Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Replication Advantage and Host Factor-Independent Phenotypes
Attributable to a Common Naturally Occurring Capsid
Mutation (I97L) in Human Hepatitis B Virus
Fat-Moon Suk,
1,2Min-Hui Lin,
1Margaret Newman,
1Shann Pan,
2Sheng-Hsuan Chen,
2Jean-Dean Liu,
2and Chiaho Shih
1*
Center for Tropical Diseases and Sealy Center for Vaccine Development, Department of Pathology and Department of Microbiology and Immunology, University of Texas Medical Branch,
Galveston, Texas 77555-0609,1and Department of Internal Medicine,
Taipei Medical University Hospital, Taipei, Taiwan2
Received 31 May 2002/Accepted 26 August 2002
Mutations of human hepatitis B virus (HBV) occur frequently within the capsid (core) protein in natural infections. The most frequent mutation of the core protein in HBV from Southeast Asia occurs at amino acid 97, changing an isoleucine (I) to a leucine (L). In our systematic study of virus-host interactions, we have examined the replication efficiency of a site-directed mutant, I97L, and its parental wild-type HBV in several different hepatoma cell lines. Interestingly, we found that this capsid variant replicated in human Huh7 hepatoma cells approximately 4.8-fold better than its parental wild-type HBV. A similar phenomenon was observed in another hepatoma cell line, J3. In addition, the level of encapsidated RNA pregenome in mutant I97L was about 5.7-fold higher than that of the wild-type HBV in Huh7 cells. Unlike Huh7 cells, no significant difference in viral DNA replication between the same I97L mutant and its parental wild-type HBV was observed in HepG2, a human hepatoblastoma cell line. This finding of a profound replication advantage for mutant I97L in Huh7 and J3 cells but not in HepG2 cells may have important implications for the emergence of this mutant in chronic HBV carriers. We speculate here that the mutation confers a host factor-independent growth advantage for the survival of HBV variants in gradually dedifferentiating hepatocytes and thus helps prolong viral persistence.
Human hepatitis B virus (HBV) can cause acute and chronic hepatitis in humans, with the latter often resulting in cirrhosis and hepatocellular carcinoma (6, 11, 55, 56). HBV is an en-veloped virus, which consists of an outer lipoprotein envelope and an inner nucleocapsid containing a 3.2-kb partially double-stranded DNA genome. The nucleocapsid, formed by a 183-amino-acid core antigen, assembles in the cytoplasm with a 3.2-kb pregenomic RNA and the viral polymerase. The encap-sidated pregenomic RNA is then retrotranscribed into viral DNA (20, 53).
Because the polymerase of HBV lacks a proofreading func-tion, HBV has a low fidelity in replication and thus tends to produce sequence variants at a high frequency. Naturally oc-curring mutations in HBV have been hypothesized to play a role in the pathology of HBV-related diseases and in the per-sistence of HBV infection (23, 27, 59, 60, 68, 69). As a major T-cell target (13, 42), the core protein accumulates frequent mutations in chronic carrier patients with active liver disease (1, 5, 8, 14, 15, 16, 27).
Within the core protein, the most frequent mutation occurs at amino acid 97 (1, 5, 14, 15, 16, 17, 21, 22, 27, 29, 33, 40, 41, 43, 48, 50, 61, 64, 65, 70). The codon 97 mutation changes the wild-type amino acid from a phenylalanine to a leucine in the
aywsubtype (F97L) or from an isoleucine to a leucine in the
adr subtype (I97L). Previously, we identified an “immature secretion” phenotype for capsid variant 97L (referring to both I97L and F97L), which is characterized by secretion of an ex-cessive amount of Dane particles containing immature single-stranded DNA intermediates in both theaywandadrsubtypes (70, 71). We further reported that this immature secretion phenotype can be rescued intramolecularly by a proline-to-threonine mutation at amino acid 130 (P130T) in the core protein (72) or intermolecularly by an alanine-to-phenylala-nine mutation at amino acid 119 (A119F) in the large envelope protein (37). An immature secretion-like phenomenon was also observed in vivo in animal models (9, 63). Although the immature secretion phenomenon of mutant 97L can be ob-served in both theadrandaywsubtypes, the mutation occurs frequently in subtypeadr(1, 5, 14, 15, 16, 17, 21, 29, 33, 61, 64, 65) but rarely in subtypeayw(40, 41, 48).
In this study, we examined the replication efficiencies of site-directed 97L mutants in both subtypes and their parental counterparts in several different hepatoma cell lines. To our surprise, we found that the adr subtype capsid mutant I97L replicated more efficiently than the wild-type virus in Huh7 and J3 cells but that they replicate almost equally well in HepG2 and Hep3B cells. This novel phenomenon consti-tutes a second strong phenotype of the capsid mutant I97L, and it implies a host factor-independent replication advan-tage for the mutant during the evolution of HBV variants in chronic carriers.
* Corresponding author. Mailing address: Center for Tropical Dis-eases and Sealy Center for Vaccine Development, Department of Pathology and Department of Microbiology & Immunology, Univer-sity of Texas Medical Branch, Galveston, TX 77555-0609. Phone: (409) 772-2563. Fax: (409) 747-2429. E-mail: [email protected].
12069
on November 8, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Plasmid constructs pWT-adr, pWT-ayw, pI97L, pI97F, and pF97L.Plasmid
pWT-aywis a tandem dimer construct of wild-type HBVaywsubtype and was
described elsewhere (70). The 3.2-kb DNA fragment of HBVadrsubtype (35)
was subcloned into theBamHI site of the PSV2A Neo vector, resulting in
plasmid pWT-adr(71). The wild-type HBV monomers of different subtype
ori-gins were used as templates to create different site-directed mutations via the Altered Site II in vitro mutagenesis system (Promega) (70). The mutant plasmids
pI97L and pI97F are of subtypeadrorigin, and the mutant plasmid pF97L is of
ayworigin.
Cell culture and transfection.Hepatoma cell lines J2, J3, and J5 (10), HCC36 (12), PLC/PRF/5 (2), FOCUS (24), HepG2 and Hep3B (34), and Huh7 (44) were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal
bovine serum at 37°C in the presence of 5% CO2. The calcium phosphate
transfection procedures were described elsewhere (54). Ten micrograms of each
HBV plasmid DNA was transfected with carrier DNA to a total of 35g for 3
⫻106 cells per 10-cm dish. Medium was changed 4 h posttransfection for
non-HepG2 cells and 12 h later for HepG2 cells. The FuGENE 6 transfection procedure was conducted for cells other than Huh7 and HepG2 according to the vendor’s suggestions (Roche, Indianapolis, Ind.) (37). Briefly, for each
transfec-tion, 3g of DNA was mixed with 9l of FuGENE reagent diluted in 200l of
Dulbecco’s modified Eagle’s medium and incubated for 45 min at room temper-ature. The DNA-FuGENE mixture was then added directly to the cells.
Preparation of intracellular HBV particles, viral DNA, and viral RNA. Intra-cellular core particles and viral DNA were prepared as detailed elsewhere (67). The HBV core particle-associated RNA was prepared with Tri-Reagent accord-ing to the manufacturer (Sigma, St. Louis, Mo.). Briefly, the core particle pellet
was first dissolved in 100l of TNE buffer (150 mM NaCl, 40 mM Tris-HCl,
1 mM EDTA [pH 7.4]) and mixed with 1 ml of Tri-Reagent. After incubation at
room temperature for 5 min, 200l of chloroform was added. The mixture was
vortexed for 15 s and incubated for 10 min at room temperature before centrif-ugation in an Eppendorf centrifuge for 15 min. The RNA was precipitated from the aqueous phase by adding 0.5 ml of isopropanol. For isolation of total RNA, we followed the vendor’s protocol (Sigma). Analysis of extracellular viral DNA was performed by gradient centrifugation as detailed elsewhere (70).
Southern and Northern blot analyses.Standard protocols for Southern and Northern blot analyses were used (70). Full-length 3.1-kb HBV DNA fragments
were purified from pWT-aywbyEcoRI digestion or from pPBR322-adr(35) by
BamHI digestion. Approximately 50 ng of the 3.1-kb DNA was radiolabeled by
the random-primed DNA labeling kit (Roche). Quantitative comparisons of HBV DNA signals on the X-ray film from Southern blots were performed with the ONE-D scan computer program (Scanalytic Co., Billerica, Mass.). The in-tensity of the overall HBV DNA level was measured by counting the signal from the 4.0-kb position to the bottom of the lanes.
Immunoblot analysis of core protein.Wild-type and mutant core proteins were prepared from cell lysates 3 days after transfection with wild-type and mutant dimer plasmids or core protein expression vectors. Protein expression was detected by standard Western blot analysis with rabbit anticore antibodies (3; M. Newman and C. Shih, unpublished data).
RESULTS
Subtypeadrmutant I97L can replicate more efficiently than its parental wild-type virus in Huh7 cells. To compare the replication efficiencies of wild-type and capsid variant 97L vi-ruses, we transfected tandem dimer replicons of either subtype
adroraywinto two different hepatoma cell lines, HepG2 and Huh7. Five days posttransfection, intracellular core-associated HBV DNA was examined by Southern blot analysis. The band-ing intensities in the lanes were measured with a densitometer in order to quantitatively compare differences in intracellular DNA synthesis of the wild-type and mutant 97L viruses.
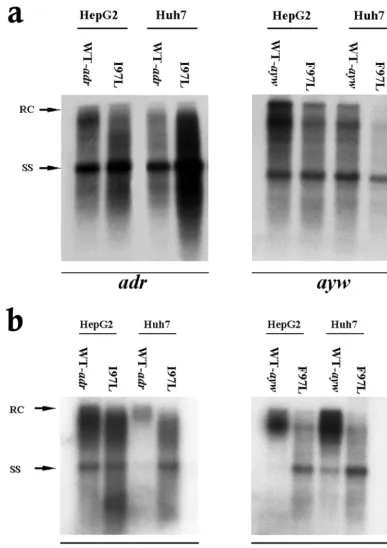
As shown in Fig. 1a, left panel, mutant I97L appeared to replicate approximately 4.8-fold better than its wild-type coun-terpart in Huh7 cells (based on five independent experiments with at least four different plasmid DNA preparations). This difference in replication between wild-type HBV and mutant I97L in Huh7 cells could originate from differences other than
the I97L mutation per se. Variable quality and concentrations of plasmid DNA preparations or the existence of unknown, unwanted mutations present in the mutant or wild-type plas-mids could also account for the differences. An argument against such trivial explanations came from the experiment in HepG2 cells. In HepG2 cells, mutant I97L replicated only slightly better than wild-typeadrby 1.7-fold (Fig. 1a, left pan-el). In contrast to subtype adr, the overall DNA replication efficiency of aywwild-type was slightly better (less than two-fold) than its mutant F97L in both HepG2 and Huh7 cells (Fig. 1a, right panel).
Subtypeadrmutant I97L can secrete more virions than its parental wild-type virus in Huh7 cells.To investigate whether the apparent intracellular replication advantage ofadrmutant I97L could also be observed at the extracellular level, we ex-amined the HBV DNA profile of secreted virions. Extracellu-lar viral DNAs were collected from the medium and analyzed by cesium chloride gradient ultracentrifugation and Southern blot analysis. Previously, we demonstrated an immature secre-tion phenotype for capsid mutant 97L in both theadrandayw
subtypes, characterized by nonselective and excessive secretion of HBV virions containing immature replicative intermediates (70, 71). As shown in Fig. 1b, capsid mutant 97L indeed ex-hibited an immature secretion phenotype in both Huh7 and HepG2 cells. In addition, we found that similar levels (1.6-fold) of viral DNA were secreted by wild-typeadrand mutant I97L in HepG2 cells; however, in Huh7 cells, mutant I97L secreted an average of 7.5-fold-more virion DNA than its parental wild-type virus (based on four independent experiments) (left panel of Fig. 1b). In the right panel of Fig. 1b, theaywwild-type HBV secreted slightly more virions than the mutant F97L in HepG2 and Huh7 cells. These findings were consistent with the results of the intracellular assay in Fig. 1a.
Replication advantage of mutant I97L over wild-type HBV occurs in another hepatoma cell line, J3. Intrigued by the replication advantage of mutant I97L in Huh7 cells, we deter-mined if the same phenomenon would occur in other hepa-toma cell lines. A number of different hepahepa-toma cell lines (J2, J3, J5, HCC36, Hep3B, PLC/PRF/5, and FOCUS) (Materials and Methods) were screened for possible replication advan-tages for the mutant. Most of these cell lines did not appear to support detectable replication of the wild-type or mutant HBVs (data not shown). As shown in Fig. 2, only the J3 cell line showed preferential replication permissivity for mutant I97L. We also found that Hep3B cells behaved like HepG2 cells, with no pronounced phenotype of replication advantage for the mutant (data not shown). In both J3 and Hep3B cells, the overall signals were much weaker than in HepG2 cells.
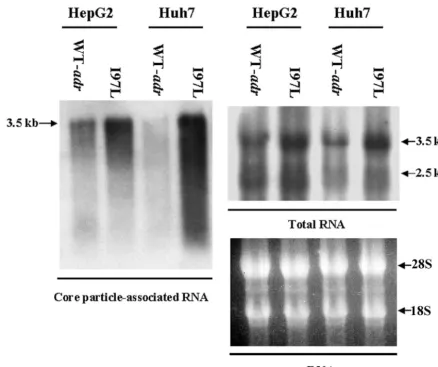
Higher level of encapsidated pregenomic RNA byadrcapsid mutant I97L. To better understand the mechanism of the increased replication efficiency of mutant I97L in Huh7 cells, we asked if there were any differences in RNA encapsidation between the mutant and the wild-type virus. Total intracellular and core-associated RNAs were collected 5 days after trans-fection and analyzed on Northern blots. As shown in the right panel of Fig. 3, the total RNA levels from cells transfected with wild-type HBV or mutant I97L were different by about twofold in HepG2 and Huh7 cells. However, in the left panel of Fig. 3, we found that the level of encapsidated pregenomic RNA of mutant I97L was significantly higher. The difference in the
12070 SUK ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 1. Significantly increased viral DNA synthesis and virion secretion of HBVadrcapsid mutant I97L in Huh7 cells but not in HepG2 cells. (a) Ten micrograms of each plasmid DNA was transfected into HepG2 and Huh7 cells. Intracellular core particles were harvested 5 days post-transfection, and the core particle-associated DNA was analyzed by Southern blot analysis with a 3.1-kbadrsubtype oraywsubtype HBV probe. In comparison with wild-type HBV (WT-adr), there was at least a fourfold increase in overall DNA signal for mutant I97L in Huh7 cells, while in HepG2 cells, wild-typeadrand mutant I97L did not exhibit any significant difference in replication (left panel). In contrast to subtypeadr, the overall DNA signal of wild-typeaywHBV was about twofold stronger than that of the mutant F97L in both HepG2 and Huh7 cells (right panel). (b) Mutant I97L secreted more HBV virions than wild-typeadrin Huh7 cells but not in HepG2 cells (left panel). This phenomenon was not observed in mutant F97L in the HBVaywsubtype (right panel). Viral particles were purified from the medium through a 20% sucrose cushion, and the HBV Dane particles were then separated by isopycnic gradient centrifugation through a 20 to 50% (wt/vol) cesium chloride gradient. Dane particles were collected according to their buoyant densities. Extracellular HBV DNA was extracted and subjected to Southern blot analysis. Full-length relaxed-circle form (RC) HBV DNA at 4.0 kb and single-stranded (SS) HBV DNA replicative intermediates at 1.5 kb are indicated by arrows.
on November 8, 2019 by guest
http://jvi.asm.org/
encapsidated RNA levels between adr wild-type and mutant I97L was more prominent in Huh7 cells than in HepG2 cells (approximately 5.7-fold in Huh7 cells and 2-fold in HepG2 cells, according to quantitative measurements of the signal intensities).
Similar stabilities of wild-type and mutant core proteins.
The contrasts in intracellular HBV DNA replication and pre-genomic RNA encapsidation between adr wild-type and mu-tant I97L could be due to the different stabilities of their respective core proteins. We therefore determined the steady-state levels of wild-type and mutant core proteins in Huh7 cells by immunoblot analysis with a rabbit anticore antibody (Fig. 4). When the tandem dimer replicons of the wild-type and mutant I97L viruses were used, we found that there was a fivefold difference in the steady-state levels of the core pro-teins, which is consistent with the data from the intracellular viral DNA replication and pregenomic RNA encapsidation assays (Fig. 1 and 3). However, when simian virus 40 expres-sion vectors carrying the wild-type and mutant core genes were used, similar steady-state levels of their core proteins were observed (Fig. 4). These findings suggest that the reduced core protein level from the wild-type HBV replicon is not caused by an intrinsic instability of the wild-type core protein in Huh7 cells. Rather, it most likely reflects the lower level of wild-type
adrRNA and DNA replication in Huh7 cells.
Increased intracellular HBV DNA synthesis of mutant I97L in Huh7 cells could be caused by acquisition of a cytosine residue at nucleotide 2191 and/or by acquisition of a leucine at core protein position 97.We screened 20 serum samples from noncirrhotic HBV carriers (60) and identified the presence of mutant 97L in 15 out of 20 samples, including mutants I97L (15 of 20) and I97F (2 of 20) (two samples contained both I97L and I97F) (F. M. Suk and C. Shih, unpublished data). While mutant I97L is caused by an A-to-C mutation at nucleotide 2191, mutant I97F is an A-to-T mutation at the same position. We asked if the phenotype was caused by the loss of an ade-nine at nucleotide 2191, or an isoleucine residue at amino acid 97, or by the acquisition of a cytosine or a leucine residue.
We created mutant I97F (A2191T) and compared its repli-cation with that of wild-type HBV and mutant I97L (A2191C) in both HepG2 and Huh7 cells. As shown in Fig. 5, Southern blot analysis of intracellular core-associated DNA revealed a similar level of overall DNA signals between wild-type, mutant I97L (A2191C), and mutant I97F (A2191T) in HepG2 cells. Interestingly, in Huh7 cells, only mutant I97L (A2191C), but not mutant I97F (A2191T), exhibited an increased level of intracellular viral DNA synthesis. These results suggest that the increased viral DNA synthesis of mutant I97L in Huh7 cells is not caused by the loss of an adenine residue at nucleotide 2191 of pregenomic RNA or by the loss of an isoleucine resi-due at core amino acid 97. Instead, it is caused by the acqui-sition of a cytosine residue at nucleotide 2191 of the pre-genomic RNA or by the acquisition of a leucine at core protein position 97.
DISCUSSION
The intriguing replication advantage of HBV core mutant I97L in Huh7 cells was observed reproducibly in a number of different assays, including viral DNA synthesis, virion
secre-tion, RNA encapsidasecre-tion, and immunoblot analyses (Fig. 1, 3, and 4). The potential mechanisms and significance of this phe-nomenon are discussed further below.
Potential explanations for higher level of encapsidated pre-genomic RNA of mutant I97L.Assembly of a replication-com-petent HBV nucleocapsid requires at least three viral compo-nents: core protein, polymerase, and pregenomic RNA. The polymerase exists in virions at a rate-limiting trace quantity during the formation of the nucleocapsid-polymerase-pre-genomic RNA complex. Without further experimentation, it is difficult to hypothesize what mechanism could be responsible for the replication advantage of mutant I97L in Huh7 cells.
[image:4.603.306.537.69.334.2]In this study, we detected a twofold difference in total RNA levels between mutant I97L and wild-type virus in both HepG2 and Huh7 cells (Fig. 3). Therefore, enhanced viral transcrip-tion or RNA stability is likely to contribute to the higher level of core protein product (Fig. 4), higher core-specific RNA transcript level as a substrate for pregenomic RNA encapsida-tion (Fig. 3), more frequent molecular interacencapsida-tions during core-polymerase-pregenomic RNA complex formation, and fi-nally the replication advantage of mutant I97L in Huh7 cells. Encapsidation of HBV pregenomic RNA is mediated by a specificcis-acting sequence, termed⌭, located at the 5⬘end of the pregenomic RNA (32). In duck hepatitis B virus, two discontinuous regions of the pregenome are required (7, 25). At present, there is no experimental evidence that an addi-tional RNA packaging signal or “packaging enhancer” may have been created and selected for by the mutation I97L in Huh7 cells.
FIG. 2. Replication advantage phenotype of mutant I97L was ob-served in another hepatoma cell line, J3. Among a number of different hepatoma cell lines tested (see text for detail), only J3 was reproduc-ibly found to behave like Huh7 cells. The Southern blot assay for viral DNA synthesis was done as described for Fig. 1a.
12072 SUK ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
Previously, we observed a significant negativeciseffect on plus-strand DNA synthesis by the codon 97 mutation in ayw
mutant F97L (70). Furthermore, amino acid position 97 is located within the frequent core internal deletion (CID) region of HBV CID variants, which have been proposed to behave like defective interfering particles (68, 69). A recent study of HBV CID variants also suggests that it may be the deleted pregenomic RNA rather than the deleted core protein that is responsible for interference (51). Taken together, these find-ings suggest there might be an unknown “ciselement” around nucleotide 2191 of the core gene which could influence HBV pregenomic RNA encapsidation and DNA replication. A pre-liminary computer-aided search for any conserved secondary or higher-order structures around nucleotide 2191 of the pre-genomic RNA has not been successful (data not shown).
In addition to interacting with the core protein (38), the polymerase is also involved in RNA encapsidation. In the pro-cess of HBV replication, polymerase binds to the stem loop of the⌭signal and initiates reverse transcription (52, 62). Since mutation I97L is located outside the polymerase open reading frame, there is no concurrent mutation within the polymerase
protein in mutant I97L. Finally, although it cannot be excluded at present, it appears unlikely that the subtle A-to-C nucleotide change in mutant I97L, which is located about 119 nucleotides upstream from the initiation codon of polymerase, could in-crease the production of polymerase.
In addition to viral factors, cellular factors such as heat shock proteins are known to be involved in RNA-protein com-plex formation and priming of DNA synthesis (4, 28). In the process of pregenomic RNA encapsidation, these cellular fac-tors could influence the pregenomic RNA encapsidation effi-ciency by binding to core proteins, polymerase, the⌭signal, or some unknown RNA structures in the vicinity of nucleotide 2191 in HBV.
[image:5.603.74.512.67.434.2]Asian adult-derived versus Caucasian child-derived hepa-toma cells.The HepG2 cell line is derived from a hepatoblas-toma from a Caucasian child (34), whereas the Huh7 cell line is from a hepatocellular carcinoma from an Asian adult (44). Hepatoblastoma cells are more related to fetal liver cells and are morphologically different from hepatocellular carcinoma cells. The ultrastructural difference may be due to different degrees of hepatic tumor differentiation (26). Moreover, the
FIG. 3. Mutant I97L encapsidated more pregenomic RNA than its parental wild-type virus in both HepG2 and Huh7 cells. Five days posttransfection, HepG2 and Huh7 cells were harvested, and total RNA (right panel) and core-associated RNA (left panel) were isolated. The entire amount of core-associated RNA from one 10-cm dish and 30g of total RNA from each sample were analyzed by Northern blot analysis with a 3.1-kb HBV double-stranded DNA probe. The difference in encapsidated RNA levels between the mutant I97L and wild-type viruses was more dramatic in Huh7 cells than in HepG2 cells. Major HBV-specific transcripts are indicated by arrows.
on November 8, 2019 by guest
http://jvi.asm.org/
content of transcription factors in these two cell lines also differs (45).
In this study, we screened several additional hepatoma cell lines for potential replication advantages for mutant I97L (Ma-terials and Methods). Another Asian adult-derived hepatoma cell line, J3 (10), appears to be supportive for mutant I97L but not wild-type HBV replication (Fig. 2). In contrast, another Caucasian child-derived well-differentiated hepatoma cell line, Hep3B (34), can support both mutant I97L and wild-type HBV at a similar level, although overall DNA replication is reduced compared to that in HepG2 cells (data not shown). This again suggests the involvement of some unknown host factors in HBV replication.
Biological significance of host factor-independent pheno-type of mutant I97L and its emergence in chronic carriers.
HBV replication is known to depend on the state of hepatocyte differentiation and can be affected by a variety of host factors (19, 30, 31, 47, 49). During the course of HBV infection, the state of hepatocyte proliferation and differentiation is altered due to chronic inflammation and liver injury, which could be accompanied by a progressive decline in some host cell factors. Progressive decline in viremia has been observed at the later stage of HBV chronic infection with liver diseases (11, 18). It could be explained by the gradual loss of host factors involved in viral replication permissivity in the dedifferentiating hepa-tocytes and by the emergence of secretion-defective HBV vari-ants (36, 70, 71). Certain host factors which can support wild-type HBV replication could be present at a higher level in HepG2 cells and absent or present at a lower level in Huh7 cells. Without these host factors, the replication efficiency of wild-type HBV is greatly reduced in Huh7 cells. We hypothe-size here that HepG2 mimics the well-differentiated hepato-cytes at the earlier stage of chronic infection, and Huh7 and J3 mimic less well differentiated hepatocytes at the later stage of chronic infections.
From this hypothesis, we suggest that the biological signifi-cance of core mutation I97L is to maintain viral replication in gradually dedifferentiating host hepatocytes and thus help pro-long the viral persistence at the middle to late stage of chronic
[image:6.603.81.498.71.215.2]infection. Indeed, emergence of mutation I97L has been ob-served in a longitudinal study of a chronic hepatitis B patient (16). We detected a significant level of mutant I97L in 75% of serum samples from chronic HBV carriers (data not shown) as well as in many liver samples from hepatoma patients in Tai-wan (27), a result consistent with its prevalence in Asia and
FIG. 5. Increased intracellular viral DNA synthesis of mutant I97L in Huh7 cells is likely to be caused by the acquisition of a cytosine residue at nucleotide 2191 or by the acquisition of a leucine residue at amino acid 97 of HBV core antigen. Plasmid DNAs of wild-typeadr
[image:6.603.305.538.394.628.2](WT-adr), mutant I97L (A2191C), or mutant I97F (A2191T) were transfected into both HepG2 and Huh7 cells. HBV core particle-associated DNA was harvested 5 days after transfection and analyzed by Southern blot analysis with an HBV double-stranded DNA probe. In Huh7 cells, mutant I97L (A2191C) but not mutant I97F (A2191T) showed increased overall HBV DNA synthesis.
FIG. 4. Comparison of steady-state levels of wild-type and mutant core proteins produced from virus replicons and protein expression vectors. Cell lysates of Huh7 cells collected 3 days posttransfection were analyzed by Western blot analysis with a rabbit anticore antibody. Plasmids SVC-WT and SVC-I97L are two core protein expression vectors of wild-type (WT) HBV and mutant I97L origins under the control of the simian virus 40 enhancer and promoter. The fact that they produced similar steady-state levels of wild-type and mutant core proteins indicated a similar degree of protein stability. Therefore, the difference in the steady-state level of core proteins expressed from the tandem dimer replicons of wild-typeadrand mutant I97L reflects their difference in intracellular viral DNA replication rather than any difference in intrinsic protein stability.
12074 SUK ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
elsewhere (1, 5, 14, 15, 16, 17, 21, 22, 29, 33, 43, 50, 61, 64, 65, 66). Finally, it should be noted that previous reports also sug-gest that core protein mutations could have an immune escape nature (5, 16, 27). The immune escape hypothesis is not nec-essarily mutually exclusive with our current replication advan-tage hypothesis regarding the emergence of the core variant I97L.
In addition to the aforementioned hypothesis, at least two alternative possibilities can be entertained. One is to assume the presence of a negative factor in Huh7 cells that is absent in HepG2 cells. Such a negative factor can selectively repress the replication of wild-type HBV but not mutant I97L in Huh7 cells. Another hypothesis is to assume that a positive factor is present in Huh7 cells but is low or absent in HepG2 cells. Such a positive factor may interact better with mutant I97L pre-genomic RNA than with wild-type HBV.
Replication efficiency of capsid variants is subtype depen-dent.Subtypeaywis prevalent in northwestern Europe, while subtypeadr is confined to southeastern Asia (39, 57, 58). An 8.3 to 9.3% nucleic acid sequence divergence is observed be-tween HBV subtypes (46). There are at least eight consistent differences in the 183 amino acids of HBV core antigen be-tween the adr and ayw subtypes (71). In this study and our previous work, we demonstrated a 1.8-fold decrease in the overall intracellular HBV DNA levels withaywmutant F97L in Huh7 cells (70). In contrast, the adr subtype in Huh7 cells showed, surprisingly, that overall intracellular DNA levels of mutant I97L increased by approximately 4.8-fold relative to its parental wild-type virus (Fig. 1). This indicates that the effect of the change to 97L on viral replication depends on the sub-type-specific context of its pregenomic RNA (Fig. 5). Our observations of the replication advantage foradrmutant I97L, as well as the replication disadvantage foraywmutant F97L, may explain why mutant I97L is more prevalent in Asia (1, 5, 16, 17, 21, 29, 33, 61, 64, 65) and predict the low prevalence of
aywmutant F97L in Europe (40, 41, 48).
Evolution and the trade-off between good and bad muta-tions.If the replication advantage of mutant I97L can be ex-tended to natural infection in vivo, then why has mutant I97L not replaced the wild-type HBV in acute infection or during the long journey of viral evolution? It is possible that it cannot outcompete wild-type virus because the well-differentiated hepatocytes in acutely infected hosts offer no significant selec-tive advantage for the mutant I97L. Presently, without an avail-able in vitro infection system, we can only speculate that im-mature secretion of mutant I97L could be a “bad” mutation which compromises infectivity by causing the virus to secrete immature genomes (i.e., negative selection).
Several naturally occurring core mutations, including P5T and L60V, have been found to result in low virion secretion (36). The biological significance of such seemingly bad muta-tions again remains unclear, since they appear to be a disser-vice to the virus per se. Since these mutations also coincide with T-cell epitopes (13, 42), they are more likely to be “good” mutations with an immune escape nature (27, 59, 60). There-fore, the coevolution of HBV variants and their host hepato-cytes probably depends on the balanced effects between good and bad mutations.
In summary, in addition to the previously identified imma-ture secretion phenotype (70, 71), the newly identified
pheno-type of replication advantage for the core mutation I97L ap-pears to be beneficial for the virus by improving its survival chance in a changing, unfriendly hepatocyte environment. Fur-ther studies on the mechanisms of the replication advantage, host factor independence, immune escape, and the evolution of mutant I97L should help clarify the biological significance of the mutant in natural infection.
ACKNOWLEDGMENTS
We thank our colleagues in C. Shih’s laboratory for careful reading of the manuscript and M. J. Chou and C. S. Yang for the J3 cell line. F.-M.S. is supported in part by Taipei Medical University Hospital and Juei-Low Sung’s Research Foundation, Taiwan. This study was supported in part by NIH grants RO1 CA 70336 and CA84217 to C.S.
REFERENCES
1. Akarca, U. S., and A. S. F. Lok.1995. Naturally occurring hepatitis B virus
core gene mutations. Hepatology22:50–60.
2. Alexander, J. J., E. M. Bey, E. W. Geddes, and G. Lecatsas.1976. Establish-ment of a continuously growing cell line from primary carcinoma of the liver.
S. Afr. J. Med.50:2124–2128.
3. Beames, B., and R. E. Lanford.1993. Carboxy-terminal truncations of the HBV core protein affect capsid formation and the apparent size of the
encapsidated HBV RNA. Virology194:597–607.
4. Beck, J., and M. Nassal.2001. Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase
domains expressed inEscherichia coli. J. Virol.75:7410–7419.
5. Bozkaya, H., B. Ayola, and A. S. F. Lok.1996. High rate of mutations in the hepatitis B core gene during the immune clearance phase of chronic hepatitis
B virus infection. Hepatology24:32–37.
6. Buendia, M. A.2000. Genetics of hepatocellular carcinoma. Semin. Cancer
Biol.10:185–200.
7. Calvert, J., and J. Summers.1994. Two regions of an avian hepadnavirus
RNA pregenome are required incisfor encapsidation. J. Virol.68:2084–
2090.
8. Carman, W. F., W. Boner, G. Fattovich, K. Colman, E. S. Dornan, M. Thursz, and S. Hadziyannis.1997. Hepatitis B virus core protein mutations are concentrated in B cell epitopes in progressive disease and in T helper cell
epitopes during clinical remission. J. Infect. Dis.175:1093–1100.
9. Chang, S. F., H. J. Netter, M. Bruns, R. Schneider, K. Frolich, and H. Will.
1999. A new hepadnavirus infecting snow geese (Anser caerulescens)
pro-duces a significant fraction of virions containing single-stranded DNA.
Vi-rology262:39–54.
10. Chang, K. S., M. J. Chou, S. Tsai, and C. S. Yang.1995. Biological charac-teristics of human hepatocellular carcinoma cell lines without prior nude
mouse passages. Chin. J. Microbiol. Immunol.28:167–178.
11. Chen, D. S.1993. From hepatitis to hepatoma: lessons from type B viral
hepatitis. Science262:369–370.
12. Chen, J. Y., T. J. Harrison, D. J. Tsuei, T. Y. Hsu, A. J. Zuckerman, T. S. Chan, and C. S. Yang.1994. Analysis of integrated hepatitis B virus DNA and flanking cellular sequences in the hepatocellular carcinoma cell line
HCC36. Intervirology37:41–46.
13. Chisari, F. V., and C. Ferrari.1995. Hepatitis B virus immunopathogenesis.
Annu. Rev. Immunol.13:29–60.
14. Chuang, W. L., M. Omata, T. Ehata, O. Yokosuka, Y. Ito, and M. Ohto.1993. Concentrating missense mutations in core gene of hepatitis B virus. Digest.
Dis. Sci.38:594–600.
15. Chuang, W. L., M. Omata, T. Ehata, O. Yokosuka, Y. Ito, F. Imwki, S. N. Lu, W. Y. Chang, and M. Ohto.1993. Precore mutations and core clustering
mutations in chronic hepatitis B virus infection. Gastroenterology104:263–
271.
16. Ehata, T., M. Omata, O. Yolosuka, K. Hosoda, and M. Ohto.1992. Varia-tions in codons 84–101 in the core nucleotide sequence correlate with hep-atocellular injury in chronic hepatitis B virus infection. J. Clin. Investig.
89:332–338.
17. Ehata, T., M. Omata, W. L. Chuang, O. Uokosuka, Y. Ito, K. Hosoda, and M. Ohto.1993. Mutations in core nucleotide sequence of hepatitis B virus
correlate with fulminant and severe hepatitis. J. Clin. Investig.91:1206–1213.
18. Evans, A. A., A. P. O’Connell, J. C. Pugh, W. S. Mason, F. Shen, G.-C. Chen, W.-Y. Lin, A. Dia, S. M’Boup, B. Drame, and W. T. London.1998. Geo-graphic variation in viral load among hepatitis B carriers with differing risks
of hepatocellular carcinoma. Cancer Epidemiol. Biomarker Prevent.7:559–
565.
19. Galle, P. R., H. J. Schlicht, C. Kuhn, and H. Schaller.1989. Replication of duck hepatitis B virus in primary duck hepatocytes and its dependence on the
state of differentiation of the host cell. Hepatology4:459–465.
20. Ganem, D., and R. Schneider.2001. Hepadnaviridae: the viruses and their
on November 8, 2019 by guest
http://jvi.asm.org/
replication, p. 2923–2970.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott Williams & Wilkins, Philadelphia, Pa.
21. Gotoh, K., S. Mima, T. Uchida, T. Shikata, K. Yoshizawa, M. Irie, and M. Mizui.1995. Nucleotide sequence of hepatitis B virus isolated from subjects
without serum anti-hepatitis B core antibody. J. Med. Virol.46:201–206.
22. Gunther, S., S. Baginski, H. Kissel, P. Reinke, D. H. Kruger, H. Will, and H. Meisel.1996. Accumulation and persistence of hepatitis B virus core gene deletion mutants in renal transplant patients are associated with end-stage
liver disease. Hepatology24:751–758.
23. Gunther, S., L. Fischer, I. Pult, M. Sterneck, N. Piwon, and H. Will.1999.
Naturally occurring variants of hepatitis B virus. Adv. Virus Res.52:25–137.
24. He, L., K. J. Isselbacher, J. R. Wands, H. M. Goodman, C. Shih, and A. Quaroni.1984. Establishment and characterization of a new human
hepa-tocellular carcinoma cell line. In Vitro20:493–504.
25. Hirsch, R. C., D. D. Loeb, J. R. Pollack, and D. Ganem.1991.cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic
RNA. J. Virol.65:3309–3316.
26. Horie, A., Y. Kotoo, and I. Hayashi.1979. Ultrastructural comparison of
hepatoblastoma and hepatocellular carcinoma. Cancer44:2184–2193.
27. Hosono, S., P. C. Tai, W. Wang, M. Ambrose, D. Hwang, T. T. Yuan, B. H. Peng, C. S. Yang, C. S. Lee, and C. Shih.1995. Core antigen mutations of human hepatitis B virus in hepatomas accumulate in MHC class II-restricted
T cell epitopes. Virology212:151–162.
28. Hu, J., D. Toft, D. Anselmo, and X. Wang.2002. In vitro reconstitution of functional hepadnavirus reverse transcriptase with cellular chaperone
pro-tein. J. Virol.76:269–279.
29. Hur, G. M., Y. I. Lee, D. J. Sun, J. H. Lee, and Y. I. Lee.1996. Gradual accumulation of mutations in precore/core region of HBV in patients with chronic active hepatitis: implication of clustering changes in a small region of
the HBV core region. J. Med. Virol.48:38–46.
30. Isom, H. C., T. Secott, I. Georgoff, C. Woodworth, and J. Mummaw.1985. Maintenance of differentiated rat hepatocytes in primary culture. Proc. Natl.
Acad. Sci. USA82:3252–3256.
31. Isom, H. C., I. Georgoff, M. Salditt-Georgieff, and J. E. Darnell.1987. Persistence of liver-specific messenger RNA in cultured hepatocytes:
differ-ent regulatory evdiffer-ents for differdiffer-ent genes. J. Cell Biol.105:2877–2885.
32. Junker-Niepmann, M., R. Bartenschlager, and H. Schaller.1990. A short
cis-acting sequence is required for hepatitis B virus pregenome
encapsida-tion and sufficient for packing of foreign RNA. EMBO J.9:3389–3396.
33. Karasawa, T., T. Shirasawa, Y. Okawa, A. Kuramoto, N. Shimada, Y. Aizawa, M. Zeniya, and G. Toda.1997. Association between frequency of amino acid changes in core region of hepatitis B virus (HBV) and the presence of precore mutation in Japanese HBV carriers. J. Gastroenterol.
32:611–622.
34. Knowles, B. B., C. C. Howe, and D. P. Aden.1980. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B
sur-face antigen. Science209:497–499.
35. Kobayashi, M., and K. Koike.1984. Complete nucleotide sequence of hep-atitis B virus DNA of subtype adr and its conserved gene organization. Gene
30:227–232.
36. Le Pogam, S., T. T. Yuan, G. K. Sahu, S. Chatterjee, and C. Shih.2000. Low-level secretion of human hepatitis B virus virions caused by two inde-pendent, naturally occurring mutations (P5T and L60V) in the capsid
pro-tein. J. Virol.74:9099–9105.
37. Le Pogam, S., and C. Shih.2002. Influence of a putative intermolecular interaction between core and the pre-S1 domain of the large envelope
protein on hepatitis B virus secretion. J. Virol.76:6510–6517.
38. Lott, L., B. Beames, L. Notvall, and R. E. Lanford.2000. Interaction between
hepatitis B virus core protein and reverse transcriptase. J. Virol.74:11479–
11489.
39. Magnius, L. O., and H. Norder.1995. Subtypes, genotypes and molecular epidemiology of the hepatitis B virus as reflected by sequences variability of
the S-gene. Intervirology38:24–34.
40. Mayerat, C., A. Mantegani, F. Spertini, and P. C. Frei.1999. Mutations in the basal core promoter and precore/core gene of hepatitis B virus in pa-tients with chronic active but not acute hepatitis B. Eur. J. Clin. Microbiol.
Infect. Dis.18:871–878.
41. McMillan, J. S., D. S. Bowden, P. W. Angus, G. W. McCaughan, and S. A. Locarnini.1996. Mutations in the hepatitis B virus precore/core and core promoter in patients with severe recurrent disease following liver
transplan-tation. Hepatology24:1371–1378.
42. Milich, D. R.1997. Immune response to the hepatitis B virus: infection,
animal models, and vaccination. Viral Hepatitis3:63–103.
43. Miska, S., S. Gunther, M. Vassilev, H. Meisel, G. Pape, and H. Will.1993. Heterogeneity of hepatitis B virus C-gene sequences: implications for
am-plification and sequencing. J. Hepatol.18:53–61.
44. Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato.1982. Growth of human hepatoma cell lines with differentiated function in
chem-ically defined medium. Cancer Res.42:3858–3863.
45. Ninomiya, T., Y. Hayashi, K. Saijoh, K. Ohat, S. Yoon, H. Nakabayashi, T. Tamaoki, M. Kasuga, and H. Itoh.1996. Expression ratio of hepatocyte
nuclear factor-1 to variant hepatocyte nuclear factor-1 in differentiation of
hepatocellular carcinoma and hepatoblastoma. J. Hepatol.25:445–453.
46. Okamoto, H., F. Tusda, H. Sakugawa, R. I. Sastrosoewignijo, M. Imai, Y. Miyakawa, and M. Mayumi.1998. Typing hepatitis B virus by homology in nucleotide sequence: comparison of surface antigen subtypes. J. Gen. Virol.
69:2557–2583.
47. Ozer, A., V. I. Khaoustov, M. mearns, D. E. Lewis, R. M. Genta, G. J. Darlington, and B. Yoffe.1996. Effect of hepatocyte proliferation and
cellu-lar DNA synthesis on hepatitis B virus replication. Gastroenterology110:
1519–1528.
48. Pollicino, T., S. Campo, and G. Raimondo.1995. PreS and core gene het-erogeneity in hepatitis B virus (HBV) genomes isolated from patients with
long-lasting HBV chronic infection. Virology208:672–677.
49. Pugh, J. C., and J. W. Summers.1989. Infection and uptake of duck hepatitis B virus by duck hepatocytes maintained in the presence of dimethyl
sulfox-ide. Virology172:564–572.
50. Radecke, K., U. Protzer, M. Trippler, K. M. zum Buschenfelde, and G. Gerken.2000. Selective of hepatitis B virus variants with amino acid substi-tutions inside the core antigen during interferon-alpha therapy. J. Med.
Virol.62:479–486.
51. Sahu, G. K., P. C. Tai, S. Banerjee, M. H. Lin, B. Tennant, J. Gerin, and C. Shih.2002. Out-of-frame versus in-frame core internal deletion (CID)
vari-ants of human and woodchuck hepatitis B viruses. Virology292:35–43.
52. Seeger, C., and J. Maragos.1991. Identification of a signal necessary for initiation of reverse transcription of the hepadnavirus genome. J. Virol.
65:5190–5195.
53. Seeger, C., and W. S. Mason.2000. Hepatitis B virus biology. Microbiol. Mol.
Biol. Rev.64:51–68.
54. Shih, C., L. S. Li, S. Roychoudhuary, and M. H. Ho.1989. In vitro propa-gation of human hepatitis B virus in a rat hepatoma cell line. Proc. Natl.
Acad. Sci. USA86:6323–6327.
55. Shih, C., P. C. Tai, W. Whitehead, S. Hosono, C. S. Lee, and C. S. Yang.
1996. Hepatitis B and C viruses and liver cancer, p. 824–834.InJ. R. Bertino
(ed.), Encyclopedia of cancer, vol. II. Academic Press, Inc., London, United Kingdom.
56. Slagle, B. L., T. H. Lee, and J. S. Butel.1992. Hepatitis B virus and
hepa-tocellular carcinoma. Prog. Med. Virol.39:167.
57. Sung, J. L.1981. Hepatitis B virus infection and its sequelae in Taiwan. Proc.
Natl. Sci. Counc. Repub. China B5:385–399.
58. Sung, J. L., and D. S. Chen.1977. Geographical distribution of the
sub-type of hepatitis B surface antigen. Gastroenterol. Jpn.12:58–63. (In
Chinese.)
59. Tai, P. C., D. Banik, G. I. Lin, S. Pai, K. Pai, M. H. Lin, G. Yuoh, S. Che, S. H. Hsu, T. C. Chen, T. T. Kuo, C. S. Lee, C. S. Yang, and C. Shih.1997. Novel and frequent mutations of hepatitis B virus coincide with a major histocompatibility complex class I-restricted T-cell epitope of the surface
antigen. J. Virol.71:4852–4856.
60. Tai, P. C., F. M. Suk, W. Gerlich, R. Neurath, and C. Shih.2002. Hyper-modification and immune escape of an internally deleted middle envelope (M) protein of frequent and predominant hepatitis B virus variants. Virology
292:44–58.
61. Takahashi, K., Y. Akahane, K. Hino, Y. Ohta, and S. Mishiro.1998. Hep-atitis B virus genomic sequence in the circulation of hepatocellular carci-noma patients: comparative analysis of 40 full-length isolation. Arch. Virol.
143:2313–2326.
62. Tavis, J. E., S. Perri, and D. Ganem.1994. Hepadnavirus reverse transcrip-tion initiates within the stem-loop of the RNA packing signal and employs a
novel strand transfer. J. Virol.68:3536–3543.
63. Tencza, M. G., and J. E. Newbold.1997. Heterogeneous response for a mammalian hepadnavirus infection to acyclovir: drug-arrested intermediates of minus-strand viral DNA synthesis are enveloped and secreted from
in-fected cells as virion-like particles. J. Med. Virol.51:6–16.
64. Tsubota, A., H. Kumada, K. Takaki, K. Chayama, M. Kobayashi, M. Koba-yashi, Y. Suzuki, S. Saitoh, Y. Arase, N. Murashima, and K. Ikeda.1998. Deletion in the hepatitis B virus core gene may influence the clinical out-come in hepatitis B e antigen-postive asymptomatic healthy carriers. J. Med.
Virol.56:287–293.
65. Uchida, T., T. T. Aye, T. Shihata, M. Yano, H. Yatsuhashi, M. Koga, and S. Mima.1994. Evolution of the hepatitis B virus gene during chronic infection
in seven patients. J. Med. Virol.43:148–154.
66. Valliammai, T., S. P. Thyagarajan, A. J. Zuckerman, and T. J. Harrison.
1995. Precore and core mutations in HBV from individuals in India with
chronic infection. J. Med. Virol.45:321–325.
67. Yuan, T. T., A. Faruqi, J. W. K. Shih, and C. Shih.1995. The mechanism of natural occurrence of two closely linked HBV precore predominant
muta-tions. Virology211:144–156.
68. Yuan, T. T., M. H. Lin, D. S. Chen, and C. Shih.1998. A defective interfer-ence-like phenomenon of human hepatitis B virus in chronic carriers. J.
Vi-rol.72:578–584.
69. Yuan, T. T., M. H. Lin, S. M. Qiu, and C. Shih.1998. Functional character-ization of naturally occurring variants of human hepatitis B virus containing
the core internal deletion mutation. J. Virol.72:2168–2176.
12076 SUK ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
70. Yuan, T. T., G. K. Sahu, W. Whitehead, R. Greenberg, and C. Shih.1999. The mechanism of an “immature secretion” phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B
virus core gene. J. Virol.73:5731–5740.
71. Yuan, T. T., P. C. Tai, and C. Shih.1999. Subtype-independent immature secretion and subtype-dependent replication deficiency of a highly frequent,
naturally occurring mutation of human hepatitis B virus core antigen. J.
Vi-rol.73:10122–10128.
72. Yuan, T. T., and C. Shih.2000. A frequent, naturally occurring mutation (P130T) of human hepatitis B virus core antigen is compensatory of
imma-ture secretion phenotype of another frequent variant (I97L). J. Virol.74:
4929–4932.