Copyright ©1993, American SocietyforMicrobiology
Wild-Type p53
Is
Not a
Negative Regulator
of Simian
Virus
40 DNA
Replication
in Infected
Monkey
Cells
ANDREASVON DERWETHAND WOLFGANG DEPPERT*
Heinrich-Pette-Institut fair Experimentelle VirologieundImmunologie,
Martinistrasse52, D-2000 Hamburg 20, Germany Received25June1992/Accepted 10November 1992
Toanalyze theproposed growth-inhibitoryfunction ofwild-typep53,wecomparedsimianvirus 40(SV40)
DNA replication in primary rhesus monkey kidney (PRK) cells, which express wild-type p53, and in the established rhesus monkey kidneycell lineLLC-MK2,which expressesa mutatedp53 thatdoesnotcomplex with large T antigen. SV40 DNA replication proceeded
identically
in both cell types during the course of infection.Endogenously
expressedwild-type
p53thus does notnegatively
modulateSV40DNAreplicationin vivo. We suggestthat inhibition ofSV40DNAreplication by wild-type p53in invitroreplicationassaysisdue togrosslyelevated ratios ofp53tolargeTantigen,thusdepletingthereplication-competentfreelargeTantigen inthe assay mixturesbycomplex formation.Incontrast,the ratio ofp53tolargeTantigenin in vivoreplication islow, leaving themajority
oflargeTantigenin afree, replication-competent state.Mutations in thetumorsuppressor genep53constitute the most common genetic alterations known in human cancer (18, 21, 29). Recent evidence suggests that p53 acts as a tumor suppressor and that mutations in p53 abolish this repressor activity (27, 30). Very little is understood about
p53 functions atthe molecular level. p53 is involved in cell cycleregulation (38),but seemingly contradictoryevidence has beenprovided forboth apositive anda negative regu-latory role forp53 inthese processes (7).Whereas there is compelling evidence for the need for p53 expression for
progression of normal and even some tumor cells through
thecell cycle (8, 33, 34, 45, 56), it has been unequivocally
demonstrated thatexpressionoftransfectedwild-typep53in tumorandtransformed cells leadstocessationof cellgrowth (36). Thisgrowth-inhibitory function ofwild-type p53 now
commonly is seen as the basis of its tumor suppressor activity (20,30). Althoughthis isanattractivehypothesis, it has to be reconciled with the set of complementary data
describing agrowth-promotingfunctionforwild-typep53in normal cells. Therefore, it seems absolutely necessary to understand both the growth-promoting and
growth-inhibi-tory activities ofwild-typep53 at themolecular level. Inthisregard, analysis of cellular transformation and lytic
replicationofavariety ofDNAtumorviruses might be very helpful, since major regulatory proteins of these viruses interact withwild-type p53 both during lytic infection and
during cellular transformation (5, 29, 42, 49). One of the
best-analyzed viral systems is simian virus 40 (SV40), in which expression of the large T antigen (large T) leads to bothcomplex formation(17, 28) and metabolic stabilization of p53 (9). In vivo as well as in vitro studies suggested a regulatory function forp53in SV40 DNA replication. It has beenshown that addition of purified wild-typep53to invitro DNA replication assay mixtures inhibits SV40 DNA repli-cation (2, 13) and that transient expression of transfected wild-type p53 in SV40-transformed monkey (Cos) cells in-hibits in vivo replication of plasmids containing the SV40
origin ofreplication (ORI) (2). Furthermore, the inhibitory effect oflargeT-p53 complexes isolated from infected
mon-*Correspondingauthor.
key cells has beendirectly demonstratedin invitro
replica-tion assays(39).Thisinhibitory functionof
p53
isdueatleast in partto aninhibition of the DNA helicaseactivityoflargeT (25, 58, 64). On the basis of these data, it has been postulated that wild-type p53playsanegativeregulatoryrole in SV40 DNAreplicationinvivo,possibly by
down-regulat-ing SV40 DNAreplicationduringlatetimes of thelytic cycle (15,25, 58,64). Thispostulated negative regulatoryeffect of
p53inSV40 DNAreplicationoftenisconsideredamodel for theproposedantiproliferativeeffect ofwild-typep53,asit is consistent with the finding that mutant p53 has lost its inhibitory effect on SV40 DNA replication inboth in vitro andin vivo assays(13, 39),inanalogytothepostulatedloss of a growth-inhibitory function of mutant p53 in cellular
proliferation (35, 37). On the other hand, the postulated
inhibitory role ofendogenously expressedwild-typep53has notyet beendemonstrated directly, i.e.,inlyticallyinfected cells, because of the lack ofa suitable cellulartestsystem. Suchasystemcould beprovided bymonkeycellsexpressing
amutant
p53,
asSV40 DNAreplicationinsuch cells should be devoid of any negative regulation byp53. That is, such cells shouldproduceenhanced levels ofSV40 DNA,asSV40 DNA replication should be released from the proposeddown-regulation byendogenouslyexpressed wild-typep53.
SV40replication usuallyis studied withkidneycells of the African green monkey (Cercopithecus aethiops), although its natural hostis the rhesus monkey(Macacamulatta) (61). The major reason for using such cells (e.g., TC7 or CV1
cells)is thatSV40 in rhesus monkey cells reportedly estab-lishes a persistent infection (41), whereas infections of African green monkey cells are lytic. Analyzing this phe-nomenon, we recently found that primary rhesus kidney (PRK) cells derived from a SV40-free monkey colony could belyticallyinfectedwith SV40 (63). As reported previously, cells of the established rhesus monkey kidney cell line LLC-MK2,on the other hand, were persistently infected by SV40 (41, 63). We found that a major cellular difference between PRK and LLC-MK2 is that LLC-MK2 cells
ex-pressed a mutant p53, whereas PRK cells, by definition, expressed a wild-type
p53.
This system of homologous monkey cellsdiffering in the phenotypes of theirp53proteins wastherefore usedtodirectly compare the effects of endog-886on November 9, 2019 by guest
http://jvi.asm.org/
enously expressed wild-type and mutant p53 proteins on SV40 DNA replication in vivo.
MATERIALSANDMETHODS
Cells and virus. TC7cells (46), LLC-MK2 cells (22), and PRKcells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal calf serum (FCS). Rhesus monkey kidneys for primary cultures were kindly provided by the Deutsches Primatenzentrum, Gottingen, Germany. Primarycultureswere preparedwith 0.2% Wor-thington collagenase type I (Seromed) in DMEM supple-mented with 20% FCS. Infection was carried out by inocu-lation of monolayer cultures of TC7, LLC-MK2, and PRK cells withSV40strain776 at aninput multiplicityofinfection (MOI)of 1.
Quantitative evaluation of rates of viral DNA synthesis. At various times postinfection, cells were labeled with
[methyl-3H]thymidine(Amersham) (30
puCi/ml/106
cells) for 1 h. Viral DNA wasthen extracted by the method of Hirt (19). DNA in thesupernatant of Hirt extracts was precipitated with etha-nol and suspended in TE buffer (10 mM Tris hydrochloride, 1 mM EDTA [pH 7.4]). [3H]thymidine incorporation intonewly synthesized viral DNA was determined by liquid scintillation counting of aliquots of DNA samples after
trichloroacetic acidprecipitation. Ratesof viral DNA repli-cationwereexpressed ascounts perminute per microgram of viral DNA per hour oflabeling.
Quantitative evaluation of amounts of total viral DNA. Amounts of total viral DNA were quantitated by dot blot
hybridization.Viral DNA fromnonlabeled infected cells was extracted by the method of Hirt (19) at various times postinfection (p.i.). Aliquots of DNA samples and defined amountsof SV40 DNAas areference were adjusted to 0.25 NNaOH(10minat2°C)and then diluted withSSC buffer to a final concentration of 0.125 N NaOH and 0.125x SSC
(0.125x SSC is18 mM NaClplus1.8 mMsodium citrate[pH 7.2]).DNAs weretransferredto aGeneScreen Plus Nylon66 membrane (New England Nuclear)with a DotBlot I Mini-fold apparatus (Schleicher & Schuell) and then hybridized
with random-primed labeled SV40 DNA.Labelingwas car-ried outby the method ofFeinbergand Vogelstein (12)as describedbyManiatisetal. (31).Toevaluate theamountof viral DNA, blots were excised and
32p
counts per minute were determined byliquidscintillation analysis.Characterization of the replicative forms of SV40 DNA.
Cellswere
pulse-labeled
at36hp.i.
with[methyl-3H]thymi-dine(Amersham)(200 ,uCi/ml/10 cells)for 15 min, and viral DNAwasextractedbythemethod of Hirt(19). Aliquotsof SV40 DNAwerefractionatedon0.8% neutralagarose
gels
inrunning buffer composed of 5 mM sodium acetate, 1 mM
EDTA, 0.03% sodium dodecyl sulfate
(SDS),
and 80 mMTris-hydrochloride (pH
7.5)
aspreviously
described(59).
Thegelswere
electrophoresed
for 48 hat a constantvoltage
of 1 V/cm. To detect steady-state levels of viral
DNA,
we stainedgelswithethidiumbromide. Forfluorography
anal-ysis, gels
weredehydrated
with acetic acid-methanol(3:1)
for4 to5h,incubated with2.5%2,5-diphenyloxazole
(PPO)
in aceticacid for12h,and soaked inwaterfor 3to5h. The
gelswerethendried underavacuumat50°Cand
exposed
on X-rayfilmsat -70°C.Determination ofvirus yields. Virus
yields
were quanti-tatedbytitration of tissue culturesupernatantsfrom infected cells harvested at various timesp.i.
for infectious units(MOI). MOIson TC7cellswere determined
by
immunoflu-orescenceanalysisfor
expression
ofSV40large
T.TC7cellsgrown on coverslips were infected with serial dilutions of virus-containing supernatants and prepared for immunoflu-orescence 24 h p.i. as described previously (43, 45). The percentageof large T-positive cells was calculated for each dilution step. The dilution step leading to 90 to 100% large T-positive cells was defined as having an MOI of 1.
Labeling of proteins. Cells were labeled as described previously (55) before extraction with [35S]methionine (Am-ersham), with 50 ,uCi per plate per ml for pulse-labeling and 100
p,Ci
per plate per ml for pulse-chase-labeling experi-ments.Extraction of proteins.Cells were washed three times with phosphate-buffered saline (140 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4 [pH 7.4]) and then extracted for 30 min at 2°C with 1 ml of lysis buffer [120 mM NaCl, 5 mM dithiothreitol, 1 mM ethylene
glycol-bis(P-aminoethyl
ether)-N,N,N',N'-tetraacetic
acid (EGTA), 1% aprotinin,0.5%Nonidet P-40,10%glycerol, 50 mM Tris-hydrochloride
(pH 8.0)]. Extracts were cleared by centrifugation(10 min, 20,000 x g, 2°C), and proteins were immunoprecipitated from thesupernatants.
Immunoprecipitation, SDS-polyacrylamide gel electro-phoresis, and Western immunoblotting.
Immunoprecipita-tions of p53 andlarge T in 1-ml cellular extract samples were
performed for 4 h at 2°C with 100 ,ul of settled protein A-Sepharose (Pharmacia) and 500 ,ul of cell culture superna-tant of the monoclonal anti-p53 antibody-producing hybri-doma cell line PAb122 (16), PAb421 (66), or PAb240 (14). Large T wasimmunoprecipitatedwith 500
pl
of cellculturesupernatant of the anti-large T antibody-producing hybri-domacell linePAb419(52).
Immunocomplexeswerewashedextensively,andproteins
wereeluted from protein A-Sepharoseand thensubjected to
SDS-polyacrylamide gelelectrophoresis asdescribed previ-ously (9). Proteins were analyzed on 10% polyacrylamide
slabgelsby the systemof Laemmli(26).Westernblotting(3)
wasperformed with 4 ml of cell culture supernatant
contain-inganti-p53 monoclonal antibodyPAb421(66)oranti-largeT monoclonal antibody PAb1O8 (16) and [3H]protein A as describedpreviously (50).
RESULTS
Down-regulation of SV40 DNA replicationin
lytically
SV40-infected monkey cells during the courseof infection. Down-regulation of SV40 DNA replication in lytically infectedmonkeycells could beanactiveprocessofviralreplication.
Alternatively, it might simply be the result of cytopathic
viral effectsonthe hostreplication machinery.To discrimi-nate between these alternatives, we analyzed the rates of SV40 DNA replication during the course of infection
([3H]thymidine incorporated per microgram of SV40 DNA perhour oflabeling) inlyticallyinfected monkeyTC7cells andrelated thisinformationtothe releaseofinfectious virus from thesecells andtothe appearance ofcytopathiceffects
(see Materials and Methods for
details). Figure
1 demon-strates that the maximal rate of SV40 DNAreplication
occurred around 36 hp.i. and that theratedeclined
sharply
from thenon.Virus release started around 36 h
p.i.
in these cells and reached its maximum about 72 hp.i.
Cytopathic
effectswere notdetectable before 60 h
p.i.
Thisanalysis
thusstronglysuggests that
down-regulation
ofSV40DNArepli-cation isnotdueto
cytopathic
effects but reflectsanactive processduringviralreplication.
Comparisonof
p53
phenotypesin PRK andLLC-MK2
cells.Analysis of the
postulated
role ofwild-type p53
inon November 9, 2019 by guest
http://jvi.asm.org/
IRATEOF VIRALDNA VIRUSRELEASE"* SYNTHESIS*
o-e __s_.
* 105cpm/pgviralDNA/1hlabeling **MOI
oI,
12 24 36 48 60 72
hp.i. FIG. 1. Course ofSV40 infection in lytically infected monkey cells. Rates of viral DNAsynthesis and virus releasewere deter-minedatthe timesindicatedasdescribed in Materials and Methods.
CPE, cytopathiceffect.
regulationofSV40DNAreplicationand the release from this down-regulation bymutant p53 invivo requires a
homolo-gouscellular system expressing authenticwild-type or
mu-tantp53 protein,respectively. Suchasystemisprovided by
PRK cells and the established rhesus monkey kidney cell lineLLC-MK2. PRKcells, by definition, expresswild-type p53,whereasp53 expressedinLLC-MK2cells ismutant,as wasdemonstrated by the following experiments.
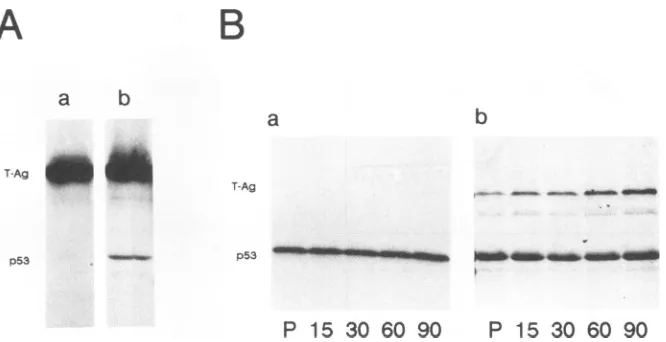
Western blot analysis of steady-state levels of p53 in LLC-MK2cellsand ofp53in PRK cells revealed thatp53in LLC-MK2 cells accumulated to relatively high levels (Fig. 2A,lane a),whereasp53 in PRKcellswasundetectable by
thisanalysis (Fig. 2A,laneb).The enhanced level ofp53in LLC-MK2cells is duetotheenhanced metabolicstabilityof this protein compared with that of p53 in PRK cells, as
demonstrated by pulse-chase analyses of [35S]methionine-labeledp53 from both cell lines.p53 inLLC-MK2 cellswas
foundtobemetabolicallystable (Fig. 2B,panel a),whereas
A
B
a b a b
P 15 30 60 90 P 15 30 6090
FIG. 2. Steady-statelevels (A) and metabolicstability(B) of p53 inLLC-MK2 and PRK cells. (A)p53 from LLC-MK2 (lane a) and PRK (lane b) cells was immunoprecipitated with PAb122 and analyzed by polyacrylamidegelelectrophoresisand then by
West-ernblotting with PAb421and [3H]proteinAfor quantitative
evalu-ationoftheprotein. (B) The metabolicstability of p53wasanalyzed
bypulse-chase labeling. LLC-MK2 (panel a)and PRK(panel b) cells
werepulse-labeled with [35S]methionine(100,uCiperplateperml)
for30 min (lanes P) and then chased for 15 to 90 min. p53 was
immunoprecipitated with monoclonal antibody PAb122.
[image:3.612.63.300.72.264.2]a
b
c
d
FIG. 3. Reactivity of p53 in PRK and LLC-MK2 cells with
monoclonal antibodies PAb240 and PAb421. Cells were pulse-labeled with[35S]methionine (50 ,uCi perplateperml)for 1 h and then immunoprecipitated with either the p53-specific antibody PAb421(lanea,LLC-MK2;lanec,PRK)orPAb240,which
recog-nizes only p53 inamutantconformation(lane b, LLC-MK2;laned, PRK).
p53in PRK cells(Fig. 2B,panel b)showed thefast turnover characteristic ofwild-type p53 (27, 29).Theenhanced level and metabolic stability of p53 in LLC-MK2 cells already
were indicative ofa mutant p53. The mutantphenotype of p53inLLC-MK2 cellswasfurthercorroborated by demon-strating that this p53 reacted with monoclonal antibody PAb240 (Fig. 3,laneb),which isspecificforp53inamutant conformation(14),whereasp53from PRK cells didnot react withPAb240 (Fig. 3, laned).
Formationofcomplexesofp53andlargeT isanimportant functional property ofwild-type p53 which is supposed to play an important role in regulating large T and/or p53 activities during lytic replication, especially in the down-regulationofSV40DNAreplication (2, 13, 25, 44, 58, 64).To analyze the ability of p53 proteins expressed in PRK and LLC-MK2 cells to form a complex with large T, infected cells were labeled with [35S]methionine for 1 h at 24 h p.i. andextracted,andlargeT-p53 complexeswere immunopre-cipitated with the large T-specific monoclonal antibody PAb419 (52). Figure 4A, lane b, demonstrates that p53 in infected PRK cellswas readilydetected in acomplexwith
large T, whereas p53 from LLC-MK2 cells was not in a
complexwithlargeT(Fig. 4A, lane a). Analysisof cellular extractsafterpulse-chase-labelingofinfectedcells with the p53-specific monoclonal antibody PAb122 (16) showed the characteristic time courseofentry oflarge T intocomplex with p53in PRKcells (Fig. 4B, panel b) and demonstrated that p53 in PRK cells became metabolically stabilized, as
reported for p53 in African green monkey kidney cells
infected withSV40 (11, 32).Incontrast,large T in LLC-MK2 cells didnot enteracomplex with thep53 in these cells,even
afterprolongedchaseperiods (Fig. 4B, panel a). Since p53 in these cellswas alsometabolically stable inuninfected cells
(Fig. 2B, panel a), a further increase inmetabolic stability uponSV40 infectionwasnottobe expected. Itis important
to notethatmetabolicstabilization of p53 inPRKcells after
SV40 infectionresulted in similar steady-statelevels of p53
at about48hp.i., asin LLC-MK2 cells (see Fig. 7B, panel
1).Theproperties of the p53 proteins expressed inPRKand in LLC-MK2cells aresummarized in Table 1.
Comparison of SV40 DNA replication in PRK and in LLC-MK2cells. Theexpression ofabona fide wild-typep53 in PRKcells andamutantp53 that doesnotform complexes with large T in LLC-MK2 cells, as described above, ren-12
10
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.382.498.72.197.2] [image:3.612.96.265.528.627.2]A
B
a
b
a
b
T-Ag 4p_
p53
T-Ag - - - o
p53 _,in14W_W_,_ 0
P
15
30 60 90
P
15 30 60 90
FIG. 4. Complexformationbyp53andlarge T (T-Ag) afterSV40 infection of PRK and LLC-MK2 cells. (A) Infected LLC-MK2 (lane a) and PRK(lane b)cells werepulse-labeled (asdescribedin the legend to Fig. 3) 24 h p.i., and large T wasimmunoprecipitated with the large T-specific monoclonal antibodyPAb419. (B) Pulse-chase-labeling (as described in the legend to Fig. 2) 24 h p.i., followed by immunopre-cipitation with the p53-specific monoclonal antibody PAb122from cellular extracts of LLC-MK2 (panel a) and PRK (panel b) cells, was performed toanalyzethe time course of entry of large T into complexes with p53 as well as the metabolic stabilization of p53.
dered these cells anideal system for the analysis ofpossible effects of endogenously expressed wild-type or mutant p53 on SV40 DNAreplication. In preceding analyses, we had established thatlarge Texpression and itsinteractions with cellular targets, especially with structural systems of the nucleus, were identical in these two types of cells (63a), thus excluding any obvious differences in viral replication.
ToanalyzeSV40 DNAreplicationin PRKand LLC-MK2
cells,thesecellswereinfected withSV40at anMOI of 1 and
labeledwith [3H]thymidine (30 ,uCi/ml) for 1 hat the times indicated in Fig. 5. SV40 DNA thenwas extracted by the method of Hirt (19) and purified. Rates of viral DNA
synthesis (counts per minute per microgram of DNA per hour oflabeling) during infection werethendetermined. In
addition,SV40 DNAwasharvested fromparallel,unlabeled culturestodetermine totalamountsof viralDNAbydot blot analysis. A graphical evaluation of these data is shown in
Fig.5. Figure5Ademonstrates identicalratesofviral DNA
synthesis in both types of cells during the course of infec-tion. As in lytically infected African green monkey cells
(compare Fig. 1and5A), DNAsynthesisstarted about 12 h
p.i.andpeakedabout 36 hp.i.inboth types of cells. Therate of DNA synthesis thendropped, regardless of whether the cells expressedawild-type (PRK)or a mutant
(LLC-MK2)
p53. It is importantto statethat the cells at36 hp.i.didnot show any sign of
cytopathic
effects, indicating that the reduction in the rate of SV40 DNA synthesis, as in TC7cells, was not simply due to cell damage induced byviral infection but reflected a cellularly or virally controlled reaction. Analysis of the total amounts of SV40 DNA
synthesized duringthecourseof infection in these cells
(Fig.
[image:4.612.145.478.72.243.2]SB) showed a similar increase in the accumulation of viral
TABLE 1. Propertiesofp53inPRK andLLC-MK2cells
Reactivity of Formation of
Cell line p53half-life p53with: withcomplexeslargeT PAb421 PAb240 byp53
PRK 30 min + - +
LLC-MK2 4h + +
-DNA in these cells until about 48 h p.i. Amounts of viral DNAin PRK cells stayed constant from then on, whereas a further increase was observed in LLC-MK2 cells. This apparentincrease in theyield of SV40 DNA, however, is due tothe fact thatmostof thenewly replicated SV40 remained in LLC-MK2 cells at the times at which it was analyzed, whereas PRK cells constantlyreleased infectious virus (Fig.
5C). Takingthis intoconsideration,the total amounts of viral DNA synthesized in the two types of cells were largely
similar.
SV40 DNAreplicationstartson superhelical form I DNA molecules within the ORI. Bidirectional replication then generates intermediate Cairns structures (ICS). Elongation ofnascentDNA on ICSproceeds until replication is about
90%complete.At thispoint, replication forks pause,
gener-ating the latest Cairns structures (LCS). The remaining parental DNA strands in the LCS arecompletely unwound, andtwocircular SV40monomers(SV40 form II* DNA) are
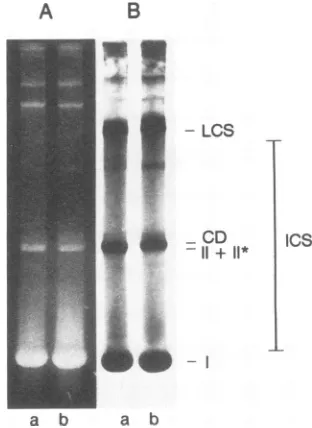
separated; each contains a short gap in the termination region of the nascent DNA strand (51; for a review, see reference 6). To characterize the replicative forms of SV40 DNAsynthesizedin PRK andLLC-MK2cells, infected cells werelabeled for 15 min with
[3H]thymidine
at 36 hp.i. and extracted by the method of Hirt (19). Viral DNA was fractionated by neutral agarose gel electrophoresis (60)and visualizedbyethidiumbromidestainingandfluorographyas described in Materials and Methods. Byapplying
theseconditions,themobilities of the various forms of SV40DNA in lyticallyinfected TC7 cells have been previously charac-terized in detail(51).The resultsareshown in Fig. 6. Inthe ethidium bromide-stainedgelsofSV40DNAfromboth cells
(lanes A), representing steady-state levels of SV40 DNA
intermediates,
the mostprominent
bandwas form I DNA(superhelical). In addition, minor bands of form II + II* DNA (relaxed circles), and catenated dimers were visible.
Fluorographic analysis of
replicating
forms of SV40 DNA(Fig. 6B) demonstrates that
pulse-labeling
of viral DNA resulted inradiolabelingof formIDNA,form II +II*DNA,
and theLCS,whileaconsiderableamountof theradiolabel was present in the ICS. ICS migrate as a smear between form I and the LCS (59, 60,
65),
representing
a randompopulation of nascent SV40 DNA at various stages of
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.62.302.659.723.2]A ?3
-G
CZ
z 0
B
co
8
0
12
-e-- ^
10
24 4X 72 h
FIG. 5. Comparison ofSV40DNA replication in PRK and
LLC-MK2 cells. (A) Rates of viral DNAsynthesis per microgram of viral
DNA wereanalyzed by measurement of 3H incorporation into viral
DNAafterpulse-labeling with 30,uCiof [3H]thymidine for 1 h at the
indicated time points (see Materials and Methods). (B) Total
amounts of viral DNA were evaluated by dot blot hybridization with
32P-labeled SV4o DNA atvarioustimes afterinfection(seeMaterials
andMethods). (C) Virus release was measured by quantitation of
theamountsofinfectious virusreleasedincellculturesupernatants
harvestedfrominfected cells at thetimepointsindicated. TheMOI
ofthe supernatants was determined as descrinbedinMaterials and
Methods.
continued replication. No difference between replicative intermediates ofSV40 DNA from LLC-MK2 cells (lane a) andthose fromPRKcells(lane b) could be detected, further supporting our conclusion that SV40 DNA replication in these two types ofcells did not show any qualitative and
quantitativedifferencesandproceeded via thesame
replica-tiveintermediates.
Large T/p53 ratios in SV40-infected cells. In SV40 DNA replication, the large T intrinsic DNA-dependent helicase activity is required forboth initiation and elongation func-tions (54). This activity is inhibited by p53 complexing to
largeT (13, 25, 58, 64).
p53,
inaddition, competes for theA
B
,-,
JA-.a.
I.;
- LCS
-CD
-11 + 11* ICS
a b a b
FIG. 6. Comparison of replicative forms of SV40 DNA repli-catedin LLC-MK2 and PRKcells. LLC-MK2(lanes a)and PRK (lanesb) cellswerepulse labeled 36hp.i.with
[3H]thymidine
(200p.Ci/106
cells) for 15 min, and viralDNAwasextracted.Aliquotsof viralDNA werefractionatedby neutralagarosegelelectrophoresis asdescribedinMaterialsand Methods. (A)Thegelswerestained with ethidiumbromide for detection ofsteady-state levels of viral DNA. (B) Fluorographic analysis revealed the different forms of pulse-labeled newly replicated SV40DNA.Replicativeforms(ICS, LCS, catenated dimers[CD],
and II + II*) and terminatedform I SV40DNAareindicated (seetext).association ofDNApolymerasewithlargeT(15). Thus,it is evident thatlargeT-dependent SV40 DNAreplication
pre-dominantly requires the activity oflarge T not
complexed
withp53 (freelargeT).Ininvivo SV40 DNAreplication,the presenceofsufficientamountsoffreelargeTis ensuredby
twoindependentmechanisms.Firstly,theamountoflargeT present in infected cells at all times of infection greatly exceedstheamountofp53
(e.g.,
Fig. 4A,laneb);
secondly, preferentiallyold andnotnewly synthesized largeT associ-ateswithp53(62) (Fig. 4B, panel b). SV40-infectedcells thus containalargeexcessof freelargeToverlarge Tcomplexedwithp53 even at48 h p.i., i.e., at atime when SV40 DNA
replicationisalready down-regulated.This isdocumentedin
Fig. 7.
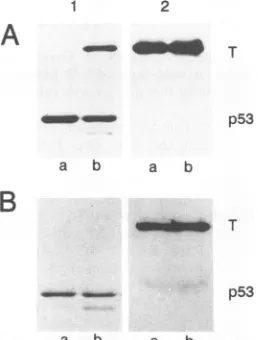
Large
T-p53
complexesfrominfectedPRKandLLC-MK2 cells at 48 h p.i. were separated from free large T bysequential immunoprecipitation,
firstwithp53-specific
mon-oclonalantibodyPAb421(Fig.7, panels1)and then withthe large T-specific monoclonal antibody PAb419 (panels 2). Aftera2-hpulse-label of infected cells, immunoprecipitation forp53demonstrated that aconsiderable amount of large T wascoprecipitatedwithp53
from PRK cells (Fig. 7A, panel 1, lane b), whereas no large T in complex with p53 wasimmunoprecipitatedfromLLC-MK2cells (Fig. 7A, panel 1, lanea). The amountsof p53 appeared to be similar in both cell typesatthat timep.i.Immunoprecipitation of free large T from clearedextracts (Fig. 7A, panel2), however, dem-onstrated thatmuch largeramountsof freelargeTthan large Tin complex with
p53
were found in PRK cells (Fig. 7A, panel 1, lane b).Western blot analysis ofsteady-state levels of p53,
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.59.301.71.472.2] [image:5.612.358.514.77.291.2]2
A
-0mT
p53
a b a b
B
T
p53
[image:6.612.119.248.77.247.2]a b a b
FIG. 7. Comparison of large T/p53ratios in infected LLC-MK2 (lanes a) and PRK (lanes b)cells. Forty-eight hours p.i., large T-p53 complexeswereseparated from freelarge T by sequential immuno-precipitation, first with anti-p53monoclonal antibody PAb421
(pan-els 1) and then with anti-large T monoclonal antibody PAb419 (panels 2). Proteins were sequentially immunoprecipitated from extracts of either 2-h pulse-labeled ([35S]methionine, 50 pCi per
plateperml) cells (A)orunlabeled cells (B).Theproteins in panelB weresubjectedtoWestern blot analysis.
tained by sequential immunoprecipitations of infected unla-beledPRKandLLC-MK2 cells (Fig. 7B, panel1), revealed thatas aresult ofmetabolic stabilizationof p53 in PRK cells,
equal levels of p53werepresentinLLC-MK2 (Fig. 7B, panel 1,lanea) and PRK(Fig.7B,panel 1, lane b) cells. However,
inbothcelltypes,thelevelsof free large T (Fig. 7, panels 2)
exceededby far the levels of total p53 (Fig. 7, panels 1)or
thelevel ofcomplexed large Tin PRK cells (Fig. 7A, panel 1,laneb). Freelarge T in LLC-MK2 cells(Fig. 7B, panel 2, lane a) reflects the total amount of large T in these cells, whereas freelargeT from PRK cells(Fig. 7B, panel 2, lane b)reflectstotal large T minus largeTcoprecipitatedwith p53 inthe first step of the sequential immunoprecipitation. The fact thatapproximately equal levels of free largeT(Fig. 7B, panel 2) were present in both cell types clearly documents that the proportion of the large T molecules blocked for replication functions by complexformationwith p53 makes
uponlyasmallfractionof the large Tmolecules availablein
infectedcells invivo. Infact,the amountof free large T in infectedPRKcells(Fig. 7, panels 2, lanes b)wasverysimilar
totheamountoffree(total) largeTinLLC-MK2 cells (Fig. 7,panels 2, lanesa).
DISCUSSION
Inthisstudy, weasked whether viral DNAreplication in
SV40-infected monkey cells would be released from the postulated negative regulatoryeffect of wild-typep53incells endogenously expressing mutated p53, i.e., whether such cells would overreplicate SV40DNA. Suchoverreplication ought to be expected if down-regulation of SV40 DNA replication in the late phase of viral infection indeed is mediatedby wild-type p53.Thisquestion isimportant inso-far as p53-mediated inhibition of SV40 DNA replication
constitutes theonlymodelsystem foranegative regulatory
role of p53 in a proliferation-associated function (DNA
replication)inwhichit ispossibletodelineate theunderlying
molecular events. Analysis of this question required a ho-mologous system of cells expressing either endogenous wild-type or mutant p53. Such a system was provided by PRK and LLC-MK2 cells. Whereas p53 in PRK cells by definition is wild type, our analysis of p53 in LLC-MK2 cells, a permanent cell line established from PRK cells (22), clearly showed that this p53 was mutant; the most prominent feature of p53 in this cell line, as far as this study was concerned,
was that thisp53failed to complex with large T in vivo. On the basis of the available data, one can assume that this mutant p53 is unable to inhibit SV40 DNA replication in vitro. Although only a limited number ofp53 mutants have been tested for their inhibitory activity on SV40 DNA replication in vitro (13, 25, 57, 58, 64), all but one of them were
inhibitory.
The only exception described so far is an oligomerization-defective mutant p53 recently constructed by Sturzbecher et al. (57). Such a mutant p53 has not yet been found among any p53 mutants arising in vivo. Given this caveat, comparison of SV40 DNA replication in LLC-MK2 cells with that in wild-type p53-expressing PRK cells should have allowed us to detect differences due to the different p53 phenotypes. However, no such differences could be detected. In particular, the comparison of the rates ofviral DNA synthesis during the course of infection dem-onstrated that these rates were similar in both types of cells throughout infection, i.e., that down-regulation of SV40 DNA replication occurred in both types of cells. As a consequence, similar steady-state levels of SV40 DNA were produced in both types of cells during late times of infection. As in lytically infected TC7 cells, a decrease in the rate ofSV40 DNAsynthesis during later times p.i. (36 h p.i.) was observed in both types of cells; therefore, this decrease was independent of whether the cells expressed a mutant or wild-typep53.Importantly, this reduction in the rate of viral DNA synthesis occurred at times after infection when no signs of cell damage were visible. Therefore, this reduction indeed reflected a cellularly or virally controlled process. However, this control apparently was not exerted by thep53
proteins expressed in the infected cells or was not directly dependent on the p53 phenotype. We thus conclude that endogenously expressed wild-type p53, although it accumu-lates to relatively high concentrations in infected cells be-cause of its metabolic stabilization after infection (Fig. 7), has no direct inhibitory effect on SV40 DNA replication in vivo. The roles ofp53and of largeT-p53 complexes inSV40 DNA replication thus remain elusive.
Animportant clue forunderstanding the apparent discrep-ancy between the analysis of the role of wild-type p53 in SV40 DNA replication described here and previous analy-ses, both in vitro and in vivo (2, 13, 25, 39, 58, 64), might be provided by considering the quantitative aspects of inhibi-tion by wild-typep53 in the latter assays. Significant inhibi-tion of in vitro SV40 DNA replication by wild-type p53
required at least equimolar concentrations ofp53 and large T, withcomplete inhibition ofSV40 DNA replication being observed only when p53was added in at least equal ratios (39, 64) or in excess over large T (2, 13, 58). Infected cells, in contrast, contain an excess of free large T even at times after infection when SV40 DNA replication was already down-regulated (Fig. 7). Free large T performs important functions in SV40 DNA replication which cannot be per-formed by large T complexed to p53 (25, 58). Therefore, a likely explanation for the inhibitory effect of wild-type p53 onSV40 DNA replication in in vitro assays might be that the added excess p53reduced the level of free large T below a threshold necessary for sustaining SV40 DNA replication.
on November 9, 2019 by guest
http://jvi.asm.org/
This explanation may also be relevant for resolving the apparentparadox thatwild-typep53 in normal cellsseems to be absolutely required for cell cycle progression (8, 33, 34, 45, 56) but reconstitution of wild-type p53 expression in tumor cells lacking p53 expression or expressing mutated p53 (35-37), on the other hand, leads to growth arrest. In fact, one has to be aware that the proposed antiproliferative effect of wild-type p53 in normal cells has not yet been demonstrated. Therefore, it seems possible that
endoge-nouslyexpressed wild-type p53 (andeven some mutantp53 proteins
[8,
56]) indeed exerts a positive regulatory effecton cellularproliferation and that reconstitution ofwild-typep53expression in tumor cellsrepresents ahighly artificialsystem for analyzing the effects of endogenous wild-type p53 on cellular DNA synthesis. Therefore, the experiments
de-scribed in this study, by analogy, may suggest that the antiproliferative effects of wild-type p53 transfected into tumorcellsdo not reflect a regulatory function of p53atthe level of cellular DNA synthesis under physiological growth conditions. Considering the strict regulation of p53 levels in
normalcells, both at the level of mRNA formation andatthe level of p53 protein synthesis and stability (8, 10, 42, 47,56), it is to be expected that reconstitution of wild-type p53 expression in tumor cells may lead very easily to
overex-pression of wild-type p53 and that growth inhibition by wild-typep53 might depend on the level of its expression (4). Since the contact sites of p53 with large T (24, 53) represent mutational hot spots on p53 (1, 23, 30, 40, 48), it is tempting to speculate that the association of p53 with large T mimics an interaction of p53 with a so far unknown cellular target. We therefore want to suggest that, in analogy to the func-tional elimination of free large T by excess p53 in in vitro SV40 DNA replication assays, which leads to cessation of
SV40 DNA replication, overexpression of reconstituted wild-type p53 might functionally eliminate an important target for p53 in cell cycle control and thereby lead to a
nonphysiological growth arrest. Although this model is
purely speculative at the moment, the system of a homolo-gous pair of cell types described in this study, differing in that one expresses wild-type p53 and the other expresses mutantp53, might be helpful in defining functions for wild-type p53 in SV40 replication. Definition of such function shouldgreatly enhance our understanding of the role(s) of
p53inproliferation of normal cells and tumor cells. ACKNOWLEDGMENTS
We thank the Deutsches Primatenzentrum for providing rhesus monkey kidneysfor preparations of primary cultures.
This study was supported by grants DFG De 212/8-1 from the DeutscheForschungsgemeinschaft and W29/90/De 1 from the Deut-sche Krebshilfe (Dr. Mildred Scheel Stiftung furKrebsforschung) and by the Fonds der Chemischen Industrie. The Heinrich-Pette-Institut is supported by the Freie und Hansestadt Hamburg and by theBundesministerium furGesundheit.
REFERENCES
1. Ahuja, H., M. Bar-Eli, S. H. Advani, S. Benchimol, and M. J. Cline. 1989. Alterations in thep53geneand the clonal evolution of theblast crisis of chronin myelocytic leukemia. Proc. Natl. Acad. Sci. USA86:6783-6787.
2. Braithwaite,A. W., H.-W.Stuirzbecher,C.Addison, C. Palmer, K. Rudge, and J. R. Jenkins. 1987. Mouse p53 inhibits SV40 origin-dependent DNAreplication. Nature (London) 329:458-460.
3. Burnette, W. N. 1981. "Western blotting": electrophoretic transferofproteins from sodium dodecyl
sulfate-polyacrylam-ide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem.
112:195-203.
4. Chen, Y., P.-L. Chen, N.Arnaiz,D. Goodrich,and W.-H. Lee. 1991. Expression of wild-type p53 in human A673 cells sup-presses tumorigenicity but not growth rate. Oncogene 6:1799-1805.
5. Crawford, L. 1983. The 53,000-dalton cellular protein and its role in transformation. Int. Rev. Exp. Pathol. 25:1-50. 6. DePamphilis, L. M., and M. K. Bradley. 1986. Replication of
SV40 and polyoma virus chromosomes, p. 99-246. In N. P. Salzman (ed.), The Papovaviridae, vol. 1. Thepolyomaviruses. Plenum Publishing Corp., New York.
7. Deppert, W. 1989. p53: onco- or anti-onco-gene. A critical review. NATO ASI Ser. Ser. H 34:399-407.
8. Deppert, W., G. Buschhausen-Denker, T. Patschinsky, andK. Steinmeyer. 1990. Cell cycle control of p53 in normal (3T3) or chemically transformed (Meth A) mouse cells. II. Requirement for cell cycle progression. Oncogene 5:1701-1706.
9. Deppert, W., M. Haug, and T. Steinmayer. 1987. Modulationof p53 protein expression during cellulartransformationwith sim-ianvirus 40. Mol. Cell. Biol. 7:4453-4463.
10. Dony, C., M. Kessel, and P. Gruss. 1985. Post-transcriptional control of myc andp53 expression during differentiation of the embryonal carcinoma cell line F9. Nature (London) 317:636-639.
11. Fanning, E., K.-H. Westphal, D. Brauer, and D. Corlin. 1982. Subclasses of simian virus 40 large T antigen: differential binding of two subclasses of T antigen from productively infected cells to viral and cellular DNA. EMBO J. 1:1023-1028.
12. Feinberg, A. P., and B. Vogelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 136:6-13.
13. Friedman,P. N., S.E. Kern, B.Vogelstein, and C. Prives. 1990. Wild-type, but not mutant, human p53 proteins inhibit the replication activities of simian virus 40 large tumor antigen. Proc. Natl. Acad. Sci. USA 87:9275-9279.
14. Gannon, J. V., R. Greaves, R. Iggo, and D. P. Lane. 1990. Activatingmutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J.9:1595-1602.
15. Gannon, J.V., andD. P. Lane. 1987. p53 and DNA polymerase compete for binding to SV40 T antigen. Nature (London) 329:456-458.
16. Gurney, E. G., R.0.Harrison,and J.Fenno. 1980. Monoclonal antibodies against simian virus 40 T antigens: evidence for distinctsubclassesof largeT antigen and for similarities among nonviral T antigens. J. Virol.34:752-763.
17. Harlow, E., D. C. Pim,and L. V.Crawford. 1981. Complex of simian virus 40 large-T antigen and host 53,000-molecular-weight protein inmonkeycells. J. Virol. 37:564-573.
18. Harris, A. L. 1990. Mutantp53-thecommonestgenetic abnor-mality in human cancer?J. Pathol. 162:5-6.
19. Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol.26:365-369. 20. Hollingsworth, R. E., and W.-H. Lee. 1991. Tumor suppressor
genes: new prospects for cancerresearch. J.Natl.Cancer Inst. 83:91-96.
21. Hollstein, M., D. Sidransky, B. Vogelstein, andC. C. Harris. 1991.p53mutations in human cancers. Science 253:49-53. 22. Hull, R. N., W. R. Cherry, and J. S. Johnson. 1956. The
adaption and maintenance of mammalian cells to continuous growth in tissue culture. Anat. Rec. 124:490.
23. Iggo, R., K. Gatter, J. Bartek, D. Lane, and A. L.Harris.1990. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet335:675-679.
24. Jenkins, J. R., P. Chumakov, C. Addison, H.-W.Sturzbecher, and A.Wade-Evans. 1988. Two distinct regions of the murine p53 primary amino acid sequence are implicated in stable complex formation with simian virus 40 T antigen. J. Virol. 62:3903-3906.
25. Kienzle, H., M. Baack, and R. Knippers. 1989. Effects ofthe
on November 9, 2019 by guest
http://jvi.asm.org/
cellular p53 protein on simian-virus-40-T-antigen-catalyzed DNAunwinding in vitro. Eur.J. Biochem. 184:181-186. 26. Laemmli,U. K. 1970. Cleavage ofstructural proteins during the
assembly of the head ofbacteriophage T4. Nature (London) 227:680-685.
27. Lane, D. P., and S. Benchimol. 1990. p53: oncogene or anti-oncogene?Genes Dev. 4:1-8.
28. Lane, D. P., and L. V. Crawford. 1979. T antigen is bound to a hostproteinin SV40-transformed cells. Nature (London) 278: 261-263.
29. Levine, A. J. 1990. Thep53protein and its interactions with the oncogeneproductsof the small DNAtumorviruses. Virology 177:419-426.
30. Levine, A. J., J. Momand, and C. A. Finlay. 1991. The p53
tumoursuppressor gene.Nature(London)351:453-456.
31. Maniatis,T., E. F.Fritsch,andJ.Sambrook. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor,N.Y.
32. McCormick, F., and E. Harlow. 1980. Association ofamurine 53,000-dalton phosphoproteinwithsimianvirus 40large-T anti-genintransformed cells.J.Virol.34:213-224.
33. Mercer, W. E., C. Avignolo, and R. Baserga. 1984. Role of p53 protein in cell proliferation as studied by microinjection of monoclonal antibodies. Mol. Cell. Biol.4:276-281.
34. Mercer, W. E., D. Nelson, A. B. DeLeo, L. J. Old, and R. Baserga. 1982.Microinjection of monoclonal antibodytoprotein p53 inhibits serum-induced DNAsynthesisin3T3 cells. Proc. Natl. Acad. Sci. USA79:6309-6312.
35. Mercer,W.E.,M.T.Shields,M.Amin, G. J.Sauve,E.Appella, J.W.Romano, andS. J. Ullrich. 1990. Negative growth regu-lation in a glioblastoma tumorcell line that conditionally ex-presses human wild-type p53. Proc. Natl. Acad. Sci. USA 87:6166-6170.
36. Mercer, W. E., M. T. Shields, D. Lin, E. Appella, and S. J. Ullrich. 1991. Growth suppression induced by wild-type p53 proteinis accompanied by selective down-regulationof prolif-erating-cellnuclearantigen expression. Proc. Natl. Acad. Sci. USA88:1958-1962.
37. Michalovitz, D., 0. Halevy, and M. Oren. 1990. Conditional inhibition of transformation and of cellproliferation bya
tem-perature-sensitivemutantofp53. Cell62:671-680.
38. Milner, J. 1991. The role ofp53 in the normal control of cell proliferation. Curr. Opin. Biol. 3:282-286.
39. Miyamoto, N., E. Kihara, T. Inada, S. Katsura, and Y. Mu-rakami.1990. Primate's p53inhibits SV40DNAreplication in vitro.Biochem. Biophys.Res. Commun. 168:604-608. 40. Nigro, J. M., S. J. Baker, A. C. Preisinger, J.M. Jessup,R.
Hostetter,K. Cleary, S. H.Bigner, N.Davidson, S. Baylin,P. Devilee,T. Glover,F.S.Collins,A.Weston,M.Modali,C. C. Harris,and B.Vogelstein.1989.Mutations in thep53geneoccur indiverse humantumourtypes. Nature(London)342:705-708. 41. Norkin, L. C. 1982. Papovaviral persistent infections.
Micro-biol.Rev. 46:384-425.
42. Oren,M. 1985. Thep53cellulartumorantigen:gene structure, expression and protein properties. Biochim. Biophys. Acta 823:67-78.
43. Pope, J. H., and W. P. Rowe. 1964. Detection of a specific antigen in SV40-transformed cells by immunofluorescence. J. Exp.Med. 120:121-128.
44. Reich,N.C.,and A.J. Levine.1982. Specificinteraction ofthe SV40Tantigen-cellular p53protein complexwithSV40DNA. Virology117:286-290.
45. Reich, N.C., andA. J. Levine. 1984. Growthregulation ofa
cellular tumour antigen, p53, in nontransformed cells. Nature (London)308:199-201.
46. Robb, J.A.,and K. Huebner. 1973. Effect of cell chromosome numberonsimian virus 40replication. Exp.Cell Res.81:120-126.
47. Rogel, A., M. Popliker, C. G. Webb, and M. Oren. 1985. p53 cellulartumorantigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol. Cell. Biol. 5:2851-2855. 48. Romano, J. W., J. C. Ehrhart, A. Duthu, C. M. Kim, E. Apella,
and P. May. 1989.Identification and characterization of a p53 gene mutation in a human osteosarcoma cell line. Oncogene 4:1483-1488.
49. Rotter, V., and D. Wolf. 1985. Biological and molecular analysis ofp53 cellular-encoded tumor antigen. Adv. Cancer Res. 43: 113-141.
50. Schirmbeck, R., and W. Deppert. 1987. Specific interaction of simian virus 40 large T antigen with cellular chromatin and nuclearmatrix duringthe course of infection. J. Virol. 61:3561-3569.
51. Schirmbeck, R., and W. Deppert. 1991. Structural topography of simian virus 40 DNA replication. J. Virol. 65:2578-2588. 52. Simanis, V., and D. P.Lane.1985. An immunoaffinity
purifica-tion procedureforSV40 large T antigen. Virology 144:88-100. 53. Soussi, T., C. C. de Fromentel, and P. May. 1990. Structural
aspectsofthep53protein inrelation to gene evolution. Onco-gene5:945-952.
54. Stahl, H., and R. Knippers. 1987. The simian virus 40 large
tumorantigen. Biochim. Biophys.Acta910:1-10.
55. Staufenbiel, M., and W. Deppert. 1983. Different structural systemsofthenucleusaretargetsforSV40 large T antigen. Cell 33:173-181.
56. Steinmeyer, K., H. Maake, and W. Deppert. 1990. Cell cycle control by p53 in normal (3T3) and chemically transformed (Meth A)mouse cells. I. Regulation ofp53 expression. Onco-gene5:1691-1699.
57. Sturzbecher,H.-W., R. Brain, C. Addison, K. Rudge, M. Remm, M.Grimaldi, E. Keenan, and J. R. Jenkins. 1992. A C-terminal-helixplus basic region-motif is the major structural determinant ofp53tetramerization.Oncogene7:1513-1523.
58. Sturzbecher, H.-W., R. Brain, T. Maimets, C. Addison, K. Rudge, and J. R. Jenkins. 1988. Mousep53blocksSV40DNA replicationin vitro anddownregulatesTantigenDNAhelicase activity. Oncogene3:405-413.
59. Sundin,O., and A. Varshavsky. 1980.Terminalstagesof SV40 DNB replication proceed via multiply intertwined catenated dimers. Cell21:103-114.
60. Sundin, O., and A. Varshavsky. 1981. Arrest of segregation leads to accumulationofhighlyintertwined catenated dimers: dissection ofthe final stages of SV40 DNA replication. Cell 25:659-669.
61. Sweet,B.H., and M. R. Hilleman. 1960. Thevacuolatingvirus. Proc. Soc. Exp. Biol. Med. 105:420-427.
62. Tack, L.C.,J. H. Wright, and E. G.Gurney.1989.Alterations
inthestructureofnewandoldforms of simianvirus 40largeT
antigen(T) defined by age-dependent epitope changes:newTis
thesame asATPase-activeT.J. Virol.63:2352-2356.
63. von der Weth, A., and W. Deppert. 1992. Lytic infection of primaryrhesus kidneycellsby simian virus 40.Virology 189: 334-339.
63a.von derWeth,A.,and W.Deppert.Unpublisheddata. 64. Wang, E. H., P. N. Friedman, and C. Prives. 1989. The murine
p53 proteinblocksreplicationofSV40DNA invitroby inhib-iting the initiation functions of SV40 large T antigen. Cell 57:379-392.
65. Weaver, D. T., S. C.Fields-Berry,and M. L.DePamphilis.1985. The termination regionfor SV40DNAreplication directs the modeofseparationfor the twosiblingmolecules. Cell 41:565-575.
66. Yewdell,J. W., J.V.Gannon,andD. P.Lane.1986. Monoclonal antibodyanalysisofp53expressioninnormal and transformed cells. J. Virol. 59:444-452.