Hepatitis C Virus-Like Particle Morphogenesis
Emmanuelle Blanchard,
1Denys Brand,
1Sylvie Trassard,
2Alain Goudeau,
1and Philippe Roingeard
1,2*
Laboratoire de Virologie1et Laboratoire de Biologie Cellulaire,2E3M-EA3250, IFR 82,
Faculté de Médecine et Centre Hospitalier Universitaire, Tours, France Received 4 October 2001/Accepted 9 January 2002
Although much is known about the hepatitis C virus (HCV) genome, first cloned in 1989, little is known about HCV structure and assembly due to the lack of an efficient in vitro culture system for HCV. Using a recombinant Semliki forest virus replicon expressing genes encoding HCV structural proteins, we observed for the first time the assembly of these proteins into HCV-like particles in mammalian cells. This system opens up new possibilities for the investigation of viral morphogenesis and virus-host cell interactions.
Hepatitis C virus (HCV) infection is a major cause of chronic hepatitis and cirrhosis and may lead to hepatocellular carcinoma. With an estimated 170 million people worldwide chronically infected with HCV, this disease has emerged as a serious global health problem since the cloning of the viral genome in 1989 (6). HCV is an enveloped RNA virus belong-ing to the genus Hepacivirus of the Flaviviridae family. Its genome is a 9.6-kb single-stranded RNA of positive polarity with a 5⬘untranslated region (UTR) that functions as an in-ternal ribosome entry site, a single long open reading frame encoding a polyprotein of approximately 3,000 amino acids (aa) and a 3⬘UTR (1). This polypeptide is posttranslationally cleaved by host cell peptidases to yield three structural proteins and by viral proteases, which generate the five nonstructural proteins. The structural proteins, which are located in the amino-terminal region of the polyprotein, include the core protein and the envelope glycoproteins E1 and E2. The non-structural proteins (NS) 2 to 5B are separated from the struc-tural proteins by the short hydrophobic polypeptide p7, the function of which is unknown (1). By analogy to related posi-tive-strand RNA viruses, replication occurs by means of a negative-strand RNA intermediate and is catalyzed by the NS proteins, which form a cytoplasmic membrane-associated rep-licase complex.
Several entire cloned HCV genomes are able to initiate infection when introduced directly into chimpanzee livers (11). However, the transfection of cell lines with constructs contain-ing these genomes does not result in HCV replication in vitro (1). Similarly, the use of infected sera to infect cell lines or primary cell cultures has yielded disappointing results because infection and replication are very inefficient (2). Molecular studies of the HCV infectious cycle in the host cell and the development of specific anti-HCV agents have been consider-ably hampered by the inability to achieve propagation of the virus in cultured cells in vitro. A significant advance in HCV research was recently made with the development of sub-genomic HCV RNAs consisting of sequences encoding non-structural proteins flanked by the 5⬘and 3⬘UTRs, which
self-replicate in hepatoma cells (4, 18). However, this model cannot be used to address the structural features of the virion or its assembly pathway. In addition, viral particles are difficult to observe by electron microscopy in the plasma or liver tissues of infected patients. A number of attempts have been made to obtain recombinant HCV particles, but only Baumert et al. (3) have reported the obtainment of HCV-like particles in insect cells, using recombinant baculoviruses expressing genes for HCV structural proteins. However, although particles contain-ing the three HCV structural proteins were generated, they were unstructured and heterogeneous.
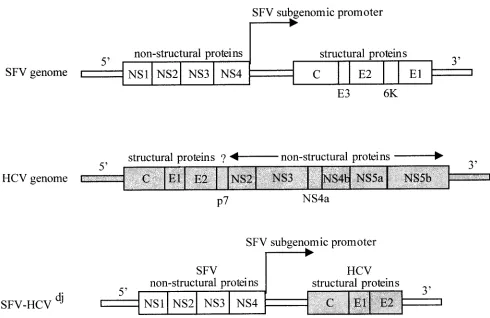
As flaviviruses and alphaviruses have similar structural fea-tures (12) and aspects of their replication strategies in com-mon, we thought that expression from an alphavirus expression vector, such as the Semliki forest virus (SFV) vector (25), might result in the production of HCV-like particles (Fig. 1). Due to the self-amplifying nature of the vector RNA, the SFV vector is constructed in a format known as a replicon, which combines a large host spectrum and the production of large amounts of proteins in transfected cells. Replicons contain both thecisandtransalphavirus genetic elements required for RNA replication and heterologous gene expression via the native subgenomic promoter. Upon introduction into various mammalian cells, the replicon RNA is translated to produce the four nonstructural SFV proteins, which together comprise the alphaviral replicase. Replication proceeds through a mi-nus-strand RNA intermediate and generates two different pos-itive-strand RNA species, corresponding to a genome-length vector RNA and an abundant subgenomic RNA encoding the heterologous proteins.
The HCV C-E1-E2 sequence was amplified from the serum of a patient chronically infected with HCV genotype 1a before antiviral treatment. Briefly, viral RNA was reverse transcribed with SuperScript II (Invitrogen, Carlsbad, Calif.) in the pres-ence of the ext 3⬘primer (5⬘ATAAACATAGGTGCCAGTA AGCG 3⬘). The products of this reaction were incubated for 20 min at 37°C with 2 U of RNase H and were then used for the first PCR with the ext 3⬘and ext 5⬘(5⬘GGAGAGCCATAGT GGTCTGCG 3⬘) primers usingTaqpolymerase (Applied-Bio-systems/Roche, Branchburg, N.J.). We used 35 cycles of 94°C for 15 s, 59°C for 30 s, and 72°C for 3 min. DNA from this first PCR was used as a template for a second reaction using
prim-* Corresponding author. Mailing address: Laboratoire de Biologie Cellulaire, Faculté de Médecine, 2 bis Boulevard Tonnellé, 37032 Tours, France. Phone: (33) 2 47 36 60 71. Fax: (33) 2 47 36 60 90. E-mail: [email protected].
4073
on November 8, 2019 by guest
http://jvi.asm.org/
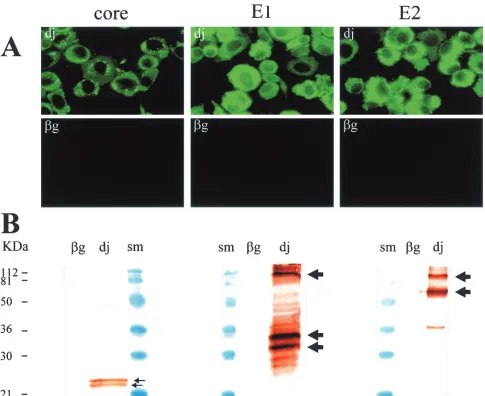
ers flanked byBamHI sites: int 5⬘(5⬘GTGGATCCTGCACC ATGAGCACGAATCCT 3⬘) and int 3⬘(5⬘GAGGATCCCCA TTACCGCCTCCGCTTGGGATAT 3⬘) containing a stop co-don introduced at the 3⬘end of the E2 protein coding region. After 35 cycles as described above, the PCR product (2,270 bp) was inserted into theBamHI site of the expression vector pSFV1 (Life Technologies, Rockville, Md.), which contains an SP6 RNA polymerase promoter upstream from the 5⬘SFV UTR. The electroporation of BHK-21 cells with RNA produced by transcription of this construct, according to standard protocols provided by the manufacturer (Life Technologies), led to the production of large amounts of the three HCV structural pro-teins. More than 90% of the cells tested positive for these proteins by immunocytochemistry performed with previously described protocols (21) and monoclonal antibodies described elsewhere (8) (Fig. 2A). Staining patterns were consistent with those of previous studies based on the expression of individual genes encoding these structural proteins in mammalian cells, showing a cytoplasmic and granular distribution for the core protein (20) and a more homogeneous cytoplasmic distribution for the E1 and E2 proteins (8). For Western blot analysis, transfected BHK-21 cells were lysed with 1% Triton X-100, 140 mM NaCl, 10 mM Tris-HCl (pH 8), 1% sodium deoxy-cholate, 0.1% sodium dodecyl sulfate, 1 mM phenylmethylsul-fonyl fluoride, 2g of aprotinin/ml, and 2g of leupeptin/ml. The lysed cells were subjected to low-speed centrifugation, and the proteins they contained were separated by electrophoresis
in a sodium dodecyl sulfate–12% polyacrylamide gel and trans-ferred to polyvinylidene difluoride membranes. Blots were in-cubated overnight at 4°C with the various monoclonal antibod-ies diluted 1/1,000 in 50 mM Tris-HCl–150 mM NaCl (pH 7.5)–0.1% Tween buffer, and antibody binding was detected by incubation with horseradish peroxidase-conjugated anti-mouse immunoglobulin G antibodies followed by diaminobenzidine staining. This Western blotting showed that the partial poly-protein encoded by the HCVdj construct was correctly
[image:2.587.46.536.67.385.2]pro-cessed by host-cell enzymes, leading to the detection of viral proteins of the expected sizes (Fig. 2B). Monoclonal anti-body directed against the HCV core protein detected similar amounts of two species, approximately 22 and 24 kDa in size. Previous studies have suggested that the larger of these two molecules is a 191-aa core precursor and that the smaller molecule is generated by cleavage of the larger molecule, at around aa 174, by a host signal peptidase on the endoplasmic reticulum (ER) (17, 24). The 22-kDa protein (p22 here but p21 in other studies) was the only protein detected in the sera of chronically infected HCV patients and is believed to correspond to the mature form of the HCV core protein (27). Monoclonal antibody directed against the HCV E1 protein detected two major bands, 32 and 35 kDa in size, correspond-ing to the two main glycosylation forms of the E1 protein (8). Some minor bands, probably corresponding to other glyco-forms of the E1 protein, were also observed in this blot. Mono-clonal anti-E2 antibody detected a major band at 70 kDa,
FIG. 1. Organization of SFV and HCV RNA genomes and description of our SFV-HCVdjDNA construct encoding HCV structural proteins.
Flaviviruses differ from alphaviruses in genome structure (structural genes situated at the 5⬘end of the genome) and in the absence of subgenomic RNA.
4074 NOTES J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
corresponding to the glycosylated form of the E2 protein, as well as a minor band at 38 kDa that probably corresponds to an unglycosylated form of E2 (8). In addition, both anti-E1 and anti-E2 monoclonal antibodies identified a band of around 100 kDa in size which may correspond to E1-E2 heterodimers, as previously described (8). This is important because the E1 and E2 proteins form not only a disulfide-linked heterodimer cor-responding to misfolded aggregates (8, 9) but also a nonco-valently linked heterodimer that probably corresponds to the native prebudding complex (7). Alternatively, we cannot ex-clude the possibility that incomplete cleavage at site between E1 and E2 in the polyprotein generated this 100-kDa band. Interestingly, the level of E1 and E2 proteins obtained with our SFV expression system in mammalian cells seems to be higher than for the baculovirus expression system that allows produc-tion of virus-like particles in insect cells (3, 19). However, the
core protein amount was much lower with the SFV expression system, confirming the previous observation that showed im-portant differences between mammalian and insect cells for the core protein expression (3).
[image:3.587.52.537.72.468.2]The most original data concerning HCV structural protein production from the SFV vector were provided by our electron microscopy (EM) analysis. Transfected cells were fixed in 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) for 48 h and were then postfixed with 1% osmium tetroxide for 1 h. They were then dehydrated in a graded acetone series, and cell pellets were embedded in epon resin that was allowed to polymerize for 24 h at 60°C. Ultrathin sections were cut and stained with 1% uranyl acetate–1% lead citrate. At low magnification (Fig. 3), major ER morphological differences were observed between most of the cells trans-fected with HCVdjRNA cells and cells transfected with-Gal FIG. 2. Production of HCV structural proteins in BHK-21 cells following electroporation. After linearization by digestion withSpeI, the SFV-HCVdjvector was used as a template for the in vitro synthesis of recombinant RNA. For the negative control, recombinant-Gal RNA, which
encodes the-Gal protein, was synthesized from the expression vector pSFV3 (Life Technologies). After electroporation and culture for 24 h, transfected cells were analyzed by immunofluorescence staining (A) and Western blotting (B) with monoclonal antibodies against HCV capsid protein (MAB8424 from Chemicon, Temecula, Calif.), E1 (A4; gift from Harry Greenberg) (7), and E2 (H52; gift from Jean Dubuisson) (7); size markers (SM) were from Bio-Rad (Hercules, Calif.).
on November 8, 2019 by guest
http://jvi.asm.org/
FIG.
3.
Electron
micrographs
at
low
magni
fication
of
ultrathin
sections
of
BHK-21
cells
electroporated
with
the
HCV
djRNA
(A)
or

-Gal
RNA
(B).
Bar,
500
nm.
Arrows
indicate
the
ER
structures,
normally
distributed
in
the
cytoplasm
in
panel
B
but
forming
areas
of
convoluted
membranes
in
panel
A.
n,
nucleus.
4076 NOTES J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
(-galactosidase) RNA. In the control cells, the ER was ho-mogeneously distributed throughout the cytoplasm (Fig. 3B), whereas areas of convoluted membranes were present in the HCVdj-transfected cells (Fig. 3A). The self-assembly of
pro-teins at these convoluted membranes was observed as electron-dense hemispherical structures. This phenomenon, which was not detected in cells transfected with -Gal RNA or in the various SFV expression experiments performed in our
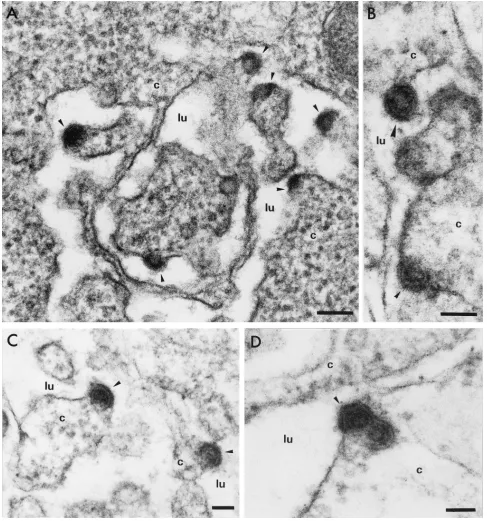
[image:5.587.49.536.73.594.2]labora-tory (5, 13), was clearly due to production of the HCV struc-tural proteins. Higher magnifications (Fig. 4) of this electron-dense material revealed budding of virus-like particles 50 nm in diameter towards the dilated ER lumen. In some electron micrographs (particularly Fig. 4B, C, and D), core-like parti-cles 30 nm in diameter surrounded by an ER-derived envelope, yielding a particle with a total diameter of 50 nm, were ob-served. In some electron micrographs (Fig. 4D), we observed
FIG. 4. Electron micrographs at high magnification of ultrathin sections of BHK-21 cells electroporated with the HCVdjRNA. Bars, 100 nm
(A) and 50 nm (B, C, and D). Arrows indicate virus-like particles budding towards the ER lumen. The two particles in panel B may represent different steps for this phenomenon, from early (small arrow) to late (larger arrow) events. c, cytoplasm; lu, lumen.
on November 8, 2019 by guest
http://jvi.asm.org/
the dual packaging of capsids into the same portion of the envelope as seen for other viruses, such as the hepatitis B virus (22). To confirm the specificity of these observations, we car-ried out immunogold labeling with the various monoclonal antibodies and the transfected cell sections. Cells were fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) for 3 h. They were then dehydrated in a graded series of alcohol solutions, and cell pellets were embedded in London resin white (Taab Lab Equipment, Aldermaston, United Kingdom). The resin was allowed to polymerize at⫺25°C for 72 h. Ultra-thin sections were cut and incubated with the various mono-clonal antibodies diluted 1/400 in phosphate-buffered saline. Immunolabeling was detected by incubation with gold-conju-gated goat anti-mouse immunoglobulin G antibodies diluted 1/100 in phosphate-buffered saline. Due to the modifications of the fixation and embedding procedures necessary for this spe-cific immunostaining method (particularly the absence of
glu-taraldehyde and osmium tetroxide), the cell structures were less well preserved than those of cells subjected to the regular EM method described above (Fig. 5). However, intense gold labeling, restricted to the convoluted ER membranes of the HCVdjRNA-transfected cells, was clearly observed with the
three monoclonal antibodies (Fig. 5A). Prebudding or budding structures at these ER membranes were strongly labeled with both anti-E1 and anti-core antibodies (Fig. 5A and C, arrows). Cells transfected with the-Gal RNA showed no gold labeling (Fig. 5B).
These EM observations demonstrate that the HCV core proteins self-assemble at the ER-membrane rather than in the cytoplasm and form a structured capsid rather than a nonstruc-tured ribonucloprotein complex. This is consistent with a re-cent report showing that, in a cell-free assay, HCV core pro-teins produced in bacteria self-assemble into nucleocapsids in the presence of viral or nonviral RNA molecules with
second-FIG. 5. Immunogold labeling of ultrathin section of BHK-21 cells electroporated with the HCVdjRNA (A and C) and-Gal RNA (B) and
stained with anti-E1 (A and B) or anti-core (C) antibodies. Arrows indicate prebudding or budding structures in which gold labeling was particularly concentrated. Bars, 200 nm (A and B) and 50 nm (C). c, cytoplasm; lu, lumen.
4078 NOTES J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.587.50.536.72.489.2]ary structures (16). It is unclear whether the SFV replication complex recently found to be associated with internal host-cell membranes (15) is involved in the assembly of the HCV capsid in a cellular system. At this time, it is also unclear whether our HCV-like particles contain RNA. As our construct contained no 5⬘HCV UTR sequence, virus-like particle assembly may in this case involve the region of the HCV RNA encoding the core, which is believed to be involved in encapsidation (26). Our observations strongly suggest that the HCV capsid ac-quires its envelope by budding through ER membranes, as for other members of the flavivirus family. This was already sus-pected because the HCV envelope proteins are retained in the ER compartment when produced by various heterologous ex-pression systems in cell cultures (8, 10), but our experiments provide the first visualization of this process.
Our EM observations also suggest that most of the particles formed in transfected BHK-21 cells have an abortive or slow budding process. Indeed, few particles were fully released from the ER membrane. This observation is consistent with the fact that no HCV structural proteins were detected in the trans-fected cell supernatant by Western blotting, even after the concentration of the supernatant by ultracentrifugation. This contrasts with the large amount of viral proteins detected within cells and the frequent observation of viral budding on cell sections. Despite the inefficiency of particle secretion, our system remains an important and original tool for studies of virus assembly mechanisms and virus-host cell interactions. We now need to identify the host cell or virus factors required for particle secretion in this system. For instance, viral accessory proteins, such as p7 or NS2 (1), and/or host cell factors, such as apolipoprotein AII (23), may be involved in virus secretion.
By analogy with another flavivirus, the Kunjin virus (KUN), it will also be of interest to investigate whether our system transcomplements the replicating HCV subgenomic RNAs de-scribed elsewhere (4, 18). In this model, virus-like particles containing a KUN RNA replicon are secreted from BHK-21 cells sequentially cotransfected with a KUN replicon RNA and an SFV vector expressing genes encoding the KUN structural proteins (14). Interestingly, transfection with an SFV vector encoding the KUN structural proteins alone does not lead to the secretion of virus-like particles in this model (14). This suggests that the presence of an efficient HCV RNA replicon may be required for particle release from the host cell in our SFV-HCVdjexpression system. The simultaneous use of these
two systems in the same cells, as for the KUN model, may be the way to establish the long-awaited in vitro culture system for HCV. However, HCV RNA replicons have been shown only to replicate in clones of the hepatoma Huh-7 cell line, and the efficiency of HCV-like particle formation in these Huh-7 clones remains to be addressed.
We thank C. Sureau for critical reading of the manuscript and help-ful discussions and F. Dubois and F. Barin for stimulating discussions. We are indebted to Harry Greenberg and Jean Dubuisson for provid-ing us with the anti-E1 antibody and the anti-E2 antibody, respectively. This work was supported by grant “Hépatite C” from the “Agence Nationale pour la Recherche sur le Sida (ANRS),” France. E. Blan-chard is supported by a fellowship provided by the Région Centre.
REFERENCES
1.Bartenschlager, R., and V. Lohman.2000. Replication of hepatitis C virus.
J. Gen. Virol.81:1631–1648.
2.Bartenschlager, R., and V. Lohman.2001. Novel cell culture systems for the
hepatitis C virus. Antivir. Res.52:1–17.
3.Baumert, T. F., S. Ito, D. T. Wong, and T. J. Liang.1998. Hepatitis C virus
structural proteins assemble into virus-like particles in insect cells. J. Virol.
72:3827–3836.
4.Blight, K.-J., A. A. Kolykhalov, and C. M. Rice.2000. Efficient initiation of
HCV RNA replication in cell culture. Science290:1972–1974.
5.Brand, D., F. Lemiale, G. Thibault, B. Verrier, S. Lebigot, P. Roingeard, L.
Buzelay, and F. Barin.2000. Antigenic properties of recombinant envelope
glycoproteins derived from T-cell-line adapted isolates or primary human immunodeficiency virus isolates and their relationship to immunogenicity.
Virology271:350–362.
6.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M.
Houghton.1989. Isolation of a cDNA clone derived from a blood-borne
non-A non-B viral hepatitis genome. Science244:359–362.
7.Deleersnyder, V., A. Pillez, C. Wychowski, K.-J. Blight, J. Xu, Y. S. Hahn,
C. M. Rice, and J. Dubuisson.1997. Formation of native hepatitis C virus
complexes. J. Virol.71:697–704.
8.Dubuisson, J., H. H. Hsu, R. C. Cheung, H. B. Greenberg, D. G. Russell, and
C. M. Rice.1994. Formation and intracellular localization of hepatitis C virus
envelope glycoprotein complexes expressed by recombinant vaccinia and
sindbis viruses. J. Virol.68:6147–6160.
9.Dubuisson, J., and C. M. Rice.1996. Hepatitis C virus glycoproteins folding:
disulfide bond formation and association with calnexin. J. Virol.70:778–786.
10.Duvet, S., L. Cocquerel, A. Pillez, R. Cacan, A. Verbert, D. Morapdour, C.
Wychowski, and J. Dubuisson.1998. Hepatitis C virus glycoprotein complex
localization in the endoplasmic reticulum involves a determinant for
reten-tion and not retrieval. J. Biol. Chem.273:32088–32095.
11.Grakoui, A., H. L. Hanson, and C. Rice.2001. Experimental models of hepatitis
C virus infection, replication, and pathogenesis. Hepatology33:489–495.
12.Helenius, A.1995. Alphavirus and flavivirus glycoproteins: structures and
functions. Cell81:651–653.
13.Hourioux, C., D. Brand, P.-Y. Sizaret, F. Lemiale, S. Lebigot, F. Barin, and
P. Roingeard.2000. Identification of the glycoprotein 41TM cytoplasmic tail
domains of human immunodeficiency virus type 1 that interact with Pr 55 gag
particles. AIDS Res. Hum. Retrovir.16:1141–1147.
14.Khromikh, A., A. N. Varnavski, and E. Westaway.1998. Encapsidation of the
flavivirus Kunjin replicon RNA by using a complementation system
provid-ing Kunjin virus structural proteins intrans. J. Virol.72:5967–5977.
15.Kujala, P., A. Ikaheimonen, N. Ehsani, H. Vihinen, P. Auvinene, and L.
Kaariainen.2001. Biogenesis of the Semliki forest virus RNA replication
complex. J. Virol.75:3873–3884.
16.Kunkel, M., M. Lorinczi, R. Rijnbrand, S. Lemon, and S. J. Watowich.2001.
Self-assembly of nucleocapsid-like particles from recombinant hepatitis C
virus core protein. J. Virol.75:2119–2129.
17.Liu, Q., C. Tackney, R. A. Bhat, A. M. Prince, and P. Zhang.1997. Regulated
processing of hepatitis C virus core protein is linked to subcellular
localiza-tion. J. Virol.71:657–662.
18.Lohman, V., F. Körner, J. O. Koch, U. Herian, L. Theilman, and R.
Bar-tenschlager.1999. Replication of subgenomic hepatitis C virus RNAs in a
hepatoma cell line. Science285:110–113.
19.Maillard, P., K. Krawczynski, J. Nitkiewicz, C. Bronnert, M. Sidorkiewicz, P.
Gounon, J. Dubuisson, G. Faure, R. Crainic, and A. Budkowska.2001.
Nonenveloped nucleocapsids of hepatitis C virus in serum of infected
pa-tients. J. Virol.75:8240–8250.
20.Moradpour, D., C. Englert, T. Wakita, and J. R. Wands.1996.
Character-ization of cell lines allowing tightly regulated expression of hepatitis C virus
core protein. Virology222:51–63.
21.Roingeard, P., S. Lu, C. Sureau, M. Freschlin, B. Arbeille, M. Essex, and
J.-L. Romet-Lemonne.1990. Immunocytochemical and electron microscopic
study of hepatitis B virus antigen and complete particle production in
hep-atitis B virus DNA transfected HepG2 cells. Hepatology11:277–285.
22.Roingeard, P., and C. Sureau.1998. Ultrastructural analysis of hepatitis B
virus in HepG2-transfected cells with special emphasis on subviral filament
morphogenesis. Hepatology28:1128–1133.
23.Sabile, A., G. Perlemuter, F. Bono, K. Kohara, F. Demaugre, M. Kohara, Y.
Matsura, T. Miyamura, C. Brechot, and G. Barba.1999. Hepatitis C virus
core protein binds to apolipoprotein AII and its secretion is modulated by
fibrates. Hepatology30:1064–1076.
24.Santolini, E., G. Migliaccio, and N. La Monica.1994. Biosynthesis and
bio-chemical properties of the hepatitis C virus core protein. J. Virol.68:3631–3641.
25.Schlesinger, S., and T. M. Dubensky, Jr.1999. Alphavirus vectors for gene
expression and vaccines. Curr. Opin. Biotechnol.10:434–439.
26.Shimoike, T., S. Mimori, H. Tani, Y. Matsuura, and T. Miyamura.1999.
Interaction of hepatitis C virus core protein with viral sense RNA and
suppression of its translation. J. Virol.73:9718–9725.
27.Yasui, K., T. Wakita, K. K. Tsukiyama, S. I. Funahashi, M. Ichikawa, T.
Kajita, D. Moradpour, J. R. Wands, and M. Kohara.1998. The native form
and maturation process of hepatitis C virus core protein. J. Virol.72:6048–6055.