).
Virus neutralization assay.The virus neutralization assay was per-formed as described previously (15). Briefly, 100l of serial 10-fold dilu-tions of purified MAb OHV-3 (0.4 mg/ml) or U14 (0.4 mg/ml) was incu-bated with 100l of virus solution at approximately 1.5⫻102to 3⫻102
TCID50at 37°C for 30 min. CBMCs or JJhan cells were then added to the
virus-antibody solution and incubated at 37°C for 1 h. After the incuba-tion, the cells were washed and cultured for 4 to 7 days. The expression of green fluorescent protein (GFP) was examined by using a fluorescence-activated cell sorter (FACS).
FIG 2Schema of the strategy for the construction of HHV-6ABAC⌬gH and HHV-6ABAC-BgH. (A) Two sequential sequences of about 20 bp downstream of the deleted gH gene sequences are labeled 1 and 2; the corresponding upstream sequences are labeled 3 and 4. The kanamycin resistance gene (aphAI) was amplified by PCR using long primers (AgH deletion F and AgH deletion R) whose sequences match the upstream and downstream sequences of gH, labeled 3 and 4 (gray) and 1 and 2 (white), respectively. The recombination at the matching sequences (b) resulted in the deletion of gH and the insertion ofaphAI(c). The I-SecI site between the gH sequence andaphAIwas cleaved (c), and the second recombination occurred (d), in whichaphAI(e) was deleted. (B) The BgH gene was amplified from HST (HHV-6B) genomic DNA by using primers BgHF1 and BgHR1 and ligated withaphAI, which was amplified by using primers BgHF2 and BgHR2-2 and flanked with HHV-6B gH sequences (dark gray) and the upstream sequences of HHV-6A gH (gray). The resulting DNA fragment, flanked with gH upstream (gray) and downstream (white) sequences, recombined with the same sequences in the HHV-6ABAC⌬gH DNA (b), which resulted in the insertion of the DNA fragment (c). The I-SecI site between the BgH sequence andaphAIwas then cleaved (c), and the second recombination occurred (d), in whichaphAI was deleted (e). The resulting construct was named HHV-6ABAC-BgH.
Oyaizu et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.44.542.68.335.2]RESULTS
Construction of the HHV-6A genome bearing HHV-6B gH.
The
HHV-6A and HHV-6B gH genes (AgH and BgH) each consist of
694 amino acid (aa) residues, with an identity of 94.3% (
Fig. 1
).
We constructed an HHV-6A genome (HHV-6ABAC-BgH) in
which the full-length AgH gene was replaced by full-length BgH
by Red recombination in
E. coli
cells (
Fig. 2
) and confirmed the
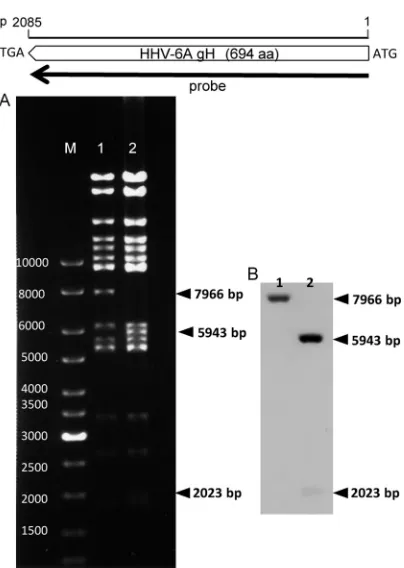
substitution by sequencing (data not shown) and Southern blot
analysis (
Fig. 3
). The resultant BAC DNA (HHV-6ABAC-BgH)
was analyzed by restriction enzyme digestion with BamHI and
compared with that of wild-type HHV-6ABAC.
As shown in
Fig. 3
A, the digestion of HHV-6ABAC-BgH (lane
2) with BamHI yielded a different pattern from that of
HHV-6ABAC (wild type) (lane 1), because there is one BamHI site in the
BgH gene but none in the AgH gene. As expected, the 7,966-bp
fragment was missing in HHV-6ABAC-BgH, and new fragments
of 5,943 bp and 2,023 bp were visible instead. The Southern blot
analysis (
Fig. 3
B) was performed by using a probe that encodes the
entire AgH sequence (
Fig. 3
, top). As expected, a positive band was
detected at 7,966 bp in HHV-6ABAC (
Fig. 3
B, lane 1), and two
positive bands at 5,943 bp and 2,023 bp were detected in
HHV-6ABAC-BgH (
Fig. 3
B, lane 2), confirming the recombination of
BgH. These data showed that we had successfully generated
HHV-6A BAC DNA in which AgH was replaced with BgH in
E.
coli
.
Reconstitution of infectious virus using HHV-6ABAC-BgH
DNA.
The reconstitution of infectious virus was performed as
described previously (
31
). Infectious viruses were reconstituted
from both HHV-6ABAC-BgH and HHV-6ABAC. We then
veri-fied the infectivity of the reconstituted viruses by performing
cell-to-cell infections. Cytopathic effects (CPEs) with green
fluores-cence were observed in cultures infected with either BAC (
Fig. 4
).
Next, to confirm viral gene expression, Western blot analysis was
performed by using anti-gB and anti-gH antibodies. As shown in
Fig. 5
, the proteins were detected in the recombinant
HHV-6A-BgH 6ABAC-HHV-6A-BgH) and recombinant HHV-6A
(rHHV-6ABAC) samples. These data show that BgH was able to
function-ally complement AgH in the HHV-6A infectious life cycle.
Growth curve of rHHV-6ABAC-BgH.
The growth curve of
each virus, rHHV-6ABAC, rHHV-6ABAC-BgH, or its revertant
virus, rHHV-6ABACrev, was determined by real-time PCR, and
the growth curves were compared. The viral genomes (
Fig. 6
A)
and gene expression levels (
Fig. 6
B) in infected cells were
quanti-fied at 0 h, 8 h, 1 day, 3 days, 5 days, and 7 days p.i. As shown in
Fig.
6
, the growth patterns of the viruses were similar; however, the
viral growth ability of rHHV-6ABAC-BgH seemed to be slightly
decreased by the replacement. Similar results were obtained when
JJhan cells were used (data not shown).
We also confirmed that the genome sequence of gL, gQ1, or
gQ2 in rHHV-6ABAC-BgH or rHHV-6ABACrev was not
changed from that of the wild type.
HHV-6B-specific neutralizing antibody blocks
rHHV-6ABAC-BgH infection.
Previously, our laboratory reported a
MAb, OHV-3, against BgH (
21
) that does not recognize AgH and
inhibits HHV-6B entry. Here we examined whether OHV-3
would inhibit rHHV-6ABAC-BgH infection and analyzed it by
fluorescence-activated cell sorter (FACS) analysis. As expected,
OHV-3 inhibited rHHV-6ABAC-BgH but not rHHV-6ABAC or
FIG 3Restriction enzyme digestion pattern of DNAs and Southern blotting. At the top, the scale bar indicates the full length of the HHV-6A gH gene, and the probe position used for Southern blotting is indicated by the black arrow. (A) Electrophoresis of digested DNAs. The 6ABAC (lane 1) and HHV-6ABAC-BgH (lane 2) DNAs were isolated fromE. coli, digested with BamHI, and separated on a 0.5% agarose gel. The bands were visualized with ethidium bromide. Specific fragments are indicated by arrowheads. Lane M, size mark-ers; lane 1, HHV-6ABAC; lane 2, HHV-6ABAC-BgH. (B) Southern blotting. The DNAs in the agarose gel were transferred onto a Hybond-N⫹nylon mem-brane and hybridized with the gH probe. Specific signals are indicated by arrowheads. Lane 1, HHV-6ABAC; lane 2, HHV-6ABAC-BgH.
FIG 4CPE and GFP expression in reconstituted virus-infected cells. Each BAC DNA isolated fromE. coliwas transfected into JJhan cells. The transfected cells were cocultured with CBMCs on the third day posttransfection. Light microscopic images are shown at the left, and GFP fluorescence images in the same microscopic field are shown at the right. (A) 6ABAC. (B) rHHV-6ABAC-BgH.
on November 7, 2019 by guest
[image:4.585.65.267.60.344.2] [image:4.585.299.543.64.257.2]rHHV-6ABACrev infection (
Fig. 7
). We also used OHV-3 in IPs
and Western blots to confirm BgH expression. gH was
immuno-precipitated by OHV-3 from rHHV-6ABAC-BgH- but not
rHHV-6ABAC-infected cell lysates (
Fig. 8
), indicating that BgH
was expressed in rHHV-6ABAC-infected cells.
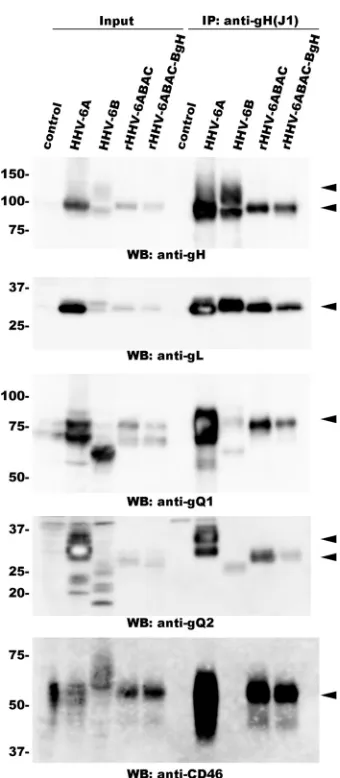
BgH forms a complex with gQ1, gQ2, and gL in
rHHV-6ABAC-BgH-infected cells.
Next, we examined whether BgH
as-sociates with gL, gQ1, and gQ2 in rHHV-6ABAC-BgH-infected
cells. HHV-6ABAC-BgH-reconstituted viruses were grown in
CBMCs, which were harvested and lysed. The lysates were
immu-noprecipitated with the anti-gH MAb, and the Western blot of the
immunoprecipitates was probed with antibodies against gQ1,
gQ2, and gL. As expected, gQ1, gQ2, and gL all coprecipitated with
BgH (
Fig. 9
), just as they did with AgH (
30
), showing that BgH
FIG 5Viral protein expression in cells infected with reconstituted virus. JJhan cells were transfected with HHV-6ABAC or HHV-6ABAC-BgH DNA and then cocultured with CBMCs to permit cell-to-cell infection. Cells were har-vested 4 days after the start of cell-to-cell infection. The samples were resolved by SDS-PAGE under reducing conditions, followed by Western blot analysis using the following antibodies: anti-gB antibody (top) and anti-gH1-1 MAb (bottom). Uninfected CBMCs or MT4 cells were used as negative controls. CBMCs infected with HHV-6A isolate U1102 and MT4 cells infected with 6B isolate HST were the positive controls. Lane 1, mock; lane 2, HHV-6A; lane 3, rHHV-6ABAC; lane 4, rHHV-6ABAC-BgH; lane 5, mock; lane 6, HHV-6B.
FIG 6Comparison of growth kinetics of 6ABAC, 6ABAC-BgH, and 6ABACrev. CBMCs were infected with 6ABAC, rHHV-6ABAC-BgH, or rHHV-6ABACrev. The cells were harvested at 0 h, 8 h, 1 day, 3 days, 5 days, or 7 days p.i. (A) To determine the number of genome copies in each sample, the viral genome copy number in each sample was quantified by real-time PCR. (B) To determine the viral gene (gB) expression level, cDNAs were constructed by using the total RNA from each sample. Viral gene expression levels were measured by real-time PCR and normalized to the expression level of beta-actin. Data represent one of three independent experiments. *,P⬍0.001 (Student’sttest).
FIG 7A BgH-specific neutralizing antibody inhibits rHHV-6ABAC-BgH but not rHHV-6ABACrev or wild-type infection. rHHV-6ABAC-BgH (BgH), rHHV-6ABACrev (Rev), or rHHV-6ABAC (wild type [WT]) was used as the virus solution. MAb OHV-3 (anti-gH) or U14 (control) was incubated with the virus solution at 37°C for 30 min. JJhan cells were then added to the virus-antibody solution and incubated at 37°C for 1 h. After the incubation, the cells were cultured for 4 days. The cells were harvested and fixed, and the expression of GFP in 104cells was analyzed by FACS analysis. Data represent
one of three independent experiments. Oyaizu et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.301.540.64.396.2] [image:5.585.79.251.66.222.2] [image:5.585.78.511.536.675.2]associates with gQ1, gQ2, and gL of 6A in the
HHV-6ABAC-BgH-reconstituted virus.
The BgH/gL/gQ1/gQ2 complex formed in
rHHV-6ABAC-BgH-infected cells binds to human CD46.
The HHV-6A gH/gL/
gQ1/gQ2 complex binds to human CD46 (
1
). Therefore, we
in-vestigated whether the BgH/gL/gQ1/gQ2 complex in
HHV-6ABAC-BgH-reconstituted virus binds to CD46. As described
previously, CD46 was coimmunoprecipitated with gH from
HHV-6-infected cell lysates (
25
). We confirmed these results by
using an anti-gH MAb (J1) that reacts with both AgH and BgH.
CD46 coimmunoprecipitated with gH from lysates of
HHV-6A-but not HHV-6B (strain HST)-infected cells (
Fig. 9
).
Next, we examined whether CD46 was coimmunoprecipitated
from the lysates of cells infected with the
HHV-6ABAC-BgH-re-constituted virus. As shown in
Fig. 9
, CD46 was
immunoprecipi-tated from these lysates, just as it was from the lysates of cells
infected with the HHV-6ABAC-reconstituted virus. Thus, the
BgH/gL/gQ1/gQ2 complex formed in cells infected with the
HHV-6ABAC-BgH-reconstituted virus bound to CD46, the
cel-lular receptor for HHV-6.
DISCUSSION
As HHV-6 variants have different characteristics, even though
their sequence homology is nearly 90%, we are searching for the
viral genes responsible for the differences. HHV-6 A and HHV-6 B
chemokines (U83) can chemoattract different cellular
popula-tions through interacpopula-tions with distinct chemokine receptors and
can play a role in cellular tropism (
11
). In the entry process,
al-though the identified cellular receptor of HHV-6 is human CD46
(
26
), several strains of HHV-6 seem not to use it (
18
). In addition,
the HHV-6A viral ligand is known to be a component of the gH/
gL/gQ1/gQ2 envelope glycoprotein complex (
1
).
Of the envelope glycoproteins, we have focused on gH, which is
highly conserved among herpesviruses and plays an important
role in virus entry, particularly in the fusion process, along with
another envelope glycoprotein, gB (
4
). Given that there is 94.3%
amino acid identity between AgH and BgH, here we investigated
whether BgH could replace AgH in recombinant, reconstituted
HHV-6A.
Here we successfully reconstituted recombinant HHV-6A
expressing BgH instead of AgH using the BAC system. The
recom-bination of gH was confirmed by DNA analysis, including
South-ern blotting, and the expression of BgH was confirmed by
IP-Western blotting using a BgH-specific MAb. The appearance of
CPEs and expression levels of the other viral proteins in
rHHV-6ABAC-BgH-infected cells were similar to those in rHHV-6ABAC
(wild-type)-infected cells. Next, virus growths were compared
among the wild type, rHHV-6ABAC-BgH, and its revertant,
rHHV-6ABACrev. Although the growth patterns appeared to be
similar among them, rHHV-6ABAC-BgH showed a decreased
growth ability compared to that of its revertant or the wild type.
Previously, our laboratory reported an anti-gH antibody,
OHV-3, that specifically neutralizes HHV-6B (
21
). Here we
exam-ined whether OHV-3 could prevent infection by
rHHV-6ABAC-BgH. Indeed, OHV-3 clearly prevented rHHV-6ABAC-BgH
in-fection but not rHHV-6ABAC inin-fection. Since OHV-3 recognizes
a conformational epitope of BgH (
27
), we conclude that the BgH
expressed in rHHV-6ABAC-BgH-infected cells maintained the
same steric conformation as that in HHV-6B-infected cells.
Previously, we found that HHV-6A could mediate fusion from
without (FFWO) in a variety of cells expressing human CD46 but
FIG 9AgQ1/AgQ2/BgH/AgL complex formation and its association with CD46. Lysates of HHV-6-infected cells (indicated at the top of the blot) or mock-infected cells (control) were immunoprecipitated with an anti-gH MAb (J1), and Western blots of the immunoprecipitates were probed with the indi-cated antibody. Arrowheads indicate specific bands.
reacts with both AgH and BgH.
on November 7, 2019 by guest
[image:6.585.337.507.61.450.2] [image:6.585.92.237.65.180.2]that HHV-6B could not (
20
). Although gH was thought to be a key
factor in FFWO, no significant difference in FFWO was seen
be-tween rHHV-6ABAC-BgH- and rHHV-6ABAC-infected cells.
Therefore, gH may require an interaction with gB, another fusion
protein, to mediate FFWO by HHV-6A. Further study will be
required to elucidate the FFWO mechanism.
Recently, we reported that HHV-6A gH/gL/gQ1/gQ2 complex
formation is required for HHV-6 trafficking and receptor binding
(
30
), indicating that the conformation of the complex itself is
important for its functions. Therefore, we examined whether BgH
as well as AgH could form a complex with gL, gQ1, and gQ2 in
rHHV-6ABAC-BgH-infected cells and, if so, whether the complex
could bind to human CD46. We found that BgH could associate
with gL, gQ1, and gQ2, forming a tetrameric complex, and that
the complex bound to human CD46.
This finding indicates that BgH can replace AgH in the folding
of the tetrameric complex and its functions in the viral life cycle,
although it slightly affects the growth of HHV-6A.
ACKNOWLEDGMENTS
We thank Gregory A. Smith (Department of Microbiology-Immunology, Northwestern University, Chicago, IL) for providing E. coliGS1783, Nikolaus Osterrieder (Department of Microbiology and Immunology, College of Veterinary Medicine, Cornell University, Ithaca, NY) for plas-mid pEP-KanS, and Ulrich H. Koszinowski (Max von Pettenkofer Insti-tute, Ludwig Maximilian University, Munich, Germany) for plasmid pHA-2. We thank Kazushige Adachi (Minoh City Hospital) and Hideto Yamada (Department of Obstetrics and Gynecology, Kobe University Graduate School of Medicine) for providing the CBMCs and Pranee Som-boonthum (National Institute of Biomedical Innovation) and Akiko Kawabata (Kobe University) for their assistance.
This study was supported in part by a grant-in-aid for scientific re-search on priority areas from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan and by a grant-in-aid for sci-entific research (B) from the Japan Society for the Promotion of Science (JSPS).
REFERENCES
1.Akkapaiboon P, Mori Y, Sadaoka T, Yonemoto S, Yamanishi K.2004. Intracellular processing of human herpesvirus 6 glycoproteins Q1 and Q2 into tetrameric complexes expressed on the viral envelope. J. Virol.78: 7969 –7983.
2.Anderson RA, Gompels UA.1999. N- and C-terminal external domains of human herpesvirus-6 glycoprotein H affect a fusion-associated confor-mation mediated by glycoprotein L binding the N terminus. J. Gen. Virol.
80(Pt 6):1485–1494.
3.Anderson RA, Liu DX, Gompels UA. 1996. Definition of a human herpesvirus-6 betaherpesvirus-specific domain in glycoprotein gH that governs interaction with glycoprotein gL: substitution of human cyto-megalovirus glycoproteins permits group-specific complex formation. Vi-rology217:517–526.
4.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ.2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol.84:12292–12299.
5.Atanasiu D, et al.2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a nec-essary step leading to cell fusion. J. Virol.84:3825–3834.
6.Aubin JT, et al.1991. Several groups among human herpesvirus 6 strains can be distinguished by Southern blotting and polymerase chain reaction. J. Clin. Microbiol.29:367–372.
7.Bates M, et al.2009. Predominant human herpesvirus 6 variant A infant infections in an HIV-1 endemic region of Sub-Saharan Africa. J. Med. Virol.81:779 –789.
8.Campadelli-Fiume G, Guerrini S, Liu X, Foa-Tomasi L.1993.
Mono-clonal antibodies to glycoprotein B differentiate human herpesvirus 6 into two clusters, variants A and B. J. Gen. Virol.74(Pt 10):2257–2262. 9.Chowdary TK, et al.2010. Crystal structure of the conserved herpesvirus
fusion regulator complex gH-gL. Nat. Struct. Mol. Biol.17:882– 888. 10. Crawford JR, et al.2009. Detection of human herpesvirus-6 variants in
pediatric brain tumors: association of viral antigen in low grade gliomas. J. Clin. Virol.46:37– 42.
11. Dewin DR, Catusse J, Gompels UA.2006. Identification and character-ization of U83A viral chemokine, a broad and potent beta-chemokine agonist for human CCRs with unique selectivity and inhibition by spliced isoform. J. Immunol.176:544 –556.
12. Dominguez G, et al.1999. Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J. Virol.
73:8040 – 8052.
13. Gompels UA, et al.1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology209:29 –51. 14. Isegawa Y, et al.1999. Comparison of the complete DNA sequences of
human herpesvirus 6 variants A and B. J. Virol.73:8053– 8063. 15. Kawabata A, et al.2011. Analysis of a neutralizing antibody for human
herpesvirus 6B reveals a role for glycoprotein Q1 in viral entry. J. Virol.
85:12962–12971.
16. Liu DX, Gompels UA, Foa-Tomasi L, Campadelli-Fiume G. 1993. Human herpesvirus-6 glycoprotein H and L homologs are components of the gp100 complex and the gH external domain is the target for neutral-izing monoclonal antibodies. Virology197:12–22.
17. Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS.2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A.107:22641–22646. 18. Mori Y.2009. Recent topics related to human herpesvirus 6 cell tropism.
Cell. Microbiol.11:1001–1006.
19. Mori Y, et al.2008. Human herpesvirus-6 induces MVB formation, and virus egress occurs by an exosomal release pathway. Traffic9:1728 –1742. 20. Mori Y, et al.2002. Human herpesvirus 6 variant A but not variant B induces fusion from without in a variety of human cells through a human herpesvirus 6 entry receptor, CD46. J. Virol.76:6750 – 6761.
21. Okuno T, et al.1990. Analysis of a glycoprotein of human herpesvirus 6 (HHV-6) using monoclonal antibodies. Virology176:625– 628. 22. Okuno T, et al.1989. Seroepidemiology of human herpesvirus 6 infection
in normal children and adults. J. Clin. Microbiol.27:651– 653. 23. Roizmann B, et al.1992. The family Herpesviridae: an update. The
Her-pesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch. Virol.123:425– 449.
24. Salahuddin SZ, et al.1986. Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science234:596 – 601.
25. Santoro F, et al.2003. Interaction of glycoprotein H of human herpesvi-rus 6 with the cellular receptor CD46. J. Biol. Chem.278:25964 –25969. 26. Santoro F, et al.1999. CD46 is a cellular receptor for human herpesvirus
6. Cell99:817– 827.
27. Takeda K, et al.1997. Identification of a variant B-specific neutralizing epitope on glycoprotein H of human herpesvirus-6. J. Gen. Virol.78(Pt 9):2171–2178.
28. Takemoto M, et al.2005. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J. Virol.79:13037–13046.
29. Takemoto M, Shimamoto T, Isegawa Y, Yamanishi K.2001. The R3 region, one of three major repetitive regions of human herpesvirus 6, is a strong enhancer of immediate-early gene U95. J. Virol.75:10149 –10160. 30. Tang H, Hayashi M, Maeki T, Yamanishi K, Mori Y.2011. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor bind-ing. J. Virol.85:11121–11130.
31. Tang H, et al.2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology407:360 –367.
32. Tischer BK, von Einem J, Kaufer B, Osterrieder N.2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques40:191–197.
33. Wyatt LS, Balachandran N, Frenkel N.1990. Variations in the replica-tion and antigenic properties of human herpesvirus 6 strains. J. Infect. Dis.
162:852– 857.
34. Yamanishi K, et al.1988. Identification of human herpesvirus-6 as a causal agent for exanthem subitum. Lanceti:1065–1067.
Oyaizu et al.
on November 7, 2019 by guest
http://jvi.asm.org/