0022-538X/93/084814-08$02.00/0
Copyright© 1993, American Society for Microbiology
The Role of Vesicular Stomatitis Virus Matrix Protein in
Inhibition of
Host-Directed Gene
Expression
Is
Genetically
Separable from Its Function in Virus Assembly
BRIANL. BLACK,tRICHARD B. RHODES,MARGIE McKENZIE, ANDDOUGLAS S. LYLES* DepartmentofMicrobiologyandImmunology, Bowman GraySchool ofMedicineof
Wake Forest
University,
Winston-Salem,
North Carolina 27157Received25 February 1993/Accepted26April1993
Recently,
the vesicular stomatitis virusmatrix(M) protein hasbeen showntobecapable of inhibition of host cell-directedtranscription in the absence of other viral components (B. L. Black and D. S.Lyles, J.Virol. 66:4058-4064, 1992). M protein is a major structural protein that is known toplay a critical role in virusassembly
by bindingthehelical ribonucleoprotein core of the virustothecytoplasmic surface of the cellplasma membrane during budding. In this study, two M protein mutants were tested to determine whether the inhibition of host transcription byMprotein is an indirect effect of its function in virusassembly
orwhether it represents an independentfunction ofM protein.Themutant
Mprotein of theconditionally
temperature sensitive (ts) vesicular stomatitis virus mutant,ts082,
was found to be defective in its ability to inhibithost-directed gene expression, as shown by its inability to inhibit expression of a cotransfected target gene encoding chloramphenicol
acetyltransferase.
Theabilityof thets082Mprotein tofunctionin virusassembly
wassimilartothatofwild-type Mprotein,as shown by its
ability
tocomplement the group IIItsM proteinmutant,tsO23. Anothermutant, MN1,whichlacks aminoacids 4 to21ofMprotein demonstrated that the abilitiesof Mproteintoinhibitchloramphenicolacetyltransferasegeneexpressionandtolocalizetothenucleus wereunaffected by deletionof this
lysine-rich
amino-terminalregion but thattheabilitytofunction invirusassembly
was ablated. Thus, the twoM protein mutants examined in this study exhibited complementary phenotypes:ts082Mprotein functioned invirusassembly
but wasdefective ininhibitionof host-directed gene expression, while MN1Mprotein functionedininhibiting gene expression butwasunabletofunctioninvirusassembly.
These data demonstrate that theroleof Mprotein in inhibition of hosttranscriptioncanbeseparatedgenetically
from its rolein virusassembly.
Vesicular stomatitis virus (VSV), the prototype
rhabdo-virus,causes a rapidand potentcytopathiceffect(CPE) (1,
35, 37). Among other effects, VSV infection results in
inhibition of transcriptionandtranslation of host gene prod-ucts.
Recently,
the viral matrix(M) proteinhas been shown to be capable of causing some of the cytopathic effectscharacteristic of VSV infection. Mproteinisamajor struc-tural protein that is known to play a critical role in virus
assembly by bindingthe helicalribonucleoproteincoreofthe
virustothecytoplasmic surface of the cellplasmamembrane
during budding. Inaddition toits role in virusassembly, M
proteinhas beenshowntoinduce theroundingofpolygonal
cells that occurs during VSV infection (3). Furthermore, using an assay in which an M gene expression vector was cotransfected withavectorencodingthe target gene,that for
chloramphenicol acetyltransferase (CAT), expression of M
proteinwasshown toinhibithost cell-directed transcription ofthe CAT gene in the absence of any other viral component
(2).Using the cotransfection assay, it was also shown that, in addition to
inhibiting
CAT gene expression, M protein inhibited its own transcription when expressed from theplasmid vector (2). This result probably accounts for the observation that M protein is difficult to express from recombinantvectorsthat rely on host cell transcription (3,
21). The inhibition of gene expression by M protein was foundtobe quite potent as Mprotein caused a more than
* Correspondingauthor.
tPresent address: Department of Biochemistry and Molecular Biology,M.D. Anderson Cancer Center, Houston, TX 77030.
10-foldinhibitionof CAT geneexpressionwhenexpressedat less than 0.2% of the level ofM gene expression inVSV infection (2). The goal of the study presented herewas to determine whether the inhibition of hosttranscription byM
proteinisanindirect effect of its function in virusassembly or whether it represents an independent function of M
protein.Thedatapresentedin this report show that the role of M protein in inhibition of host transcription can be separatedgeneticallyfrom its role in virusassembly.
A previously described conditionally
temperature-sensi-tive(ts)mutantofVSV, tsO82,wasreportedtocomplement ts mutants of all five VSV complementation groups (6), indicatingeither thatoneof theVSV genes is bicistronicor that one of the VSVproteins has two or more genetically separablefunctions. ThetsO82M genewasfoundtocontain asinglepointmutationresultinginamethionine-to-arginine changeatposition51in theproteinsequence(6).Inaddition,
tsO82 virus exhibits little or no CPE at the nonpermissive temperature in chicken embryo fibroblast cells (6). This observation raised the possibility that M protein plays an important role in the CPE of VSV infection and that the cytopathicandassembly roles of M protein represent sepa-rate functions. This hypothesis is supported by results
presentedhere inwhich the tsO82 M proteinwasfoundtobe defective in its ability to inhibit host cell-directed gene
expressionbutnotinitsabilitytofunction invirusassembly. Deletion of thecodingsequencefor the amino-terminal 32 amino acids from the M gene results in a protein fragment
whoseexpressionis similartowild-typeMprotein in that it is difficultto express in transfected cells (3).This suggests 4814
on November 9, 2019 by guest
http://jvi.asm.org/
that such a deletion fragment might be able to function in
inhibition of host cell-directed gene expression like wild-type Mprotein. The amino terminus of M protein is highly basic, as it contains eight lysine residues from amino acids 5
through 19. This region of M protein has been postulated to be involved in virus assembly by binding to the cell mem-brane (20) or bybindingto the viral nucleocapsid (27, 28, 32). An M protein mutant, MN1, which lacks the positively charged amino-terminal region was constructed to investi-gate the cytopathic and assembly functions of this region of M protein. The data presented in this study show that the
abilityof Mprotein to inhibit gene expression was unaffected by deletion of the lysine-rich region but that the ability to
function in virus assembly was ablated by the deletion. Thus, the MN1 and tsO82 mutants have complementary
phenotypes.These results establish the genetic separation of Mprotein's role in virus assembly from its ability to inhibit host cell-directed transcription.
MATERIALS ANDMETHODS
Viruses. Originalstocks of the San Juan (wtSJ) and Orsay (wtO) strains (wt, wild type) of VSV (Indiana serotype) and the ts M protein mutanttsO23 were prepared as described
previously (15). Vaccinia virus recombinant vTF7.3 which expresses the bacteriophage T7 RNA polymerase (9) and a
vaccinia virus recombinant which expresses the VSV N
protein (24) were obtained from Bernard Moss (National Institutes of Health). Virus stocks were prepared in CV-1
cells and assayed asdescribed previously (4).
Plasmids. The following plasmids have been described
previously: pSV2.Neo (33), pSV2.CAT (11), and two
plas-mids, pSV2.M(+)and pSV2.M(-), containing the M gene of thewtSJ-strain of VSV(Indianaserotype) (31), under control ofthe simian virus 40 (SV40) early promoter in the coding andnoncodingorientations, respectively (2).
The MN1 mutant M gene was constructed by
oligonucle-otide-directedmutagenesis(16) of the wtSJ strain M gene in
M13mpl8 described previously (2). The oligonucleotide
5'-CCTCITTCATAAGGGGGTGGTGCGATCGAACTCATGA
TGAATGGATTGGGATAAC-3'
was used as a primer todelete thecodingsequencefor-amino acids 4 to 21 in the M
protein sequence. The MN1 M gene was removed from
M13mpl8bydigestionwithHindlIland was subcloned into theHindIII site of the plasmid pSV2.Neo to create plasmid
pSV2.MN1(+).
The M gene of the wtO strain of VSV was reverse tran-scribed from virion RNA with the oligonucleotide5'-CCCC
AAGCITAACAGATATCACGATCTAAGTGTTATC-3'
as a primer and then amplified by polymerase chain reaction withthe same primer together with the oligonucleotide5'-
CCCCAAGCTTGACAGGATATTAGTIGTTCGAGAGGC-3' as described previously (5). The amplified DNA was
di-gested with HindIII and cloned into the HindIII site of
M13mpl8 with the 3'end of the M geneorientednear the PstI siteofM13mpl8. Theoligonucleotide 5'-GACAAATCCTATT
TTGGAGTTGACGAGAGAGACACTCATGATCCG-3'
was usedforoligonucleotide-directed mutagenesis (16) of this con-struct tochange the codingsequencefor amino acid 51 from methionine toarginine, recreating the M protein sequence of the ts virus tsO82 (6). The wtO and tsO82 M genes were subcloned into the HindIII site of pSV2.Neo in thecod-ing orientations to create the plasmids pSV2.OM(+) and
pSV2.082.M(+), respectively.
ThewtSJ strain and MN1 mutant M genes were cleaved from M13mpl8 constructs with EcoRI and PstI and then
subcloned into thecorrespondingsites of plasmid pGEM-3Z (Promega) in the coding orientations relative to the
T7
promoter. The wtO strain and
tsO82
mutant M genes were removed from plasmidspSV2.OM(+) and pSV2.082.M(+), respectively, by digestion withHindIII
and then subcloned into theHindIII site of plasmidpT7/T3al9
(GIBCO BRL) in the coding orientations relative to theT7
promoter.DNA transfection. BHK cells were transfected with the Lipofectin reagent (GIBCO BRL) according to the manufac-turer's directions with minor modifications as previously described (2). Briefly, cell monolayers were washed once with serum-free Dulbecco modified Eagle medium (DMEM) (Flow Laboratories, Inc.), and then 15
,g
of Lipofectin reagent and 3 pLgof DNA were added in 1.0 ml of Optimem reduced-serum medium (GIBCO BRL) per106
cells and incubated for 5 h, except in transfections for complementa-tion assays, when 25pg
of Lipofectin reagent and 10p,g
of DNA were added. Two volumes of DMEM with 15% fetal calf serum were then added to the monolayers, and the cells were incubated for the indicated times before harvesting.CAT assays. Cells were harvested, and extracts were prepared by three freeze-thaw cycles as described previ-ously (2). Cell lysates were then quantitated for total protein, and an equivalent amount of cell lysate (normalized for total protein [22]) was assayed for CAT activity as described previously (11) with 0.1
,uCi
of['4C]chloramphenicol
(58.2 mCi/mmol; Dupont, NEN Research Products) and acetyl coenzyme A (5.3 mM) in a final reactionvolume
of 150p,l.
Reactions were conducted at37°C
for 5 h. The reactions were stopped and analyzed by thin-layer chromatography as previously described (11), and conversion to monoacety-lated and diacetymonoacety-lated species was then quantitated by radioanalytic imaging (AMBIS Systems, Inc., San Diego, Calif.).Since the extent of acetylation of chloramphenicol by CAT is not linearly related to the amount of CAT enzyme, data on percent acetylation were converted to units that are linearly related to CAT activity (relative units [RU]) in which 1 RU is the amount of CAT enzyme that will acetylate 50% of the chloramphenicol in the standard assay we used. The conversion of percent acetylation to RU is accomplished by comparison with a series of dilutions of a standard amount of CAT enzyme such that the activity of an extract in RU represents the extent to which it would have to be diluted to give 50% acetylation. Oneinternationalunit of purified CAT
enzyme has an activity of approximately 1 RU when added to untransfected cell extracts and processed in parallel with samples from transfected cells in the standard assay used (2). When the extent of monoacetylation is above 95%, mono-acetylated chloramphenicol begins to be mono-acetylated at its second site, giving rise to diacetylated chloramphenicol. Diacetylation remains linear to greater than 55 RU of CAT
engzyme
(over 20% conversion to the diacetylated form).35Sradiolabeling, immunoprecipitation, and sodium
dode-cyl sulfate-polyacrylamide gel
electrophoresis.
Cells were labeled at 37°C with 100p,Ci
of[3
S]methionine
(1,000 Ci/mmol; Dupont, NEN Research Products) in 0.5 ml of DMEM without methionine plus 2% dialyzed fetal calf serum for 1 h and processed for immunoprecipitation and electrophoresis as described previously (19) with anti-M monoclonal antibody 23H12 or 24H6 (23). Labeling of M protein for pulse-chase analysis was performed as described above for 30min.Labeled cells were then chased for 0, 1.5, or 3.5 h in unlabeled DMEM with 10% fetal calf serum, then harvested and immunoprecipitated as described above.Immunofluorescence
analysis.
BHK cells (3 x104)
wereon November 9, 2019 by guest
http://jvi.asm.org/
A. 40 45 50 55 60
Orsay Wild-Type ...PIDKSYFGVDE®DTHDPNQLRYE...
40 45 50 55 60
ts082 Mutant ...P1DKSYFGVDE[DTHDPNQLRYE...
B. 5 10 15 20 25
San JuanWild-Type MSSLKKILGLKGKGKKSKKLGIAPPP...
1 23 4 56
MN1 Mutant MSS APPP...
FIG. 1. Amino acid sequences of wild-type and mutant M pro-teins used in thisstudy. Mutationsweregenerated by oligonucleo-tide-directed mutagenesis of thewtOand wtSJM genes.Sequences areindicatedwith thesingle-letter amino acid code.(A)Sequence of amino acids40 to 62 ofthe wtO and tsO82mutant Mproteins. The circle and square at position 51 highlight the single amino acid difference between the wild-type and mutant sequences. (B) Se-quenceofamino acids 1 to26 of the wtSJ and1 to 8of the MN1 mutantMproteins. The line in the MN1 sequence represents the deletion of amino acids4 to21 from thewild-type sequencewhich generated theMN1 mutant.
seeded onto glass coverslips in 24-well plates. Cells were then infected with either vaccinia virus recombinantvTF7.3
which expresses the bacteriophageT7 RNA polymerase or
withavaccinia virus recombinant which expresses the VSV nucleocapsid (N) protein (24) at a multiplicity of infection of 50 for 2 h. Cells infected with vTF7.3werethen transfected asdescribed above with 300 ng ofplasmidDNAand 1.5 ,ul of
Lipofectin reagentin 200 ,ul of Optimem.At8 h
posttrans-fection, the cellswere fixed, permeabilized, andprocessed
for immunofluorescence labeling with anti-M monoclonal
antibody 23H12 or anti-N monoclonal antibody 10G4 as describedpreviously (23).
Complementation assays. BHK cells (2 x 106) were in-fected withvacciniavirusvTF7.3 at amultiplicityof infec-tion of 50 PFU per cell for 2 h and then transfected, as described above,with 10 ,ug ofplasmidDNAand 25 ,ug of
Lipofectinreagent in 1 mlofOptimem. At5h posttransfec-tion, the cells were washed once with serum-free DMEM and infected withtsO23orwild-typeVSVat amultiplicityof infection of10 PFU per
cell
for 1 h at 39°C, washedtwicewith serum-freeDMEM, and then incubatedat39°Cfor16 h in 2 ml of DMEMplus2%fetal calfserum.Virusyieldwas determinedby plaque assay of the supernatants with BHK cells at 33°C.
RESULTS
Effect ofM gene mutations on inhibition of target gene expression. We have shown previouslythat VSV M protein inhibits host-directed transcription of target genes in vivo
(2). In this study, two different M protein mutants were constructed to determine whether the mutations altered the
abilityof Mprotein to inhibit gene expression (Fig. 1). In the first mutant, arginine was substituted for methionine at amino acid 51 to recreate the M protein of the ts virustsO82
(6). The second mutant, MN1, was generated by deletion of the coding sequence for amino acids 4 to 21 of the M gene andis similar to theAl mutantstudied by Blondel et al. (3). The tsO82 mutation was introduced into the wtO strain M protein sequence because it was the parental strain from which tsO82 virus was isolated (6). The MN1 mutation was
-'
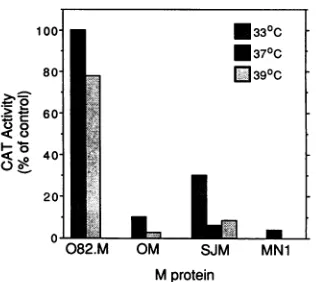
60-00
040-20-
l
0
082.M OM SJM MN1
Mprotein
FIG. 2. Effect of wild-type andmutantMproteinsonCATgene expression. BHK cells were cotransfected at a 20:1 ratio of M protein-encoding plasmids to the CAT plasmid, pSV2.CAT. The cotransfected M plasmids were pSV2.082.M(+) (082.M), pSV2.OM(+)(OM), pSV2.MN1(+) (MN1), and pSV2.M(+) (SJM). Cellswereincubatedateither33, 37,or39°C,asindicated, for24h. CATactivity of cellextractswasdeterminedby the conversion of [14C]chloramphenicol to acetylated forms and analyzed by thin-layer chromatography. Thin-thin-layer chromatographswerethen quan-titated byradioanalytic scanning, andRU werecalculated. The data are expressed asthe percentage of the RU generated in control assaysperformedatthe indicatedtemperaturein which the
cotrans-fectedM gene was inthenoncoding orientation. Results shownare
fromarepresentative of threeseparateexperimentscomparing wtO (OM), wtSJ (SJM), and tsO82(082.M)M genes. Intheexperiment shown,theCATactivity of the negative controlwas5.0 and 1.8RU at 33 and 39°C, respectively. The wtSJ and MN1 M genes were
comparedin twoseparate experiments. Inthe experiment shown, theCATactivityofthenegativecontrolwas5.0RU.
introduced into the wtSJ strain Mprotein sequencesothat the results obtained with thismutantcould becomparedwith those of Blondeletal.(3).Both wtO and wtSJarestrainsof the Indiana serotype of VSV whose Mproteins onlydifferby
5 of 229 amino acids (26, 31). Each of these mutants was tested for its abilityto inhibit gene expression by cotrans-fectingBHKcells with the M genes togetherwith a target geneencodingCAT(2).
Briefly, each mutant M gene was subcloned into plasmid pSV2.Neo (33) under control of the SV40 early promoter,
creatingtheplasmidspSV2.MN1(+)andplasmid pSV2.082.M (+).BHKcellswerethen cotransfectedwith either of these
plasmids together with plasmid pSV2.CAT (11). The cotransfected cells wereincubated for 24 h at 37°Cfor the MN1 mutant or at thepermissive (33°C) ornonpermissive (39°C)temperaturefor thetsO82mutant. Cellextracts were then harvested, and CATactivitywas determinedby mea-suring conversion of chloramphenicol to its acetylated
forms.
Figure2 shows therelative level of CAT geneexpression
in the presence of each of the mutant Mproteins andtheir
wild-type parent M proteins. The data are expressed as a percentageof therelative units of CATgeneratedin control cotransfectionsattheappropriatetemperaturewithplasmids
in which the M gene is in the noncoding orientation. The tsO82 Mprotein was ineffective atinhibiting CAT expres-sionatboth thepermissiveandnonpermissivetemperatures. In contrast, the MN1 Mprotein and both of the wild-typeM proteins (wtOorwtSJ)causeda10-foldorgreaterreduction in the amount of CAT enzyme compared with control transfected cells.Asexpected,the wtO and wtSJMproteins weresimilarly effective at inhibitingCAT gene expression,
on November 9, 2019 by guest
http://jvi.asm.org/
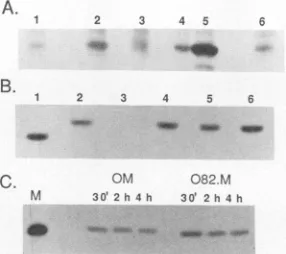
[image:3.612.356.515.68.209.2] [image:3.612.65.288.71.190.2]A.
B.
C.
3 4 5
1 2 3 4 5 6
lo AM _a _
OMA 082.M
M 30' 30t 2h4h
-W w - V
FIG. 3. Immunoprecipitation analysis ofM protein expression. (A) Expression of M proteins fromSV40vectors. BHK cellswere
transfected with SV40 plasmid vectors encoding either tsO82 M protein(lane 2), wtOMprotein(lane 3),orwtSJ M protein(lane 6)
or wereuntransfected (lane 1). Transfected cellswereincubatedfor
23h and then incubated for 1h in medium containing [35S]methio-nine. Lysateswere immunoprecipitated with the M protein
mono-clonalantibody 24H6. In lanes 4and 5, untransfected control cell lysatewasmixed with lysate from VSV-infected cells (labeled from 5to 6hpostinfection)ata1:1,000 ratio(lane4) andat a1:100 ratio
(lane5)toshow 0.1and1%of the level of M protein inVSV-infected
cells, respectively. (B) Expression of M proteins from vaccinia
virus-T7vectors. BHK cellswereinfected with vTF7.3, avaccinia
virus recombinant expressing the T7 RNA polymerase, and then
transfected with T7 expression plasmids encoding either MN1 M
protein (lane 1), wtSJMprotein(lane 2),vectoralone (lane 3),tsO82 Mprotein (lane 4),orwtO M protein (lane 5) for 18 h. In lane 6,
untransfected control cell lysatewasmixed with lysatefrom cells
infected with VSV for6hata1:10 ratiotoshow 10% of the levelof
Mprotein in VSV-infected cells. Cellswere incubated for 2 h in
medium containing [35S]methionine. Lysateswere
immunoprecipi-tated with the M protein monoclonal antibody 23H12. (C) Pulse-chase analysis of the stability of thetsO82 and wtO M proteins.
BHKcellswereinfected withvTF7.3and then transfected withT7 expression plasmids encoding either the tsO82 (082.M) or wtO
(OM)Mprotein. At 24 h posttransfection, cellswerelabeled for 30
min in medium containing [35S]methionine and then incubated in
unlabeled medium for either0, 1.5,or3.5 h and immunoprecipitated
with the M protein monoclonal antibody 23H12. The time from the
beginningof the pulse is indicated.Lane M,[35S]methionine-labeled virion M proteinas amarker.
indicating littleor nodifferencebetween thesetwo strainsin thisfunction. The datafrom Fig. 2 indicate thatthe mutation inthetsO82 Mgene reduces the ability of the M protein to
inhibit host-directed gene expression, whereas the MN1 mutation has littleor noeffecton inhibition of host-directed
gene expression.
The level of expression of the mutant M protein from plasmidpSV2.082.M(+)wascompared withthe level of M protein in cells transfected with each of the wild-type M plasmids, pSV2.M(+) and pSV2.OM(+), by radioimmuno-precipitation analysis (Fig. 3A). In this experiment, BHK cellsweretransfected,incubatedat37°C,and then labeledat
23hposttransfection for 1 h with [35S]methionine,lysed,and subjected toimmunoprecipitation analysis witha
monoclo-nalantibodyspecific forMprotein. These experimentswere
performed at 37°C for each of the M proteins since no
differences in the phenotype of either thetsO82 orwtO M
proteinwere observedbetween 33 and 390C. The ts082 M protein (Fig. 3A,lane 2)is madetoahigher levelthaneither
thewtO (lane 3)orwtSJ (lane 6) M protein. Expression of the wild-type M proteins was barely detectable above the
nonspecificbackgroundprecipitation ofaproteinwith
elec-trophoretic mobility similar to that of M protein (Fig. 3A, lane 1). Lanes 4 and 5 are controls in which radiolabeled lysates from VSV-infected cells at 6 h postinfection were mixed with lysates from uninfected, untransfected control cells at a 1:1,000 ratio (lane 4) and at a 1:100 ratio (lane5).
Thus, lanes 4 and 5 represent 0.1 and 1% of the level of expression of M protein, respectively, produced by VSV-infected cells. The level of expression of the
tsO82
M protein produced from the SV40 vector (Fig. 3A, lane 2) can be determined by comparison with lanes 4 and 5 to be approx-imately 0.2% of the level of M protein produced by 6 h in VSV infection. Each of the wild-type M proteins was pro-duced at less than 0.1% of the level in VSV-infected cells. The MN1 M protein was also produced at less than 0.1% of the level in VSV-infected cells and was barely detectable above background in separate experiments (data not shown). The low level of wild-type M protein produced from the SV40 vectors has been shown to be due to M protein-induced inhibition of its own transcription (2). Thus, the higher level of expression of thetsO82 M protein is consis-tent with a reduced inhibitoryeffect on its own transcription from the SV40vector.The low level of the M proteins produced from the SV40
vectors was at the limit of detection in the immunoprecipi-tation assay and was insufficient to accurately compare the efficiency of translation and the stability of the
tsO82
M protein with the wild-type M proteins. To achieve higher levels of M protein expression, both the wild-type M genes and the mutant M genes were recloned under control of the bacteriophage T7 promoter and transfected into cells in-fected with the vaccinia virus recombinant vTF7.3, which expresses the T7 RNA polymerase (9, 21). This system has been shown to express proteins to a very high level and offers the advantage that the T7 RNA polymerase system may not be as sensitive to M protein-mediated inhibition of transcription as vectors that require nuclear function.Figure 3B shows the results of a radioimmunoprecipitation analysis of cells expressing the MN1,tsO82,wtO, and wtSJ M proteins under control of the T7 promoter. BHK cells were infected for 2 h withvTF7.3 vaccinia virus and then transfected with each of theT7 promoter-M gene constructs, incubated for 22 h, labeled for 2 h with [35S]methionine,
lysed, and subjected to immunoprecipitation analysis with a monoclonal antibody specific for M protein. All the M proteins were expressed equally well to approximately 10% of the level produced during VSV infection (Fig. 3B, lane 6). This level of M protein expression represents a 100-fold increase in expression over that obtained with the SV40 vector. These data indicate that there is little or no difference in the level of expression of the different M proteins when expressed from a vector that does not rely on the host transcription apparatus.
Using the vacciniavirus-TI7 expression system, we com-pared the stability of thetsO82 M protein and its wild-type parent, wtO M protein, in a pulse-chase analysis (Fig. 3C). BHK cells were infected with vTF7.3 and transfected with plasmids encoding either the wtO or thetsO82 M protein. At 24 h posttransfection, the cells were pulse-labeled for 30min in medium containing [35S]methionine. The labeled cells were then chased in unlabeled medium for 0, 1.5, or 3.5 h, lysed, and subjected to immunoprecipitation analysis with a monoclonal antibody specific for M protein. This experiment shows that both thetsO82 and wtO M proteins exhibit little degradation during the 4-h time course of the experiment. Densitometric analysis showed that the amount of label remaining in the wtO andts082 M proteins was 69 and 76%,
on November 9, 2019 by guest
http://jvi.asm.org/
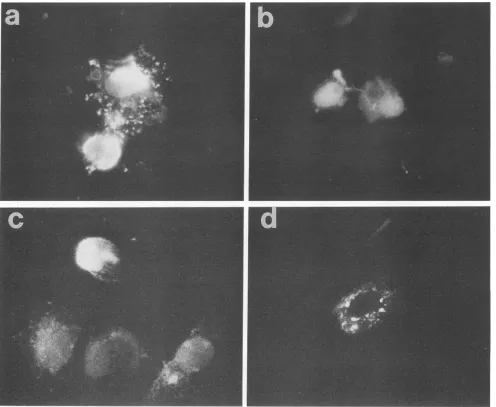
[image:4.612.119.262.73.200.2]FIG. 4. Immunofluorescenceanalysisof theintracellulardistributionofmutantMproteins.Inpanelsa,b,and c,BHKcellswereinfected withvTF7.3, avaccinia virus recombinantexpressingtheT7RNApolymerase,for2 hand then transfected with T7expressionplasmids encodingeitherts082(a),MN1(b),orwtSJ(c)Mprotein.Inpanel d,BHKcellswereinfected withavaccinia virus recombinantexpressing theVSVNprotein.At10hpostinfection, cellswerefixed andpermeabilized with1%Triton X-100.Cellswerelabeled with either monoclonal antibody 23H12, specific for Mprotein (a, b, andc), or monoclonal antibody10G4, specific forNprotein(d), andthen labeled witha
fluorescein-conjugated rabbit anti-mouseimmunoglobulin G.
respectively, at the end of the chase compared with that present atthe beginning(mean oftwo experiments). These
results, alongwith the results shown inFig. 3A,demonstrate that the lack of inhibition of CAT gene expression by the tsO82 M protein seen in Fig. 2 was not due to a lack of
synthesisof thetsO82mutantM protein, norwas it dueto instabilityof the mutantMprotein.
Intracellular localization of mutantMproteins.Mprotein is present in both thenucleus and cytoplasm of VSV-infected cells(23).Theintracellulardistribution of both the MN1 and tsO82 Mproteins was examined to determine whether the nuclear localization of either of the mutantM proteinswas altered. In theexperimentshown inFig.4, BHK cellswere infected with vaccinia virus
vTF7.3
for 2 h and then trans-fected with theplasmids encoding the tsO82, MN1, or wtSJ M proteins. Cells were also infected with a vaccinia virusrecombinant which expresses the VSV
nucleocapsid
(N) protein (24) as a control for a viral protein that is only observed in the cytoplasm of infected cells (23). At 8 hposttransfection, the cells were fixed,
permeabilized,
andsubjected to analysis by immunofluorescence microscopy
with a monoclonal antibody specific for M protein or N
protein and a fluorescein-conjugated rabbit anti-mouse
im-munoglobulinG.
Boththe tsO82(Fig. 4a)andthe MN1(Fig. 4b)Mproteins
were presentin the nuclei of transfected cells and exhibited patterns of fluorescence similar to those seen with the wild-typeMprotein (Fig. 4c).Cells that expresshighlevels of Mproteinfrom thevaccinia virus-T7systemshowamuch greater degree of cell rounding than cells infected with vaccinia virus aloneasdescribedbyBlondeletal. (3) using a differentexpression strategy.Examples of such
on November 9, 2019 by guest
http://jvi.asm.org/
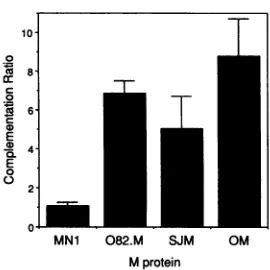
[image:5.612.55.551.69.476.2]0
88
E
-D4-E
0
2-0.L
MN1 082.M SJM OM
M protein
FIG. 5. Complementation oftsO23 by M protein mutants.BHK cellswere infectedwithvaccinia virusrecombinant vTF7.3 for 2 h,
transfected with 17 expression plasmids encoding either wtSJ
(SJM), wtO (OM), tsO82 (082.M), or MN1 M protein, and then
coinfectedwith the VSVtsMproteinmutanttsO23at5 h
posttrans-fection.Cellswerethen incubated at39°Cfor 16 h. Yields oftsO23 virusweredeterminedbyplaqueassayofthesupernatants at33°C.
Complementationratiosweredeterminedby dividing the virus yield
fromthe M protein-expressingcells by the virus yield obtainedfrom negativecontrolassays.Aratioof1 indicatesnocomplementation.
The data are the average offive independent experiments. Error
bars indicate the standarderrorof themeanfor thefive experiments.
The yieldoftsO23virus obtained from thenegative control cellswas
(2.6±1.2) x 104PFU/ml. The yield of wtSJ virus from cells infected
with vTF7.3, transfected with plasmidvector lacking an M gene,
andcoinfectedwith wtSJ VSVwas (1.25 ± 0.65) x 108PFU/ml.
logically altered cells include the cell shown nearest the bottom of Fig. 4a, the cell on the right in Fig. 4b, and the
brightlylabeled cellnearthe topinFig. 4c. The nuclei of all these cells were heavily labeled with antibody against M protein. However, the presence of M protein in the cell nucleus can be seen more clearly in cells that were not so
rounded. Examples include the cell expressing thetsO82 M protein nearest the top in Fig. 4a, the cell on the left
expressing the MN1 M protein in Fig.4b, and the three cells
near thebottom of the panel in Fig. 4cwhich were
express-ing wild-type M protein. The cell expressing N protein in Fig. 4dexhibitsadistinct lackof nuclear staining, confirming
that neither thevaccinia virus infection northe
permeabili-zation procedure was opening the nucleus to nonspecific nuclearlocalization in this experiment.These data show that neitherofthe mutations in theMgenealtered the ability of
the mutant Mproteins to enter host cellnuclei. This result indicates that the inefficiency of the tsO82 M protein in inhibition ofgeneexpression isnotdue to a failureto enter
cell nuclei. Also, the results with the MN1 mutantindicate that the lysine-rich amino terminus of M protein is not
required for nuclear localization and that M protein must
therefore have nuclear localization determinants carboxy terminal to amino acid position 21.
AbilityofMprotein mutantstofunctioninvirusassembly. The abilityof thetsO82 andMN1 M proteins tofunction in virus assembly was tested by their ability to complement growth of the group III (M gene) ts mutant tsO23 at the nonpermissive temperature with the vaccinia virus-T7
ex-pression system (18, 21, 36). BHKcellswere infectedwith vaccinia virus vTF7.3 for 2 h and then transfected with wild-type or mutant M genes under the control of the T7
promoter. At5 hposttransfection,the cellswerecoinfected
with tsO23 virus and incubated at 39°C for 16 h. The supernatantswereharvested, andthe growth oftsO23virus
wasdetermined by plaque assaysat33°C.
The data shown in Fig. 5 are the yields oftsO23virus in the presence of vector-encoded M protein divided by the backgroundtsO23virus yield obtained from cells transfected with vector alone (no M protein). A complementation ratio of 1 indicates the absence of complementation. A comple-mentation ratio of 2 or higher is generally considered to indicate that a significant level of complementation has occurred (30). The data shown are the average of five
independent experiments. The results in Fig. 5 show that the tsO82 M protein (average complementation ratio of 6.9) functions as well as the wild-type M proteins (average ratios of 5.0 and 8.8 for wtSJ and wtO M proteins, respectively) in complementation of tsO23 virus growth. In contrast, the MN1 M protein is unable to function in virus assembly (average complementation ratio of 1.1). This result was expected, since the amino-terminal region of the M protein has been implicated in binding of M protein to cellular membranes during virus assembly (20) and indirectly affects the binding of M protein to nucleocapsids (15). While the level of complementation obtained in these experiments is still about 500-fold lower than the yield of wild-type VSV, it is similar to the complementation ratios reported by other investigators for complementation by M protein expression in single-cycle growth experiments (21). Complementation by M protein expression appears to be much less efficient than complementation of group V (G gene) ts mutants by expression of G protein in this system (18, 36). Furthermore, the difference in complementation between the tsO82 and MN1 mutants was highly reproducible. The MN1 mutant did not display a significant level of complementation in any of the individual experiments. The level of complementation by the tsO82 and wild-type M proteins varied among experi-ments, but the ratios obtained with thetsO82 mutant were always comparable to those obtained with wild-type M protein and always indicated that significant complementa-tion hadoccurred. These data indicate thattsO82 M protein functions as effectively as wild-type M protein in virus assembly, while MN1 M protein cannot function in virus assembly.
DISCUSSION
Data presented in this study show that the role of M protein in the inhibition of host-directed gene expression and its role in virus assembly represent two independent, genet-ically separable functions. Two M protein mutants, tsO82
and MN1, have complementary phenotypes: tsO82 M pro-tein functions in virus assembly but is defective in inhibition of host-directed gene expression, while MN1 M protein is functional in inhibiting gene expression but cannot function in virus assembly (Fig. 2 and 5). The identification of M gene mutations that genetically dissociate the functions of M protein in virus assembly and inhibition of gene expression is important for reinforcing the idea that M protein may play a major role in virus-induced inhibition of host transcription. Expression of M protein in the absence of any other viral geneproduct was found to inhibit transcription of a cotrans-fected CAT gene by Northern (RNA) hybridization and nuclear runoff transcription analyses (2). It had been shown previously that it was difficult to express M protein with recombinant vectors (3, 21), and in the cotransfection exper-iments, it was demonstrated that vector-encoded wild-type M protein inhibited its own expression in addition to CAT gene expression (2). Since the tsO82 M protein failed to inhibit host cell-directed gene expression, it might be ex-pected not to inhibit its own expression and would therefore
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.110.245.66.201.2]bemadeto ahigherlevelthan thewild-typeMproteins.This expected result was obtained in the immunoprecipitation
analysis shown inFig. 3A.
The tsO82 virus is able to complement the growth of mutantsinall fiveVSVcomplementation groups,including
the group III Mproteinmutants(6).Thisimpliesthatoneof theVSVgenesis bicistronicorthatoneofthe VSVproteins
has more than one genetically separable function. Further-more, the tsO82 mutant is temperature sensitive for virus
growth inchickenembryofibroblastcells and in HeLacells,
but not in BHKcells, and thus is aconditionallyts mutant (6). Our datado notdirectly address the mechanism ofthe
temperaturesensitivity of virusreplication. However, rever-sion of tsO82 virus totemperaturestabilityis accompanied
by reversion of arginine 51 in the tsO82 M protein to the
methionine found in the wild-type sequence (6), making it likely that this mutation is responsible for the temperature
sensitivity ofvirus growth. While the tsO82 mutant is not defective forvirus growth in BHK cells (6), the tsO82 M
protein was defective for inhibition of host-directed gene
expression atboth the permissive and nonpermissive tem-peratures in BHK cells. These data demonstrate that the
mutation in thetsO82Mproteinislikelytobethe mutation responsiblefor thefailureof tsO82 virustoinhibithost cell RNAsynthesis.
Previous data haveimplicatedthe VSVleaderRNAin the
virus-induced inhibition ofhost celltranscription(12-14, 25,
34).
Other studies have shown, however, thatleader RNA alone cannot be responsible for inhibition of host RNAsynthesis in vivo (7, 29). The observation that M protein
alone iscapableof
inhibiting
host-directedtranscription
does not rule out the possibility thatduring VSV infection bothleader RNAandMproteinareinvolved. Suchacooperative
role would be consistent with the observation that during infection Mprotein (23) and leader RNA(17) are the only
VSV gene productsfoundinthe cell nucleus insubstantial
quantities. Likewild-type Mprotein,both of themutantM
proteinsweredistributed throughoutthecytoplasmand the
nucleus of transfected cells, and there were no obvious
differencesbetweenwild-typeandmutantMproteinsin the extentofnuclear localization (Fig. 4). This result indicates thattheinefficiency ofthetsO82 Mprotein in inhibition of geneexpression is not due to a failure to entercell nuclei.
Also, the results with the MN1 mutant indicate that the
lysine-richamino terminus of Mprotein isnotrequired for nuclear localization. The possibility remains that this se-quenceisanuclearlocalizationsignal, asit issimilartothe
positively chargednuclear localizationsignals found inmany
proteins (reviewedin reference 10). However,ifthisregion
is a nuclear localization signal, then M protein must have additional sequences sufficient to direct transport to the nucleuswhichare carboxy terminal to aminoacidposition
21.Since deletion of thelysine-richamino terminus failedto ablate nuclearlocalization, the results with this mutant do notaddress thequestion ofwhether ornot Mproteinmust enterthe nucleustoinhibithost-directedtranscription. Since VSVcan replicateinenucleate cells (8),it remainsa
possi-bilitythat theeffects of M protein on host-directed transcrip-tion aremediated indirectlythrough interaction with
cyto-plasmiccomponents and thatlocalization to the host nucleus occursforsomeother reason.
While thelysine-richamino-terminalregionof Mproteinis notrequired for nuclearlocalization, it isrequired for virus
assembly, as demonstrated by the inability of the MN1 mutanttocomplementtsO23 virusgrowth(Fig.5). Therole of the amino terminus of M protein in virus structure has
beeninvestigated previously,yetitsexactrolein assembly
remains controversial. Early studies suggested that this sequence wasinvolvedinbindingtoviral nucleocapsids (27, 28,
32).
However, more recent data suggested that thisregion ofMproteinbinds to the virusenvelope (20). Other recent data suggest that this region of Mprotein doesnot bind directly to nucleocapsids but rather is indirectly in-volved in the binding (15). Regardless of the nature ofthe
role of the Mprotein amino-terminal region in virus struc-ture,the dataobtained with the MN1mutantpresentedhere support anessentialrole for this sequence in virusassembly.
The amino-terminal region of M protein may also be in-volved in other functions during VSV infection(28, 32),but
it is clear from the data in Fig. 2 that this region is not required for inhibition ofhost-directed gene expression. This observation also makes itlikelythatthe failure of the MN1 mutant tofunction in virus assembly is not due to production of a misfolded, completely nonfunctional, or degraded M protein since this mutant maintains its full function of inhibiting host-directed gene expression (Fig. 2).
A role for M protein in the cytopathogenesis of VSV infection was first demonstrated by Blondel et al. (3), who showed that M protein is responsible for the characteristic cellrounding of VSV-infected cells. Cells that expresshigh levels of Mprotein from the vaccinia virus-T7 system also show a much greater degree of cell rounding than cells infected with vaccinia virus alone. No differences were noted between wild-type and mutant M proteins in their ability to induce cell rounding. It is interesting that the tsO82 M protein caused cell rounding tooccur and yet failed to inhibit gene expression, suggesting that inhibition of host-directed transcription may be an independent CPE from cell rounding. Alternatively, it is possible that the tsO82 M protein is less effective than the wild type in causing cell rounding, but the 100-fold increase in expression obtained from the vacciniavirus-T7 expression system over the SV40 vector system couldobscure thisdifference.
In summary, the data presented here provide further support for the idea that Mprotein plays a major role in the cytopathogenesis of VSV infection. Theassembly and cyto-pathic functions of Mprotein describedhere are genetically separable, as demonstrated by thetsO82and MN1 mutants, which havecomplementaryphenotypes. Theremay be other mutations in the M protein which would also demonstrate
thegeneticseparation of theassemblyandcytopathic func-tions of Mprotein. Inparticular,there may be many poten-tial mutations in the Mprotein that would abolish the ability to inhibit geneexpressionwithoutaffectingvirusassembly. Asdiscussed previously (2, 3), M gene mutationsmay play a role in generation of viruses with reduced CPEs such as those thatariseduringpersistent virus infections. The tsO82
mutation may be one example of many mutations of this
type. It remains to be determined whether M protein is involved in other aspects of the cytopathology of VSV
infection,in addition to cellroundingand inhibition of RNA synthesis, and whether the multiple roles in cytopathogene-sisrepresent related orindependentfunctions of Mprotein.
ACKNOWLEDGMENTS
We thank Bernard Moss for the vaccinia virus recombinants expressingT7RNApolymeraseand the VSVNproteinandGriffith Parksand PaulinaKaptur forcritical reviewofthe manuscript.
This work wassupported by Public Health ServicegrantAI15892 from the National Institute of Allergy and Infectious Diseases. Oligonucleotide synthesiswasperformedintheDNAsynthesiscore laboratory ofthe Comprehensive Cancer Center ofWake Forest
on November 9, 2019 by guest
http://jvi.asm.org/
University andwassupportedinpartby Public Health Service grant CA 12197 from the National Institutes of Health. B.L.B. was supportedbytraininggrantAl07401 from the National Institutesof Health.
REFERENCES
1. Baxt, B., and R. Bablanian. 1976. Mechanisms of vesicular stomatitis virus-induced cytopathic effects. II. Inhibition of macromolecular synthesis induced by infectious and defective-interferingparticles. Virology 72:383-392.
2. Black, B. L., and D. S.Lyles. 1992. Vesicular stomatitis virus matrix proteininhibits host cell-directed transcription of target genesinvivo. J. Virol.66:4058-4064.
3. Blondel, D., G. G. Harmison, and M. Schubert. 1990. Role of matrix protein incytopathogenesis of vesicular stomatitis virus. J. Virol.64:1716-1725.
4. Bowman, M. R., D. S. Lyles, and J. W. Parce. 1987. Possible mechanisms by which the H-2K"'3 mutation may decrease cytotoxic T-lymphocyte recognitionofvesicular stomatitis virus nucleoprotein antigen. J.Virol. 61:1992-1998.
5. Caldwell, J., P. McElhone, J. Brokaw, R. Anker, and B. A. Pollok. 1991. Co-expression of full-length and truncated Ig ,u-chains in human B
lymphocytes
results from alternative splicingof asingle primary RNA transcript. J. Immunol. 146: 4344 4351.6. Coulon, P., V. Deutsch, F. Lafay, C. Martinet-Edelist, F. Wyers, R. C. Herman, and A. Flamand. 1990. Genetic evidence for multiple functionsofthe matrix protein ofvesicular stomatitis virus. J. Gen. Virol.71:991-996.
7. Dunigan, D. D., S.Baird, andJ. Lucas-Lenard. 1986. Lack of correlation between the accumulation of plus-strand leader RNA and the inhibition of protein and RNA synthesis in vesicular stomatitis virus-infected mouse L cells. Virology 150:231-246.
8. Follett, E. A.C., C. R. Pringle, W. H. Wunner, and J. J.Skehel. 1974. Virus replication in enucleate cells:vesicular stomatitis virus andinfluenzavirus. J. Virol. 13:394-399.
9. Fuerst, T. R., P. L. Earl, and B. N. Moss. 1987. Useof a hybrid vaccinia virus-T7 RNA polymerase system for expression of targetgenes. Mol. Cell. Biol.7:2538-2544.
10. Garcia-Bustos, J.,J. Heitman, and M. N. Hall. 1991. Nuclear proteinlocalization. Biochim. Biophys. Acta 1071:83-101. 11. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982.
Recombinant genomeswhich express chloramphenicol acetyl-transferase inmammalian cells. Mol. Cell. Biol. 2:1044-1051. 12. Grinnell, B. W., and R R.Wagner. 1983.Comparative
inhibi-tionof cellulartranscription by vesicular stomatitisvirus sero-types New Jerseyand Indiana:role ofeach viral leader RNA. J. Virol.48:88-101.
13. Grinnell,B.W., and R. R. Wagner. 1984.Nucleotide sequence and secondarystructure of VSV leader RNA andhomologous DNAinvolved in inhibition ofDNA-dependent transcription. Cell 36:533-543.
14. Grinnell, B. W., and R. R Wagner. 1985. Inhibition of DNA-dependent transcriptionby theleader RNA of vesicular stoma-titis virus:role of specific nucleotide sequences and cell protein binding. Mol. Cell. Biol. 5:2502-2513.
15. Kaptur, P. E., R. B.Rhodes, and D. S. Lyles. 1991. Sequences of the vesicular stomatitis virus matrix protein involved in bindingtonucleocapsids. J. Virol.65:1057-1065.
16. Kunkel, T. A., J. D.Roberts,and R. A. Zakour. 1987. Rapid and efficient site-specific mutagenesiswithoutphenotypic selection. Methods Enzymol. 154:367-382.
17. Kurilla, M. G., H.Piwnica-Worms,and J. D.Keene. 1982. Rapid and transientlocalizationof theleader RNA ofvesicular stoma-titis virusin thenuclei ofinfectedcells. Proc.Natl.Acad. Sci. USA79:5240-5244.
18. Lefkowitz, E. J., A. K.Pattnaik,and L. A. Ball. 1990. Comple-mentation of avesicular stomatitisvirusglycoproteinGmutant with wild-typeproteinexpressedfrom either a bovinepapilloma
virus or a vaccinia virus vector system. Virology 178:373-383. 19. Lefrancois, L., and D. S. Lyles. 1982. The interaction of anti-body with the major surface glycoprotein of vesicular stomatitis virus. I. Analysis of neutralizing epitopes with monoclonal antibodies. Virology 121:157-167.
20. Lenard, J., and R. Vanderoef. 1990. Localization of the mem-brane-associated region of vesicular stomatitis virus M protein at theN terminus, using the hydrophobic, photoreactive probe 125I-TID. J. Virol. 64:3486-3491.
21. Li, Y., L. Luo, R. M. Snyder, and R. R. Wagner. 1988. Expression of the M gene of vesicular stomatitis virus cloned into various vaccinia virus vectors. J. Virol. 62:776-782. 22. Lowry,0. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall.
1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275.
23. Lyles, D. S., L. Puddington, and B. J. McCreedy. 1988. Vesic-ular stomatitis virusM protein in the nuclei of infected cells. J. Virol. 62:4387-4392.
24. Mackett, M., T. Yilma, J. K. Rose, and B. Moss. 1985. Vaccinia virus recombinants: expression of VSV genes and protective immunization of mice and cattle. Science 227:433-435. 25. McGowan, J. J., S. U. Emerson, and R.R. Wagner. 1982. The
plus-strand leader RNA of VSV inhibits DNA-dependent tran-scription of adenovirus andSV40 genes in a soluble whole-cell extract. Cell 28:325-333.
26. Morita, K., R Vanderoef, and J. Lenard. 1987. Phenotypic revertants of temperature-sensitive M protein mutants of vesic-ular stomatitis virus: sequence analysis and functional charac-terization. J. Virol. 61:256-263.
27. Ogden, J. R., R. Pal, and R. R. Wagner. 1986. Mapping regions of thematrix protein of vesicular stomatitis virus which bind to ribonucleocapsids, liposomes, and monoclonal antibodies. J. Virol. 58:860-868.
28. Pal, R., B. W. Grinnell,R M. Snyder, and R. R. Wagner. 1985. Regulation of viral transcription by the matrix protein of vesic-ular stomatitis virus probed by monoclonal antibodies and temperature-sensitive mutants. J. Virol. 56:386-394.
29. Poirot, M.K.,W. M. Schnitzlein, and M. E. Reichmann. 1985. The requirement of protein synthesis for VSV inhibition of host cell RNA synthesis. Virology 140:91-101.
30. Ramig, R F. 1990. Principles of animal virus genetics, p. 95-122. In B. N. Fields and D. M. Knipe (ed.), Virology. Raven Press, New York.
31. Rose, J. K., and C. J.Gallione. 1981. Nucleotide sequences of the mRNAs encoding the vesicular stomatitis virus G and M proteins determined from cDNA clones containing the complete coding regions. J. Virol. 39:519-528.
32. Shipley, J. B., R Pal, and R. R. Wagner. 1988. Antigenicity, function, and conformation of synthetic oligopeptides corre-sponding to amino-terminal sequences of wild-type and mutant matrix proteins of vesicular stomatitis virus. J. Virol. 62:2569-2577.
33. Southern,P.J., and P. Berg. 1982. Transformation of mamma-lian cells to antibiotic resistance with a bacterial gene under control of theSV40
early
region promoter. J. Mol. Appl. Genet. 1:327-341.34. Weck, P. K., A. R. Carroll, D. M. Shattuck, and R. R. Wagner. 1979.UseofUVirradiation to identify the genetic information of vesicular stomatitis virus responsible for shutting off cellular RNA synthesis. J. Virol. 30:746-753.
35. Weck, P. K., and R. R. Wagner. 1978. Inhibition of RNA synthesis in mouse myeloma cells infected with vesicular sto-matitisvirus. J. Virol.25:770-780.
36. Whitt, M. A., L. Chong, and J. K. Rose. 1989. Glycoprotein cytoplasmic domain sequences required for rescue of a vesicu-larstomatitis virus glycoprotein mutant. J. Virol. 63:3569-3578. 37. Yaoi, Y., H. Mitsui, and M. Amano. 1970. Effect of U.V.-irradiated vesicularstomatitis virus on nucleic acid synthesis in chick embryocells. J. Gen. Virol. 8:165-172.