Vol.60,No. 2 JOURNALOFVIROLOGY, Nov. 1986, P. 729-742
0022-538X/86/110729-14$02.00/0
Copyright © 1986, American Society for Microbiology
A
Bovine Papillomavirus
Type
1-Encoded
Modulator Function
Is
Dispensable for
Transient Viral Replication
but
Is
Required
for
Establishment of the Stable
Plasmid State
MONIKA LUSKYt AND MICHAEL R. BOTCHAN*
Department ofMolecular Biology, University of California, Berkeley, California 94720 Received5 June1986/Accepted6 August 1986
Abovinepapillomavirus (BPV) type 1-encodedfunction (M)which isanegativeregulatorof viral plasmid replication has been described elsewhere(Bergetal. Cell, inpress;Roberts and Weintraub, Cell,in press). We reporthere thatexpression ofM, which isa repressorof transient BPV replication and is notrequired as a
positive factor in theseassays,is required for the establishment ofthe viralgenome as astable nuclear plasmid.
This function is encoded inpart by the5' portionof the BPV Elopenreading frame, whereas the3' partof this open reading frame encodes a positive replication function (R). The R function is required for early replicationevents. We usedtransient replicationassaystodefinethe phenotypes ofmutantsin both theRand Mgenesandcomplementation teststoshowthat RandMdefinetwo separategenes.We showed that R- and M- mutants could also complement each other in stableassays. Incotransfection experiments,M- mutants hadalethaleffectonthe growth of G418-resistantcolonies, and inadditiontheirmorphological transformation efficiencieswere reduced. Therarecolonies which didappearcontained themutantDNAintegrated intothe cellular genome. R- mutants transformed with
wild-type
efficiency, and the mutant DNA was also found integrated. Whencotransfected, R- andM- mutantscouldeachbeestablishedasunrearranged plasmids. Most of the viral information involved in latent bovinepapillomavirustype 1 (BPV-1)replicationandmorphological transformation is localizedto a5.4-kilobase (kb)subgenomic
fragment termed the 69% transforming fragment (20) (see
Fig. 1). Regulatorycis-actingsignals and trans-acting
func-tions implicated in viral replication have been identified withinthis region. Forexample, two discrete plasmid main-tenance sequences (PMS-1 and PMS-2) havebeen defined, either of which can support replication of recombinant plasmids in cellsthatprovide viral trans-acting factors (22).
PMS-1 has been localizedto the viral upstream regulatory
region termed the URR (A. Stenlund, G. L. Bream, and M. R. Botchan, submitted for publication) and consists of
two domains (24). Domain 1 is located just 5' to the 69% transforming fragment and has enhancer activity, and do-main 2, mapped by electron microscopy (39)overlapswith theviral
origin
ofreplication. PMS-2 is located withintheElopen
reading
frame(ORF)
(23). Acis-acting siteimplicated
in
negative
control ofreplication
hasrecently been showntooverlap withdomain2of PMS-1. Asecond
negative
control ofreplication
has beenmapped
to sequences just 5' toPMS-2 (32a).
Severaloverlappingsplicedandunspliced
polyadenylated
transcripts
all transcribed fromone strand have been iden-tified (1, 15, 37, 41). Whereastheysharea common3'end,
at least three different 5' ends have been
mapped
(37,41;
Stenlund et al.,
submitted).
The start site for two RNAspecies which contain parts of the El
region
have beenlinked both in vivo and in vitro to a promoter,
P1,
which overlaps domain2ofPMS-1 withintheURR(Stenlund
etal.,
submitted).
Genetic approaches to
identifying
viral geneproducts
required
for latentBPVreplication
have beenguided
by
the* Correspondingauthor.
tPresent address: ZMBH,
University
ofHeidelberg,
D6900 Heidelberg, FederalRepublicofGermany.location of eight ORFs withinthe69%transforming fragment as deduced from the DNA sequence (5). However, this approach bears two inherent problems. (i) Splicing can
combine ORFs in complicated ways. For instance, an
out-of-framespliceeventjoinspartof theBPVE6ORF withpart
ofthe BPV E7 ORF to create the E6/E7 gene (3, 41). (ii)
Many ofthe ORFs present in theviralgenome overlap(Fig.
1).Complementation analysishelps to resolve thisproblem,
since mutations in different ORFs that complement each
other canbeassignedtodifferentgenes.Therefore,by useof smallmutationsaffecting onlysingle ORFs, several comple-mentationgroups thatplayarole in viralplasmidreplication
havebeen identified(2a, 3, 9, 13, 23, 24, 33). A geneproduct encoded in part by the 3' portion of the El ORF has been showntobeabsolutelyrequired for early replicationevents
(24). Moreover, mutations in this region lead invariably to
integration of the mutant viral DNA into the host cell
genome in stable assays (13, 23, 33). The mutant function
canbe
complemented
in transby
awild-type (WT)
gene or mutantsin othercomplementation
groups(23, 24).In an attempt to genetically define the BPV El ORF in
more detail, we introduced frameshift mutations by
using
linker insertions throughout this ORF. The mutants were
analyzed intransientand stableassays. Our results showed
that the BPV El region contains coding information for at least two
complementation
groups. The 3' part of the ElORFencodesa
positive
replication
function(R).
The 5'partoftheElORF encodesamodulator function
(M),
described earlier(2a, 32a),whichcannegatively
regulate
viralreplica-tion. Evidence is
provided
here that shows that themodu-latorfunction,
although
notrequired
for transientreplication
ofBPV DNA, is
required
for its establishment as a latentnuclear
plasmid.
We showed that R- and M- mutantscomplementeach other in stable and transientassays.
Thus,
M- mutants can
provide
Rintransandaccordingly
allow fortransient
replication
of both genomes in C127 cells. R-mutantscanprovide
Mintransand thusnegatively
regulate
729
on November 10, 2019 by guest
http://jvi.asm.org/
IE6
H -I T HinCtlI
7
6959, PMS1, I
l
I E8
EZWII
I E7
I
El
I
I
775 576 EI-Sma d1l0 i2113-2 d1211
711
I .
Sia Bpfit
945
15T'
PMS2EcoRI
2113 2878 3089Ncol
Hpol P1
URR
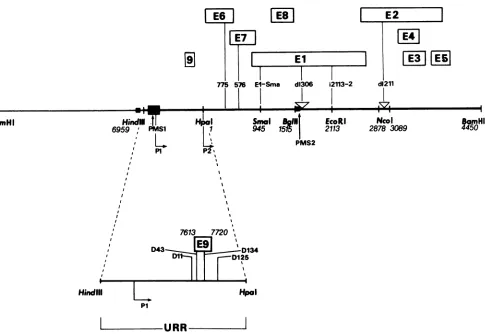
FIG. 1. Organizationof theBPV-1genomeand location of mutations within the viral DNA. Thetopline shows thephysicalmapofthe
BPV-1genomeopenedattheuniqueBamHI site withsomerestriction sites indicated aslandmarks. Theplasmidmaintenancesequences PMS-1 and PMS-2 andtwotranscriptionalpromotersareindicated. The ORFs withinthe 69%transforming fragmentdeduced from theDNA
sequence (5)are shown above themap. The positionsof several BPV mutantsare shownbyvertical barsconnectingthe DNA withthe
particularORF affected. Below themapof thefull-lengthgenome,the location of the E9 ORFwithin the viral URRis shown. Thepositions of mutations in thisregionareindicatedbyvertical bars.
M- mutants in transient assays. In stable transformation assays, bothgenes arerequiredforplasmid replication.
MATERIALS ANDMETHODS
Recombinant plasmids. The plasmidpMLBPV5 (WT)and mutants thereof, dl2ll
(E2-),
BallS (E2- E5-), i2113-2(El-),
dl306(El1-),
have been previously described (23). Since thesteps takentoproduce the Ball5mutanthavenot beencompletely described, wereportthemhere. pMLBPV-5 DNAwas digested and linearized with KpnI. The DNA was then subjected tolimited digestion with Bal 31. XhoI linkerswereaddedtothe ends of thedigested DNA, and the molecules were recloned. Analysis of one clone chosen showed that the 5' border of the deletionextendedto BPV position 2694. This was measured by electrophoresis of small restriction fragments, including HhaI, Sau3A, and SphI. The 3' border of the deletion extended pastthe pMLSalIsiteatpBR position 650. This DNAwaslinearized again with XhoI at the linker site, and the small PstI-BamHI BPV-1fragment which spans the BPV early 3'poly(A) site andenhancerwasblunt end ligatedtothefilled-inXhoI ends. Both XhoI sites were lost upon cloning; however, the BamHI sitewas reformed. For this study, tocompare this
construction with other BPV-1 mutants inserted in pML-1, weexcised the DNA from the plasmid vector with BamHI
and reclonedit into WTpMLDNAattheunique BamHIsite ofthe vector. The 5' boundary of the deletion in mutant Ball5 is then nucleotide 2694 (±5 basepairs [bp]),and the 3' boundaryis definedbythe BPVPstI siteatnucleotide 4173. Mutant d1576 (E6/E7-) has been previously described (23), and mutant 775 (E6-) has a 25-bp deletion between positions 445 and 470, accompanied by insertion ofanXhoI linker (34) and was provided by J. Schiller. Mutants BPV Dll (PWT), BPV D43 (Rsa43), D134, and D125 have a BamHI linker inserted atpositions 7609(Dll), 7620(D43), 7673 (D134), and 7760 (D125). We had initially introduced these mutations intothe BPVsequencesby usingaplasmid carryingthe BPVXbaI-SmaIfragment (24). Full-length BPV genomes were reconstructed by replacement of the WT XbaI-SmaIfragment with each of themutantfragments. We did not change the names of these mutants in this study. Therfore,Dll, D43, D134, and D125 in thispaperrefertoa full-length BPV genome with a single linker insertion, whereas inourpreviousreport(24)thesesame names were given to abacterial plasmid containing only a mutant frag-ment of BPV. Mutant D144 is a full-length BPV genome which has a BamHI linker insertedat position 883 to 884. Similarly, the El Smamutant isidenticaltothe WTexcept fora4-bp insertionatthe SmaI site (2a). A partof the El ORFwas mutagenized by random linker insertion (14). The
BPVSmaI-EcoRI fragment(945to2113)wasinserted intoa
BomHI
I E4 I
1E311E51
40HI
HindIll
A
a
i I I I
I
i
I I
I
I
i I I
I
I I I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.71.556.63.397.2]BPV-1-ENCODED MODULATOR FUNCTION 731
1089
E8S
1479 859
II
I
I
El
I
I|
lI
l813 2663
i i i
Smol Hindl
945 1008 1515SgIII 811D11
EicoRl 2113
C127ceIs
Rplication
-D144
-El-S -D112
-D8
-D28
-D9 D29
I i D18
D13
iI~- -- Dill
II D31
_~~~~~~~~~~~~~~~~-D35i
-d130
-D4
D26
-i211l
4 883/884 +
Sma 945 +
+1001-1006 +
A 952-1030 +
1043-1071 + 113-1121 + +1123-1132 +
A12381279
+1-A1280-1287
+1-I A1361 -1416
+1-A1406-1430
-i+1404-1417
-X61515-1811
+1-A1709 +18071830
3-2 2113
-WT
91% 90% 84%
92%
74% 82% 92% 4% 18% 22%
0 0
9%y
0 0 0
[image:3.612.67.560.64.339.2]100%
FIG. 2. Deletionand linker insertion mutations within the BPV El ORF:mappositions and transient replication properties in C127 cells. Thetopline showspartof the physicalmapofBPV-1 DNA.Thelocations ofORF E7, El, and E8areindicated above. Vertical bars within
theElORF indicatethe positionsofinternal ATG codonsatpositions 849, 1506, 1596, 1632, 1794, 1938, 1980, 2169, and2619.Thepositions ofthe mutations described here areshown below. A slash (/) betweentwonucleotide positions (D144)refers toasimple linkerinsertion
betweenthesetwopositions. Linker insertionsaccompanied by deletionof adjacentsequences areindicated byaA(delta) followed by the
deletedsequences;for example, inD9 thenucleotides 1113to1121,and thus9bp,aredeleted. Duplications ofsequences areindicated by
a + followed by the nucleotides whichare duplicated; for example, linker insertion in D112 occurred with aduplication of6 bp of the nucleotides 1001to1006.The replicationproperties ofallmutantDNAsin transientassays areshownonthefar right.Theautoradiograms
obtainedfromreplicationassays werequantitatedbydensitometry. Replicationofmutantsis expressed relativetoWTpMLBPV, andzero
indicates thatnoDpnI-resistant supercoils could be detected. The results presentedaretheaverageofthreeseparateexperiments, and results
variedby ±5%.Inno case,however,wasreplication detected for themutantsshowntohave0% replication potential.
pML2-derived vector, pp3 (21, 24), between the SmaI and EcoRIsites of thevector,giving risetopp3SR.pp3SRDNA was randomly cleaved with DNase, and the double-stranded,syntheticoctamer5'CGGATCCG3' (New England BioLabs) encoding the BamHI recognition sitewasinserted at the double-stranded breaks. Sequence analysis of the linker mutationswas done by standard procedures (2, 26). The map and nucleotide positions for the inserted BamHI linker in the individual mutants are shown in Fig. 2. Each
mutantfragmentwas then usedtoreplace theWTfragment in thefull-length BPVgenome.
Cellsand DNAtransfections. C127(10), 576, and775 cells were maintained atlow cell densityin DME plus 10%fetal calfserum.576 cells(3, 23)areC127derivatives and contain one to five copies of the BPV 576 (E6/E7) mutant and, similarly,775cells containonetofivecopiesof the BPV 775 (E6)mutant.For stable and transienttransfections,0.5to20 ,ug of plasmid DNAwasappliedto2 x 105to3 x
105
cellsper 60-mm (diameter) dish in the presence of 30 ,ug of Polybrene (18) per ml, followed by a 22.5% dimethyl sulfoxideshock 6 h after transfection. Forfocus assays, the medium was changed once per week. For selection of G418-resistant colonies, the cells from each 60-mm dish were trypsinized 48 h after transfection and split onto two 100-mm (diameter) dishes each. Selection was done in the
presence of 500 ,ug of G418 per ml (6). Foci and
G418-resistant colonies werecounted after 3 weeks unless other-wiseindicated.
Isolation and analysis of cellular DNA. Total DNAs from morphologically transformed and G418-resistant cells were prepared by the standard modifications (25) of the procedure of Thomasetal. (38). The DNAswere analyzed uncleaved or cleaved with the indicated restriction enzymes. For transientreplicationassays,low-molecular-weight DNAwas extracted from Hirt (16) supernatants at 24-h intervals, starting either 24 or 72 h after dimethyl sulfoxide shock. DNAsamples from theHirt supernatantsweredigestedwith DpnI, which cleaves unreplicated (methylated) DNA (31). DNAanalysis by gel electrophoresis and Southern blotting were aspreviously described (22 and referencestherein).
RESULTS
DeletionofBPV3'earlyORFs and transientreplication. A transient assay for BPV replication has been previously describedwhich allows formeasurementofviralreplication ofcellswithout selection (24). We havepreviously reported that coding information 3' to the BPV El ORF was not absolutely required for plasmid maintenance. A mutant (BallS) lacking ORFs E2, E3, E4, and E5 was maintained stablyin C127 cells(22). A mutant
(dl211)
defectiveonly in the E2 ORF could, under certain conditions (G418selec-U I
0i i
i
VOL.60, 1986
on November 10, 2019 by guest
http://jvi.asm.org/
M3 BaI5 r----
---1
40
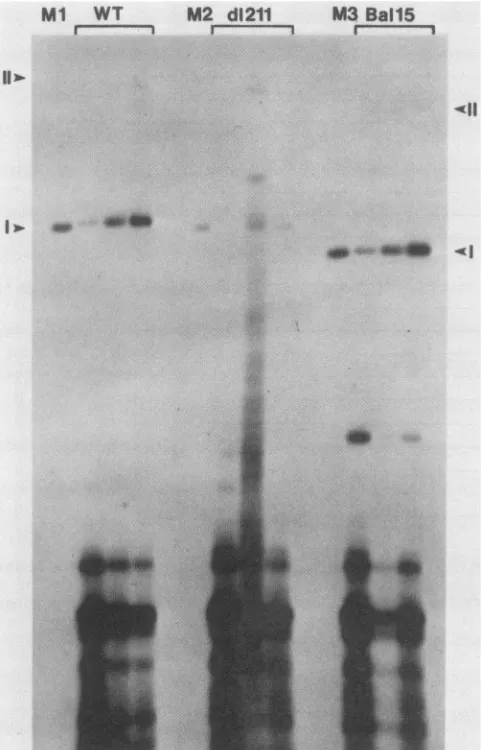
...FIG. 3. Transient
replication
ofWTpMLBPV5
and themutantsd1211
and Bal15in C127 cells. After DNAtransfection, DNAwasisolated from Hirt supernatants every 24 h,
starting
72 h afterdimethyl
sulfoxide shock(e.g.,
72,96,and 120h,lefttoright
in eachpanel). Equal
fractions of eachsample
wereanalyzed.
Beforegel
electrophoresis,
the DNAs were cleaved withDpnI
todigest
theunreplicated (methylated) input
DNA. Aftergel
electrophoresis
through
0.9% agarose and transfer tonitrocellulose,
hybridization
wasdone with106countsof
32P-labeled,
nick-translatedpMLBPV5
DNA(specific
activity,
2 x 108to x 108dpm/,ug
ofDNA).Thisprobe
wasused for allexperiments.
LanesMland M3 contained 200 pgeach ofWTpMLBPV5
andBall5plasmid
DNAs,respectively.
LaneM2contained 20pgof
d1211
as amarker. Arrowheadstothe left andright
indicate thepositions
of form I(supercoiled)
and II(nicked circle)
WTand BallSDNAs,respectively. DpnI-sensitive
DNA
(unreplicated)
was at the bottom of thegel. Hybridization
throughout
the lane ofthe secondtimepoint
inthed1211 panelwas duetopartial DpnI
digestion.
tion),
be maintained as aplasmid; however,
when selectedfor
morphological transformation,
the DNAwasfoundinte-grated
(23).
Tosubstantiateourprevious
resultsandfurtherprobe
thereplication phenotypes
of thesemutants,itwasof interesttocomparethe initialreplication
properties
of bothmutants side
by
side. WTpMLBPV5,
BallS,
andd1211
DNAs were transfected into C127 cells
(1
,ug of DNA per 60-mmdish).
Low-molecular-weight
DNAwas extractedat 24-h intervals anddigested
withDpnI,
which cleavesonly
nonreplicated
(methylated)
DNA(31).
Thus,
accumulationofsupercoiled DNA over time is a measure of DNA
repli-cation. Three time
points
fromeach transfection(72, 96,
and 120h)
are shown inFig.
3. Theexperiment
showed thatreplication
of mutant BallS isindistinguishable
from that seenwith WT BPVDNA,
whereasmutantdl211
replicates
at a much lower rate(see
also reference24).
Densitometricscanning
of theautoradiogram
showed that accumulation ofBallS DNA was 97% of that seen with WT BPV
DNA,
whereas d1211 DNA accumulated to
only
11% ofthe level seenwith WT DNA. From thesedata,
weconclude that this mutationsolely
within the E2 ORF affects therateofearly
replication events, whereasdeletion ofthe entiresequences
E2throughE5 didnot
impair
theinitialreplication
properties
of the BPVgenome.
Only
partof the ElORF isrequired
for transientreplica-tion. Atrans-actingfunctionencoded within theEl ORF had
beenimplicatedin stable
plasmid
maintenance(13,
23,
33).
Furthermore,aBPV mutant,i2113-2
(23)
(Fig. 1),
within this complementation group had been shown tobe defective in transient replication(24),
indicating
that the functionaf-fectedwas
required
forearly
replication
events. Toanalyze
the El ORF in more
detail,
we chose to introduce smallframeshift mutationsthroughoutamajorpartof theEl ORF. Synthetic BamHIlinkers were inserted atrandom
positions
(14) within the BPV Smal-EcoRI
fragment (see
Materialsand Methods). The nucleotidepositions of the linker
inser-tion mutants described here are shown in
Fig.
2. Eachmutant fragment was used to replace the appropriate WT fragment in the full-length BPV genome. The
replication
propertiesof thenewly created linker insertionmutantswere assayedinitiallyin transientexperiments. AllmutantDNAs were transfected into C127 cells. Their behavior in the transient assays is summarized in
Fig.
2.Representative
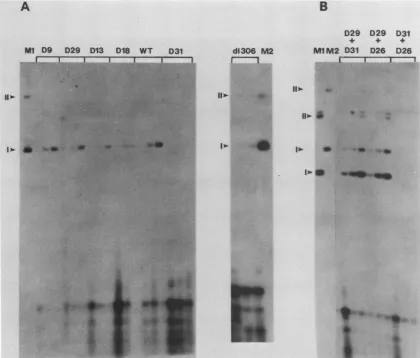
replication assays for the mutants D9, D29,D13,D18,
D31,
and d1306 are shown in Fig. 4A. Three time
points
(72,96,
and120 h) for eachtransfection areshown. Toour
surprise,
wefound that mutants D144 through D29replicated in the
transient assay in a way similar to thatof WT BPV. Mutants D13and Dlll werealso able toreplicate; however, therate of DNAaccumulationseenwiththesemutantsseemedtobe
delayed andquantitatively less whencompared with that of
the WT. Both D13 and Dlll are in-frame
deletion-substitution mutants, and thus, partial activity of a trans-actingfactor may beexpected.However, mutant D18,which
does have aframeshift mutation, alsoreplicated at alower
ratethan WT BPV and mutants that scored positive. Since
theframe shift in D18 would lead to deletion of information for this gene encoded 3' to the mutation (see below), we believe that an internal ATG may be used to initiate a fragment of the protein which has partial activity. The sequence and position of the frame shift for mutant D18
predictsatermination codon at BPV nucleotide 1480, and a potential reinitiation codon in frame with the rest of El is
positioned just5' tothis point at nucleotide 1506. Peabody
and Berg (29, 30) have recently presented compelling data
consistent witha reinitiation (as opposed to scanning)
mech-anism for eucaryote translational use of internal AUGs. However, theyhave shown that the position of the termina-torwith respect to the AUG is critical. If the terminator is too far from the AUG, reinitiation is notdetected. In this regard, it is interesting that our mutantsD31and D35, which are completely negative for replication, have frame shifts
which predict termination at nucleotides 1460 and 1426,
respectively. Therefore, a simple resolution to the apparent
quantitativedifferences between these mutants and D18 may be that thetermination codons for mutants D31 and D35 are
Ml WT
m- M2 d1211
11I
I
_4I4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.60.301.68.443.2]BPV-1-ENCODED MODULATOR FUNCTION 733
A
B
Ml D9 D29 D13
I I I
B WT D31
I[ I' .
dl1306
MJ2
.
D29 D29 D31
MIMU2
D31 D26 D26XI Xh . I....
If . ...
..)~ :. ...2SE'..::
,
.8
...,
rs....
..K It
FIG. 4. (A)Transient DNA replication of BPV ElmutantsD9,D29, D13, D18, D31,d1306, and WT pMLBPV5 in C127 cells. Time points ofDNAisolationand analysiswere asdescribed in the legendtoFig.3. Lanes MlandM2contained200pgeachofpMLBPV5andd1306
DNAs, respectively. Forms I andIIareindicated by arrowheads.(B)TransientDNAreplication of BPVElmutantsincotransfectionassays:
D29plus D31, D29 plus D26, and D31 plus D26. The molar ratio ofthecotransfectedDNAswas1:1 (0.5 ,ug ofuncutplasmid DNA plus 0.4 ,ugofcutplasmid DNA). For cotransfection of D29 plus D31, the BPV DNA in D29wasremoved from thepMLvectorplasmid by partial BamHI cleavage; for the othertwo cotransfections, the BPV DNAin D26was removed from the vector sequences in the same way.
Therefore,thecotransfectedDNAswereofdifferent sizes. Lanes Ml and M2 contained 2,ugof cellularID13DNAand 200pgofpMLBPV5 plaSmid DNA, respectively; forms I and II of the marker DNAsareindicatedbyarrowheads.
too far from the AUG at 1506 to allow for significant reinitiation. Clearly, further work,particularlyidentification of the protein encoded by this gene, will be needed to substantiate thishypothesis. Mutants D4 andD26 failed to showanyDpnIresistance and thusreplicatedDNA. Mutant d1306 was able to replicate in this assay, although with slower kineticsandto alowerlevel than did the WT. Using stableassays, wehave earlierreportedthat this mutant, an in-framedeletion, haspartial activity (23).
From the data desctibed above it is not possible to distinguish whether mutationswhich do notreplicate lie in cis-acting signals oraffect trans-actingfactors. To address thisissue, weperformedcomplementationtests. Ifdifferent trans-acting functions areaffectedbymutationsthatscored positive and those that scored negative, then a positive
mutantshouldrescuethereplication deficiencyofanegative
mttant.The mutantswerepairwise cotransfectedwith each other. The result ofarepresentative experimentis shown in Fig. 4B.The molar ratio of thecotransfectingDNAswas1:1
foreachexperiment.Forcotransfectionof D29plus D31,the
BPV DNA in D29 was removed from the pML vector by partial BamHI cleavage; for the othertwo cotransfections, the BPV DNA in D26 was removed from the vector se-quences in the same way. Thus, the cotransfected DNAs wereof different sizes. Three time points (72, 96, and120h) were taken from each transfection. The experiment shows that cotransfection of D29 plus D31 and D29 plus D26 resulted in replication ofboth transfected DNAs. In con-trast, cotransfection ofD26plus D31 resulted inno
appear-anceofreplicatingDpnI-resistantDNA ofeither mutant. Weconcluded that thereplication deficiency of the repli-cation-negative mutants (D31 through D26) can be comple-mentedintrans. Mutants i2113-2 and i2405-5(23, 24)did not replicatein transientreplicationassays anddid not comple-mentmutantD31for transientreplication (datanotshown); therefore, they are all part of the same complementation group. MutantsD144through D29definea second comple-mentationgroup within theEl ORF (see below). The func-tion encoded bythis complementation groupis dispensable forinitialreplication events.
11 VOL.60, 1986
....f
It.t.
:,*.,;on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.105.525.70.428.2]5762-cells
775-cells
M1 M2 M3 WT D43 D144 D43 D144 WT
I I I I I I
1I1) ID- ^
d
<IIIresident
DNA
s~~~~~~~~~~~~~~~V
...'
zeS-FIG. 5. Transient replication assay of BPV mutants D43 (E9) and D144 (5' El) and WT pMLBPV5 in 576 and 775 cells. Before transfection, all BPV DNAswereremovedfrom thevectorplasmidby complete(WT)orpartial (D43andD144)BamHI
cleavage.
Therefore, transfectedDNAs wereofadifferent size(8kb)than residentmutantDNAs(10.6kb).AfterDNAisolation(time
points
were asdescribed in thelegendtoFig.3), eachsamplewasdigestedwithDpnIand PvuI beforegelelectrophoresis,resultingindigestion
of allunreplicated
DNA (DpnI)and linearization of theresidentd1576and 775 DNAs(PvuI).ThePvuIsite is located in thepMLvectorsequencesandwastherefore not present in the transfected DNAs. Thus, replication can be scored as accumulation of supercoiled DNA. Gel electrophoresis and hybridizationwere asdescribed inthelegendtoFig.3. LanesMl,M2,andM3contained2,ug of cellularID13DNAand 200pg each ofuncut(M2) and linear (M3) pMLBPV5DNAasmarkers.FormsI, II, andIIIof the markerDNAsareindicatedbyarrowheadsontheleft,and the arrowheadontheright indicates linear residentmutantBPV DNAs(dl576and775).
5' El mutants and mutations within the URR define a negative modulator function in transient assays. In results
presented elsewherewe have shown thattransient
replica-tion ofsupertransfectedWT BPV DNAisrepressedincells
(576 and 775) carrying a
low-copy-number
mutant of BPV(2a). Establishment of high-copy-number WT genomes in thesecells isalsoinhibited(2a, 3). Surprisingly,it was found that a frameshift mutant,
El-Sma
(containing a4-bpinser-tion atthe BPV SmaI site), could replicate in these cells.
Furthermore, sinceWT DNA whencotransfected with this
particular mutant inhibits replication of both genomes, we reasoned that BPV encodes for a negative modulator
func-tion(M) whichcan actin trans. The results presented here extend theseobservations.
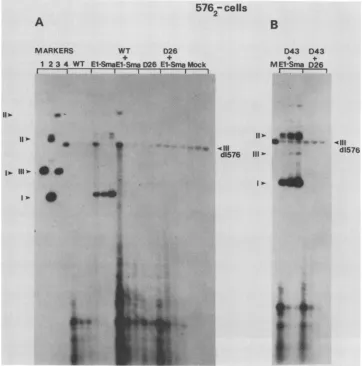
Wefound thatmutations(D43 and D134[Fig. 1]) in a small ORF(E9) within the URR also affect this 5' El gene (see
below).Asdescribed elsewhere(2a), aphenotype of mutants
in this 5' El gene is that they replicate transiently in cells
harboring BPV low-copy-number mutants. This assay dis-tinguishes a unique phenotype of this gene in a quick and convenient way and is therefore usedas a primarymethod forscreening mutants which may be members of the same complementation group. A representative experiment is shown inFig.5. As can be seen with WTBPV,noreplicated supercoils couldbe detected in transient assays after trans-fection of either 576 or775 cells (see also2a). In contrast, mutants D43 (E9-) and D144 (5' El-) replicated equally wellin either cell line. Identical results were obtained with mutantsD134, D112, D28, D9, and D29. Avarietyof other mutants, including Dll, D125, d1576, 775, and Bal15, be-haved as did the WT in these cells; i.e., they did not
replicate. As weexpected, all mutants that are R- also did notreplicate in these cells (data not shown). To determine whether the E9 and 5' El mutation affected a trans-acting
function, we contransfected themtogether with 3' El mu-tants (R-) into 576 cells. We reasoned that both types of ..
I" jijz 4
.4 A&
r
I A:...
auki
IV. B.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.139.474.68.455.2]BPV-1-ENCODED MODULATOR FUNCTION 735
5762-
cells
A
B
MARKERS WT D26
+b 4
1 23 4 WT EI-SfaEl-SmaD26 El-SmaMock
I I I I I
D43 043 MEl-Sma
a_
D26I I
_ .
11W
a
0 t+
-v * 4111
d1576
1l)-or '0W.
(III d1576 Iw. 111)-.
1w .
1'
.4
_..-P.
^--'.. ..
I
I
FIG. 6. Complementation of BPV mutants in transient replication assays in 576 cells. (A) Complementation of the El-Sma mutant (5' El) by WT pMLBPV and D26 (3'El).Before transfection, allBPV DNAs were removed from the plasmid vector by complete (WT andE1-Sma) orpartial (D26)BamHIcleavage. The molar ratio of thecotransfected DNAs was 1:1. Time points were taken as described in the legend to Fig. 3. Each DNA sample was cleaved either with DpnI plus PvuI (WT and D26lanes;see thelegend to Fig. 5) orDpnIplusSmaI (E1-Sma, WT+ El-Sma,and D26 +E1-Smalanes).SmaIdid notcleaveE1-SmaDNA but linearized theWT, D26, and residentd1576DNAs.Thus, replication could be measured as accumulation of supercoiledE1-SmaDNA and accumulation oflinear WT or D26 DNA in the cotransfection experiments.Gelelectrophoresis and hybridization were as described in the legend to Fig. 3. The lanes marked Mock contained DNA samples of timepoints taken after mock transfection of 576 cells and cleaved withDpnIplusPvuI.The markerlanes contained 2 ,ug ofHindIll-digested cellular ID13 DNA,resulting in linearization of the resident viralBPV DNA(Ml);2 ,ugof uncut celluar ID13 DNA(M2);and 200 pgeach ofuncut(M3) and linearized pMLBPV5 DNA(M4). Forms I, II, and III of viral BPV DNA resident in ID13 cells and forms I andIIof pMLBPV DNAare indicated by arrowheads onthe left; the arrowhead onthe right indicates form III of the resident d1576DNA. (B) Replicationof El-Sma and D43mutantsandcomplementation ofmutantD43bymutantD26in 576 cells.Beforetransfection,allBPVDNAs wereremoved from thevectorsequences (seeabove). After DNAextraction, each samplewascleaved withDpnIplusSmaI(D43 + E1-Sma lanes)orDpnIplus PvuI (D43 + D26lanes). The marker lane contained 200 pg of linearizedpMLBPV5DNA.The arrowheadtotheright indicates linear resident576 DNA.The arrowheadstothe left indicateformsIandII ofreplicatingEl-SmaDNAandformIIIofreplicating D43 DNA.
genome should be inactivatedfor
replication
ifR- mutantsencode
for
theputative negative modulator function,
M.Theresults of these experiments are shownin Fig. 6.
Cotrans-fection of D26 (R -) with the E1-Sma mutant
(Fig.
6A) abolished replication of El-Sma DNA.Similarly,
cotrans-fection ofD26 with D43 (E9-)
(Fig.
6B) led to shutoff of replication of D43DNAin576 cells. Thesameresultswereobtained with the 5' El mutants
D144, D28,
and D29(data
not shown). In contrast, cotransfection of D43 DNA with
El-Sma DNA resulted in
replication
of both mutant genomestogetherin thesecells.Thus,
this assayestablishedthatE9and5'El mutations affectthesamegenes.
Further-more,sinceD43and El-Smamutantsboth could
replicate
in C127 cells(Fig.
7),they
mustbeR+.We concludedfromthese
experiments
that BPVencodes anegativetrans-acting
factor. 3' El mutants which cannotreplicatecan provide this factorin trans and are therefore
R- yet M+. Pairwise cotransfections with 3' El mutants
defined E9 mutants and mutants
D144,
El-Sma, D28,
and D29asM-.Transfectionof E9 and 5' Elmutantsinto 576or C127 cells ledtoreplication; thus, they
areR+ yetM-.M- mutants aredefectivefortransformation andlethalto cellgrowth.The
experiments
described abovedemonstratedoneaspectoftheM-
phenotype-the
ability
of M- mutants VOL.60, 1986on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.131.493.68.434.2]FIG. 7. TransientreplicationofmutantsD43(E9),D144(5' El), El-Sma (5' El), and WT pMLBPV5 in C127 cells. After DNA transfection, low-molecular-weight DNAwas isolated every 24 h starting 24 h aftertransfection (e.g., 24, 48, 72, and 96 h). DNA analysiswas asdescribedinthelegendtoFig.3. Lane Mcontained 200pgof WTpMLBPV5 plasmidDNA. FormsI,II,andIII(linear
DNA)areindicatedbyarrowheads onthe left.
to replicate transiently in an environment where WT BPV does not. It was of interest to determine whether these mutants could establish themselves stably in transformed cells. When stable transformation assays were performed,
TABLE 1. Focusformation of URR and Elmutants
inC127 cells
No.of foci/jig ofDNA
Plasmid Mutation 1 lig 10,ug 20,ug
Expt ia Expt 2a Expt 2 Expt 1 Expt 2
pMLBPV5 None 136 143 TMTCb TMTC TMTC
(WT)
D1l (qWT) URR 145 141 TMTC TMTC TMTC
D43 URR-E9 0 0 1 1 2
D134 URR-E9 0 0 2 2 3
D125 URR 123 117
D144 El 0 0 0 2 1
El-Sma El 0 0 2 3 1
i2113-2 El 105 TMTC TMTC
aExpts 1 and 2 referto two separateexperiments performed ondifferent
days. Each number is theaverageof the colonies scoredontwoplates. Foci
werecounted20 days aftertransfection.
bTMTC, Toomanyto count.
wefoundtooursurprisethattheabilities ofM- mutantsto
induce foci were reduced at least 100-fold
compared
with that ofthe WT(Tables
1 and 2). Fromthese results,
twointerpretations
werepossible.
Either the E9 and 5' El mutationsimpaired
a function directly required foronco-genic
transformation
orthey definedanimportant
regulatory
genewhoseabsence would render
establishment
of
the BPVgenome
impossible.
Forinstance,
in the absence of the negative modulatorfunction,
detectedby
the transient as-says(seeabove), BPV infection couldbe
lethal. The resultsof
cotransfection experiments
with M- mutants with anintegrating marker, the neor gene, are consistent with this latter view. In these
experiments
M- mutants D43,D134,
D144, E1-Sma, D8, D28, and D29 were
cotransfected
with theneormarker, and the number ofG418-resistant colonies
wasscored. Parallel
cotransfections
of the neor marker withWTBPV anda mutantDll
(PWT
[Fig. 1])wereperformed.
Representative results are shown in Fig. 8. In all of the experiments, the absoluteamountof neor marker DNAwas
kept constant (0.5 p,g of DNA per 3 x
105
cells), and increasingamountsofmutant orWTDNAwereadded
tothe transfection mixtureattheindicated
molarratios.Figure 8A showsthat, with increasingconcentration
ofWT BPVard
TWT, the number of G418-resistant colonies actually in-creased
slightly.
In contrast,increasing
the dosage of M-mutant DNAsclearly
interfered with the growth of G418-resistant colonies. Atratiosof 20:1 (BPVmutantDNA-neor
DNA), we
observed
atleast a 200-fold difference between the number of coloniesobtained
with WT versus mutant DNA.Theincompletenatureof interference atlowerdoses iscertainlythe sumof several differenteffects,including the frequency ofcotransformation (which isapproximately
80%,
this sets a fivefold upper limit on the effect at 1:1
ratios).
Furthermore, this
lethality
isclearlyaslowkineticphenom-enon
(Fig.
8B). At 12days
afterselection, the difference in the number ofappearing
G418-resistant colonies between the WT and mutants was not nearly as dramatic as that observed 24 days after selection. Thus, although G418-resistant colonies initially appeared, they died before 3weeks. This could mean that a few cell doublings were
allowedtooccurbefore
cell
deathbecame manifest, which inturn implies that the lethality may not be absolute. There-fore, atthelevelsofBPV DNAused in the1:1mixture with
TABLE 2. Focusformation ofEl
mutant§
inC127 cellsPlasmid No. of foci/
5 jigof DNA' WTb.*---'---'--- 157C
D12... 1
D8... 0
D28... 3
D9... 1
D29
...0
D18... 4
D13... $6
Dlll... 71
D31... 95
D35... 98
dl306... 165d D4... 142
D26... 131
i2113-2... 129
aFociwerecounted20daysaftertransfection.
bThe
WT
carriesnomutation.cNumber of foci per1,ugofDNA.
dNumber offoci per0.1 ,ugofDNA.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.316.556.529.699.2]BPV-1-ENCODED MODULATOR FUNCTION 737 the neormarker, the absolute amount ofBPV DNA maynot
begreat enough to elicit complete lethality.
To examine the state of the mutant BPV DNAs in the surviving G418-resistant cells, we picked colonies three
weeks after selection and expanded them into cell lines. Attemptsto isolate colonies and expand them into cultures before this time inevitably failed because of cell death. Figure 9 shows the results of Southern blots obtained with mutants D43 (E9-, lanes A to D) andD144(5' El-, lanesE to H). Thecellular DNAs were analyzed uncut. In no case
could supercoiled plasmid DNA be detected, but rather all
hybridizing BPV DNA migrated with the
high-molecular-weight cellular DNA. The same results were obtained with mutantD134 (E9-) and the 5' El mutant El-Sma (data not
shown). Furthermore, the integrated state of these mutant BPVDNAs in stable assays was independent of the molar ratio of neor DNA to BPV DNA used for transfection. These
A
200
z
0
0
0 0
100
0L t
Ci t
--b- WT
-D1I IVWTI
0-D144 _- EI-Sma
O-043
1:1 1:2 1:5 110
D43+
neoD144+neo
M
A B
C
D
E
F
G H
I
1II
[image:9.612.317.559.65.326.2]I
FIG. 9. Southern blot analysis of total cellular DNAs from G418-resistant C127 cells derived by cotransfection ofpNEO5' DNAplus the mutants D43 (E9) and D144 (5'El). Cotransfection was done at a 1:10 molar ratio of marker DNA to mutant BPV plasmidDNA with0.5,ugofpNEO5'DNA.Total cellular DNA was isolated from individual G418-resistant colonies which had been picked and expanded into cell lines 26 days after transfection. Gel electrophoresis and blot and hybridization analyses were as de-scribed in thelegendtoFig.3.LanesAthrough H eachcontained10
jig
ofundigested cellularDNA. Lane Mcontained 100 pg (5 copy equivalent) ofpMLBPV5plasmid DNA. The arrowheads on the left indicate thepositionsofDNAforms IandII.MOLAR RATIO pNEOS'NA BP DNA
B
WT
pMLIPV
043
12days iBdeas 24days
FIG. 8. Cotransfection of BPV E9 and 5' El mutants with the neomarkergeneon pNEO5'. (A)Effect ofincreasing BPV DNA
dosageonthenumber of G418-resistantcolonies. TheDNAswere cotransfectedattheindicatedmolar ratios with 0.5 ,ugofpNEO5' DNA. The values of each curve represent the average of two independent experiments for each cotransfection. Colonies were counted 20 days after transfection. (B) Representative plates of
G418-resistant C127 cellsstained12,18,or24daysaftertransfection
with the indicated DNA. The cellswere fixed withglutaraldehyde and stained with 1%methyleneblue. Cotransfectionwasdoneata
pNEO5'DNA-BPVplasmidDNA molar ratio of 1:10.
results showed that the M- mutants could not be stably maintainedas
plasmids.
Weconcludedthat the Mfunction isrequired
for establishment of latentBPVplasmid replication.
Todeterminewhether theseparticular phenotypes of M-mutants
(lethality
andintegration)
wereindeed duetoloss of function ofatrans-acting factor,
weperformed
complemen-tationassays. Theresultsareshownin Tables 3 and4.
First,
variousM- mutants were
cotransfected
withmutantsin the E6/E7 and E6complementation
groups(d1576
and775,
respectively),
andability
to induce foci was measured.Mutants D43,
D134, D144,
and E1-Sma could allcomple-mentthemutantsd1576 and775fortransformation
(Table 3).
Thetransformation
efficiency
wasatWTlevels,
demonstrat-ing that the M- mutants were not dominant lethal. In contrast,cotransfection ofM-mutantswith each other
only
induced few fociat
high
DNAconcentrations.Todetermine whetherthelethalphenotype
ofM- mutants could also berescued by an R- mutant we
performed
thefollowing
transfection
experiments.
The mutantsD43, D134,
D144,
and E1-Sma
(M-)
were each cotransfected with mutanti2113-2
(R-) andtheintegrating
neormarkergene.Withtheappropriate
transfectionmixes,
WTlevels ofG418-resistant colonieswere detected(Table 4). Futhermore,
observationofthe
appearing
coloniesovertime didnotshowanylossinnumber or cell
death,
and the colonies werereadily
ex-panded
into cell lines. Notunexpectedly,
crosses between twoM- mutantsdidnotleadtocomplementation (Table 4).
VOL.60, 1986I
AWWL14 #
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.59.301.252.626.2]TABLE 3. Complementationof BPV mutants in C127 cells-focusformation
No. offoci/,ugof DNAb Plasmida Mutation
Expt1 Expt2
pMLBPV5 (WT) None 156 123
d1576 E6/E7 10 12
775 E6 12 9
D43 + dl576 E9 x E6/E7 101 95
D43 + 775 E9 xE6 124 112
D134 + d1576 E9 x E6/E7 98 100
D134 + 775 E9 xE6 105 111
D144 + d1576 5'El x E6/E7 125 113
D144 + 775 5'El x E6 112 110
E1-Sma + d1576 5'El x E6/E7 125
ElSma + 775 5' Elx E6 128
D43 + D134c E9 x E9 2 3
D43 + D144c E9 x 5' El 1 0
D144 + El-Smac 5'El x 5'El 2 2
aSee Table1for the transformationefficiencyof each individual E9orEl
mutant.
bThe molarratio ofcotransfecting DNAswas1:1. Fociwerecounted 20 days after transfection.
cNumberof foci per 10
pLg
ofDNA.For example, cotransfection of D43 with El-Sma and
pNEO5'
ledto atleasta20-fold reductionin G418-resistant colonies. Thesameresultwasobtainedwhen D43 DNAwaslinked to the neor gene (D43-neo) and cotransfected with El-Sma (Table 4). Lack of
complementation
between M-mutants wasalsomanifestwhen BPV DNAwasanalyzed
inthe rarely appearing G418-resistant colonies.
Figure
10B shows the DNA analysis of G418-resistant cells from thecotransfection experiment with D43-neo plus El-Sma. The
Southern blot showed that bothmutant DNAs appeared to
existasintegrated copies in genomicDNA. In contrastand consistentwith ourresultsdescribed in transientassays,Fig.
10Ashowsthat R-
MI
x R+ M- crosses(e.g., i2113-2 x D43 andi2113-2
x D144)yielded cell lines whichcarrybothmutantDNAs asunrearranged plasmids.
Thus, in stable assays M- mutants D43, D144, and El-Sma all integrated as did R- mutants. Together, they complemented each other and established themselves as
latentreplicating plasmids.
DISCUSSION
The results presented abovedescribe a new BPV gene (M)
required forestablishment of stable plasmids in transformed cells. We showed that this gene is encoded in part by sequenceswhichmap to the 5' end of theElORF.
Hereto-fore,
onehadassumed that theentireEl
ORF was involved inencodingasinglefunction, since its size and location areintact and conserved among all of the papillomavirus genomes sequenced which have replication potential (7).
This new gene is clearly distinct from the positive factor
previously defined which is required in trans for transient BPV DNA replication (24). The positive factor (R) is en-coded in part by the 3' sequences of this sameORF. We are
confidentthat the two genes, R and M, must define separate
polypeptides, since a series of frameshift insertions at the 5' end ofEl do not destroy R activity. We used a series of assays to demonstrate the uniqueness of eachfactor and to
probe theirphenotypes. These assays are summarized be-low.
(i) WT BPV DNA or mutants in other ORFs do not
replicate in transformed cells which carrylow-copy-number
mutants (2a,
3).
Mutations in the 5' part ofEl, however,
allowed for
replication
aftersupertransfection
into these cells.Furthermore,
these mutantsreplicated
in uninfected C127 cells.(ii)
Stablemorphological
transformation wasseverely
re-duced with mutants that
interrupt
thecoding
sequences inthe 5'
portion
oftheElORF,
and thesemutantsinterfered incotransformation
experiments
with theoutgrowth
of G418-resistantcolonies. Incolonies that could be established with suchmutants, the DNAwasintegrated
in cellular DNA.(iii)
Mutations in El ORF sequences 3' to this gene, incontrast, did not
replicate
in transient or stable assays and didnotaffecttransformation efficiencies.The
phenotypes
described in i and ii define theproperties
ofM-mutants,whereas those in iii describe the
phenotypes
ofR- mutants. We used the
following
complementation
tests toshow thatR and Mdefine two separate genes.
R-mutantsdidnot
replicate
transiently
ineitherC127
cells orlow-copy-number
cellsbut,
cotransfectedtogether
with M-mutants, both genomesreplicated
inC127
since the latter classofmutantprovided
thepositive
replication
function.Inlow-copy-number cells,
neither genome couldreplicate
when cotransfected because R- mutants
provided
theneg-ative
regulator.
In stable transformation assays, neithergenome was
carried
as a freeplasmid
when transfectedseparately,
buttogether they
complemented
each other.The outlines of a model for BPV
replication
control,
asdescribed
by Berg
et al.(2a),
provide
the basis forunder-standing
theassays used here todefine these twogenes. Inparticular,
thismodelprovides
ahypothesis
useful indesign-ing
futureexperiments
aimed atprobing
the role ofM in stable transformation. Theexperiments
described herecer-tainly
suggest that this new geneplays
some role in theestablishment
(and
perhaps
maintenance)
of theplasmid
state and transformation.
Briefly,
the model suggeststhat,
afterinfectionortransfection of naive
cells,
BPVundergoes
slow but transient
amplification;
thisamplification
is fol-lowedby
amaintenance mode ofreplication
which ischar-acterized
by
coordinate cellcycle
replication
of viralplas-mids.The M
protein
eitherdirectly
orindirectly
assuresthis modeofreplication
evenin thepresenceofanexcessofR.Wedonotknow how theM
protein
worksbuthypothesized
that it is a direct
DNA-binding protein
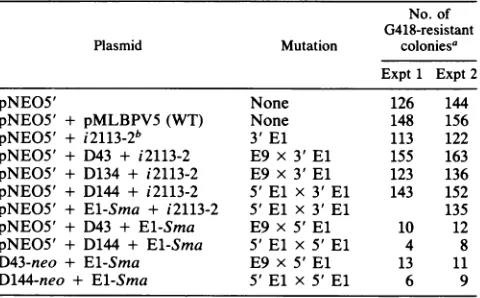
(see below). SomeTABLE 4. Complementationof BPVmutantsinC127 cells
No. of G418-resistant
Plasmid Mutation coloniesa
Expt1 Expt2
pNE05' None 126 144
pNE05' + pMLBPV5 (WT) None 148 156
pNE05' + i2113-2b 3'El 113 122
pNE05' + D43 + i2113-2 E9 x 3'El 155 163 pNE05' + D134 + i2113-2 E9 x 3'El 123 136 pNE05' + D144 + i2113-2 5' El x 3'El 143 152 pNE05' + E1-Sma + i2113-2 5' El x 3' El 135
pNE05' + D43 + E1-Sma E9 x 5' El 10 12
pNE05' + D144 + E1-Sma 5' El x 5' El 4 8
D43-neo + El-Sma E9 x 5' El 13 11
D144-neo + El-Sma 5' El x 5' El 6 9
aThe molar ratio of
pNEO5'
DNA to mutant DNA was1:10;theratio ofthemutantDNAstoeachotherwas1:1(e.g.,pNEO [0.5 ,ug] + mutant 1[5jig]
+ mutant 2 [5 ,ug]). G418-resistant colonies were counted 20 days after
transfection.
bi1223-2DNAwasremovedfromthe vectorsequencesby cleavage with
BamHIin allexperiments.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:10.612.320.560.527.676.2]BPV-1-ENCODED MODULATOR FUNCTION 739
uncut D43
BamHI
I
D144 D43 12113-2
B D43-neo+EI-Sma
uncut B&mHI
m
Im-
ID144 i2113-2
A B C M1M2 D E F
m
m.-s
F G H I K
- 8kb
44.8kb 44.3kb 3.5kb 3.1 kb
42.6kb
FIG. 10. Southern blot analysis of cellularDNAsfrom G418-resistant C127 cellsderivedupon cotransfection ofpNEO5' DNA with
pairwisecombinations of E9, 5'El,and 3'ElmutantBPV DNAs. (A)Cotransfectionsof D43 (E9) plus i2113-2(3' El) andD144(5' El)plus
i2113-2.The molarratio of the cotransfectedmutantBPVplasmid DNAswas1:1. Theratio of pNEO5' DNAtototalBPVplasmidDNAwas
1:10. We cleaved i2113-2 DNA withBamHI before transfection to remove the BPV sequences from the vector DNA, creatinga size
differentialbetween the cotransfecting DNAs. Individual G418-resistant colonieswerepicked 20 days after transfectionand expanded into celllines, and total cellular DNAwasisolated. Gelelectrophoresis and blot analysisweredoneasdescribed in thelegendtoFig. 3. Lanes Athrough E each contained 10 ,ug of undigested cellular DNA.Lanes FthroughK eachcontained10,ugof thesamecellular DNAs cleaved
with BamHI.Forexample,lanesAandForBand G analyzed thesamecellular DNA. Witht2113-2DNA(removedfrom the plasmidvector),
BamHIcleavage resulted inafragment of 8 kb (full-lengthBPV).Cleavage ofmutantDNAsD43 and D144,both linkedtothe pMLvector, gaverisetofragments of 4.8 and 3.1 kb (D43) and 4.3 and 3.5 kb (D144), respectively, andacommon2.6-kbfragmentwhich isthe pMLvector. Lanes Mland M2contained 100pgof WT pMLBPV5 DNA and 2,ug of cellular DNA isolated from ID13 cells(18)whichcontain viral BPV DNA.Thepositions of formsI and IIofpMLBPV5DNAand viralBPV DNAresidentinID13 cellsareindicated by arrowheadsonthe left. Thearrowheadsontheright indicate the sizes of DNAfragments obtained after cleavage of themutantDNAswithBamHI.(B) Cotransfection of BPVmutantD43 (E9) linkedtopNEO5'(D43-neo) plus E1-Sma (5' El). Cotransfection wasdoneatamolar ratioof 1:1. Establishment ofcelllines,isolationofDNA,and DNAanalysisweredoneasdescribedin thelegendtoFig.6A. LanesA, B,and Ceachcontained 10 iLg
ofundigested cellular DNA isolated fromthree individual clones. LanesD, E, and F contained the same DNAsdigestedwith BamHI.
Arrowheadstotheright indicate the positions of the BamHI fragmentsof thetwomutantDNAs.Digestionof themutantDNAs with BamHI gaverisetotwofragments of 8 and2.6 kb fortheE1-Sma mutant,whereasD43-neoDNAwascleavedinto threefragmentsof 5.1(pNEO
DNA), 4.8, and 3.4 kb.
system marks resident plasmids toassure such replication which is coordinate with the chromosome. This is shown clearly in Fig. 5 and 6; notice that, even though the input DNAsreplicated and amplified, the resident plasmids didnot join this replication pool.Thetransientassaysprovedthat M isanegative regulator. Asanaturalextension of thesedata, we speculated that M plays some role in the marking process.
Thus, according tothis model, when WT DNA enters a low-copy-number cell lineit does notfindan excesspoolof R(seeBergetal. [2a] forfurtherdiscussion)provided bythe resident genome. In these cells, the WT DNA can only expressR and Matlevels coordinate with cell cycle
repli-cation. However, an R+ M- mutant enters such cells, producesonly R, and thus canamplify.Akey featureofthe modelis that theresidentplasmidsinlow-copy-numbercell lines provide only enough factors to allow for stable repli-cation ofthemselves and thatM,once bound, is noteasily released. The differences in the dynamics of replication betweenanaive andatransformed cellmustthen bedictated by thestate of the cell itself and the levels of Rprotein.
Thetransientassays showed that the M factor is capable ofnegativeregulation,andthe stableassaysshowed thatthis factor is requiredfor plasmid establishment. A strong pre-diction of the model is that, upon removal of M from the system,the BPV genome should notbe able toregulateits
A
i2113-2 i2113-2 M1M2 A B C D E
I I
:..''
I1.
1I)
_
i,
8 kb
'5.1kb c4.8kb
-'3.1kb
4c2.6kb VOL.60, 1986
on November 10, 2019 by guest
http://jvi.asm.org/
[image:11.612.91.534.67.406.2]replication, andrunaway replication would ensue. Lethality
maythenbe the resultofan excess ofBPV DNA or agene product whose expression is normally limited by the WT
replication system. Here an analogy to other small but
unrelated DNA tumorvirusesmay be relevant. With simian
virus 40 or polyomavirus, transformation in permissive
systems can onlybe achieved with large-T-antigen mutants
(see, for example, reference 11). In special cases in which
thereplication origin is specifically mutated, large-T antigen plus permissive lines can be obtained (11). In contrast, however, in cellsystemsin which only limited replicationis
detected, transformation frequencies have been reported to
rise with temperature-sensitive large-T-antigen mutants
when thetransformation wasassayed atpermissive
temper-atures (8). In these latter cases, presumably, the effects of limitedreplicationwere notdeletions for establishment, and the increased persistence ofthe viral DNA afforded by the
limited replication increased the chance for stabilization.
Therefore,
different effects of any particular M- or R-mutant on transformation might be expected for BPV,de-pending on the cell state. In other words, uncontrolled replication may be deleterious, but limited replication may
be advantageous. In this contextit is interesting thatd1306,
an in-frame R deletion mutant, transformed C127 cells at
frequencies
aleast 10-fold greater than did theWT(Table2)(23).
The lethal phenotype ofthe M mutants will probably be
themost difficultaspect of the phenomenon described here tostudy further. In ourtransfection experiments, only
10-4
to
10'
cells would eventually becometransformedalthough 1 to 10% of transfected cells transiently expressed andreplicated
BPV (24). Thus, events proceeding toward stablereplication
andtransformation may happen in only a smallfraction of transfected cells. The cell lethality reported, of course, only measuredevents in this subpopulation.
There-fore,
relevant virus-cell interactions mediated by the Mprotein
in the transition from transient amplification to theestablishedstatemaybedifficulttofollow becausetransition occurs in this minor fraction of cells.
We reliedoncomplementationtests todocumentmany of
the
points
made inthis study. Our results are notconsistent with recombination playing a significant role. Two distant mutants, e.g., D43 (E9-) and D29 (5' El-), did notcomple-menteach other, whereas two closely spaced mutants such asD29 andD31did. Furthermore,in stable assays no sign of rearrangement was detected by Southern blots ofthe two
complementation
genomes. This is in contrast to a recent report (13) in which a high frequency of recombination betweentwoBPV mutants was readily detected. It is known that transfecting DNA presented to cells in large calciumphosphate
precipitates is built into large oligomeric struc-tures, forming "peckalosomes" (32). With methods that do not form such precipitates, recombination frequencies arereportedly low. Forexample, in mixedtransfections oftwo
different simianvirus 40 circles withDEAE-dextran, almost allofthedimers detectedin vivo werehomopolymeric (12),
and high frequencies ofrecombination are only detected if two circles are first linked in vitro (40). The Polybrene methodusedin our studies does not depend on a precipita-tion step fortransfection.
The genetic analysis provided here for R and Mprobably does not define the structure of the entire genes. For example, mutations which interfere with geneexpression,as well as protein structure,can be scored as M- orR-. The productswork in trans, and any BPVgenome containinga
wild-type gene complements a defective gene whether the
defect affects a cis function
required
forexpression
or onerequired forprotein information. A summary ofthe
genetic
definitions
provided
in this paperis shown inFig.
11.The E9ORF which is part of theMgene is small
(only
108bp
[Fig.
1]),but this
region
is encodedinto stableRNA(A.
Stenlund,
J. Choe,and M.Botchan,
unpublished data)
and islocated3'toapromoter,
P1, expressed
in transformed cells(Stenlund
et al., submitted). Furthermore, frameshift mutantsDll and D125, which map
just
5' and 3' tothisORF,
donothave the M- phenotype(Table
1;Fig.
8A).Thus,
whereas the E9 region may be a cis-acting regulatory site needed for Mexpression, it seems possible that it is part of the
protein
encoding information. Interestingly, anRNA species with a
splice donor at BPV nucleotide position 1234 has been reported in an earlier study of Stenlund et al. (37). Recent
unpublished studies from our laboratory detected at least two RNAspecies (1.4 and1.8kb) which could encodeforthe
M protein and, indeed, analysis of cDNA clones showedthat
a splice between 1234 and 3224 is present in a
relatively
abundant RNA. It is intriguing that the genetic data
summa-rized in Fig. 11 indicate an endpoint of one exon ofthe M
gene around position 1300 in the BPV genome.
The putative M protein may work by itself or require cellular machiriery to manifest negative regulation. The available genetic data eliminate the need for any of the encoding information 3' to the El ORF as required for modulation in transient and stable assays. For example, Ball5 is established as a stable nuclear plasmid, and in an
independent assay system Roberts and Weintraub (32a)have
also shown that these ORFs are dispensable for negative regulation. From their data one can also rule out any role for M action that requires the R protein simultane-ously.
Whereas R and M play pivotal roles in the regulation of BPV replication, their function interacts in unknown but important ways with other viral genes. For example, the E6 and E6/E7 gene products (2a, 3, 23) are required to maintain high levels of viral DNA, perhaps by keeping the pool of R protein high (2a). A role for the 3' ORFs (E2, E3, E4, and E5) is also indicated by several results; however, much of these data are at present confusing. For example, mutant d1211 has a significantly reduced replication rate in transient assays (Fig. 2), implying a role for E2. Consistent with this observation, others have found that E2 mutants integrate in stable assays (9, 13; P. Howley, personal communication). However, BallS, which deletes all of the 3' ORFs including E2, displays WT activity with respect to transient replication (Fig. 2) and stable plasmid maintenance (22). Furthermore,
Grof
and Lancaster (13) have reported that small E5 mu-tants affect stable assays, whereas others have not detected such effects (D. DiMaio and P. Howley, personal communi-cation). Several explanations present themselves, includingthe following. (i) The presence of E5 (or any downstream
ORF) makesthe functions of other ORFs, for example, E2, obligatory (13). (ii) The mutations affect the structure or expressionofothermRNAs, and the particular mutant may
ormay not affect the activity of a variety of other genes by alterations of a class of mRNAs. (iii) The mutants all manifest their phenotypes because of an indirect effect on replication, and results are highly influenced by
experimen-tal protocols. Forside-by-side differences between mutants BallSandd1211, we believe that some aspects of explanation
i or iimay be relevant. However, because different groups
have obtained conflicting results with the identical deletion
mutant (9, 13; P. Howley, personal communication), it
seems likely that differences in cell conditions or
on November 10, 2019 by guest
http://jvi.asm.org/
BPV-1-ENCODED MODULATOR FUNCTION 741 P1
Hindlil
I
P2
HpaI
II1
1E91
Smal EcoRI
I
El
I
11
-M
D43x D134 x
D144
R*M-
El-Sma
D28 D9 029 D31
R-M
i2113-2
*-2405
M
R
x
x
x
x
(X)
FIG. 11. Summary of genetic data. Mutants D43 and D134 define the most 5' region of the M gene, and mutantsD144through D29 define another part of this same gene. Mutant D31 defines the 5' border of the R gene, whereas mutant i2113-2 is close to the 3' border of this gene. AnotherEl mutant at position 2405 described previously (23) did not maintain itself as a plasmid in stable assay and did not complement mutant i2113-2for plasmidreplication. Mutant i-2405 did not complement mutant D31 for transient or stable replication. Therefore, we concluded that i-2405, i2113-2, and D31 are all part of the same complementation group and are R-. We did not test i-2405 for the M phenotype, hence the parentheses in the figure. The solid bars show our conclusion about the position of the M gene; the open bar denotes the region encoding R.
tion protocolshave influencedsomeofthese results. This is particularlytruefor E2 since thisgeneisapositive transcrip-tional activator (35) and the requirements for such viral genesinothersystemscanbeinfluenced by the multiplicity ofinfectionorthetypeof cellused (27, 28, 35; see also the unpublished observations discussedin reference 17).
Anotherpuzzle thatwemustaddressconcerns conserva-tion of the El region as an intact ORF in all papil-lomaviruses, ifin fact it encodes fortwodiscrete genes. A simple resolutiontothisparadoxicalsituationmaybe solved by understanding the complete structures of RNAs which encode the protein information. By linking donors and acceptors forthe two genes in complex ways, deletions or substitutions which would change reading frames may be impossible. On the other hand, latentreplicationof BPV in C127 cells provides no information regarding vegetative replication. It is, of course, possible that this El ORF encodes anothergenethatuses the ORF inanotherway or intact atthis stageof its replication cycle.
ACKNOWLEDGEMENTS
WethankA.Stenlund forcommentsonthemanuscript. This work was supported by Public Health Service grant CA 30490 from theNational Cancer InstituteandgrantMV-91 from the
American CancerSociety.
LITERATURECITED
1. Amtmann, E., and G, Sauer. 1982. Bovine papillomavirus transcription: polyadenylatedRNA speciesandassessment of thedirection oftranscription.J. Virol. 43:59-66.
2. Bencini,D.A.,G. A.O'Donovan, and Y. R. Wild. 1984.Rapid chemicaldegradation sequencing.Biotechniques4:1-3.
2a.Berg, L., M. Lusky, A. Stenlund, and M. R. Botchan. 1986.
Repressionof bovinepapillomavirusreplicationis mediatedby
avirallyencoded trans-actingfactor. Cell46:753-762.
3. Berg,L.J., K.Singh,and M. Botchan. 1986.Complementation ofa BPVlow-copy-number mutant: evidence for atemporal
requirement of the complementing gene. Mol. Cell. Biol. 6:859-869.
4. Botchan, M. R., L. J. Berg, J. Reynolds, and M. Lusky. 1986. Thebovine papilloma virus replicon, p.53-67. In D. Evered and S. Clark(ed.), Papillomaviruses. John Wiley & Sons, Inc., New York.
5. Chen, E. Y., P. M. Howley, A. D. Levinson, and P. Seeburg. 1982. The primary structure and genetic organization of the bovine papilloma virus type 1 genome. Nature (London) 299:529-534.
6. Colbere-Garapin, F., F.Horodniceanu, P. Kourilsky, and A.-C. Garapin. 1981. A new dominant hybrid selective marker for higher eukaryotic cells. J. Mol. Biol. 150:1-14.
7. Danos, O., L. W. Engel, E. Y. Chen, M. Yaniv, and P. M. Howley. 1983. Comparative analysis of the human type la and bovine type1 papillomavirus genomes. J. Virol. 46:557-566. 8. Della Valle, G., R.G. Fenton, andC. Basilico. 1981. Polyoma
largeTantigen regulates theintegration of viralDNAsequences into the genomeof transformed cells. Cell 23:347-355. 9. DiMaio,D. 1986. Nonsensemutation in openreading frameE2
of bovinepapillomavirus DNA. J. Virol. 57:475-480.
10. Dvoretzky, I., R. Shober, and D. Lowy. 1980. focus assay in mousecells for bovinepapillomavirus. Virology 103:369-375. 11. Gluzman, Y.,and B. Ahrens.1982.SV-40earlymutantsthatare
defective for viralDNA synthesis butcompetent for transfor-mation of cultured rat and simian cells.Virology 123:78-92. 12. Goff, S. P., and P. Berg. 1977. Structure and formation of
circular dimers of simian virus40 DNA.J. Virol. 24:295-302. 13. Groff,D.E.,and W. D.Lancaster. 1986. Geneticanalysisofthe
3' early transformation and replication functions of bovine papillomavirustype1. Virology150:221-230.
14. Heffron, F.,M.So, andB.McCarthy. 1978.Invitromutagenesis
ofacircularDNAmoleculebyusingsyntheticrestriction sites. Proc. Natl.Acad. Sci. USA 75:6012-6016.
15. Heilman, C. A., L. Engel, D. R.Lowy, and P. Howley. 1982. Virus-specific transcription in bovine papilloma virus trans-formedmousecells.Virology119:22-34.
16. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmousecell cultures.J. Mol. Biol. 26:365-369. 17. Imperiale, M. J., L. T. Feldman, and J. R. Nevins. 1983. VOL. 60, 1986
on November 10, 2019 by guest
http://jvi.asm.org/
[image:13.612.60.554.64.278.2]Activation ofgene expression by adenovirus and herpesvirus regulatorygenes actingintransandbyacis-acting adenovirus enhancer element.Cell 35:127-136.
18. Kawai, S., and M. Nishizawa. 1984. Newprocedure for DNA transfection with polycation anddimethyl sulfoxide. Mol. Cell. Biol. 4:1172-1174.
19. Law, M.-F.,D.R. Lowy, J. Dvoretzky, and P. M.Howley. 1981. Mousecellstransformed by bovinepapilloma virus contain only extrachromosomal viral DNAsequences. Proc.Natl. Acad.Sci. USA 78:2727-2731.
20. Lowy, D. R., J. Dvoretzky, R.Shaber,M.-F.Law, L. Engel, and P. M. Howley. 1980. In vitro tumorigenic transformation by a defined subgenomic fragment of bovinepapilloma virusDNA. Nature (London)287:72-74.
21. Lusky, M., and M. Botchan. 1981.Inhibition of SV40 replication in simian cells by specific pBR322 DNA sequences. Nature (London) 293:79-81.
22. Lusky, M., and M. R. Botchan. 1984. Characterization of the bovine papillomavirus plasmid maintenance sequences. Cell 36:391-401.
23. Lusky, M., and M. R. Botchan.1985.Genetic analysis of bovine papillomavirus plasmidtype1trans-acting replication factors.J. Virol.53:955-965.
24. Lusky, M., and M. R. Botchan. 1986. Transient replication of BPV-1plasmids:cisandtransrequirements. Proc. Natl. Acad. Sci. USA 83:3609-3613.
25. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: alaboratory manual. ColdSpringHarborLaboratory, ColdSpring Harbor,N.Y.
26. Maxam,A.M., andW. Gilbert. 1980. Sequencingend-labeled DNAwithbase-specific chemical cleavages. Methods Enzymol. 65:499-560.
27. Nevins, J. R. 1981. Mechanism of activation of early viral transcription by the adenovirus ElA gene product. Cell 26:213-220.
28. Nevins,J.R.1982.Induction of the synthesis ofa70,000-dalton mammalian heat-shock protein by the adenovirus ElA gene product. Cell 29:913-919.
29. Peabody, D. S., and P. Berg. 1986. Termination-reinitiation occursin the translationof mammalian cellmRNAs.Mol.Cell. Biol. 6:2695-2703.
30. Peabody, D. S., S. Subramani, and P. Berg.-1986. Effect of
upstream readingframes ontranslation efficiencyinsimianvirus 40recombinants. Mol. Cell. Biol. 6:2704-2711.
31. Peden, K. W. C., J. M.Pipas, S. Pearson-White, and D.Nathans. 1980. Isolation ofmutantsofananimal virus in bacteria.Science 209:1392-1396.
32. Perucho, M., D. Hanahan, and M. Wigler. 1980. Geneticand physical linkage of exogenous sequences in transformedcells. Cell 22:309-317.
32a.Roberts, J. M., and H. Weintraub. 1986. Negative control of DNA replication in composite SV40-bovine papilloma virus plasmids.Cell 46:741-752.
33. Sarver, N., M. S. Rabson, Y.-C. Yang, J. C. Byrne, and P. M. Howley. 1984. Localization and analysis of bovine papil-lomavirustype1 transforming functions. J. Virol 52:377-388. 34. Schiller, J. T.,W.C.Vass,andD. R.Lowy. 1984.Identification
ofa seconutransforming region in bovine papillomavirus DNA. Proc. Natl. Acad.Sci. USA 81:7880-7884.
35. Shenk, T.,N.Jones,W.Colby,and D.Fowlkes.1979.Functional analysis of adenovirus type 5 host range deletion mutants defective fortransformation ofrat embryo cells. Cold Spring HarborSymp. Quant. Biol. 44:367-375.
36. Spalholz, B. A., Y.-C. Yang, and P. M. Howley. 1985. Transactivation of a bovine papilloma virus transcriptional regulatory element by the E2geneproduct. Cell42:183-191. 37. Stenlund, A., J. Zabielski, H. Ahola, J. Moreno-Lopez, and U.
Pettersson. 1985. The messengerRNAsfrom the transforming region of bovine papilloma virus type 1. J. Mol. Biol. 181:541-544.
38. Thomas,C.A., K. J. Berns, Jr., and T. Kelly, Jr. 1966.Isolation ofhigh molecular weightDNAfrom bacteria and cellnuclei, p. 535. InG. L. Cantoni andD. R. Davies (ed.), Procedures in nucleic acid research. Harper & Row, Publishers, Inc., New York.
39. Waldeck, W., F.Rosl,and H.Zentgraf. 1984.Origin of replica-tion in episomal bovine papilloma virus type 1 DNAisolated from transformed cells. EMBO J. 3:2173-2178.
40. Wake, C. T.,andJ.H. Wilson. 1980. Defined oligomeric SV40 DNA: a sensitive probe of general recombination in somatic cells. Cell21:141-148.
41. Yang, Y.-C., H. Okayama, and P. M. Howley. 1985. Bovine papilloma virus contains multiple transforming genes. Proc. Natl. Acad. Sci. USA 82:1030-1033.