JOIURNALOFVIROLOGY,JUlY1975,p.
Copyrigit01975 American Society for Microbiology Printed in U.S.A.Vol. 16, No. 1
Resolution of the DNA
Strands of the
Specialized
Transducing
Bacteriophage Xh80C1857dargF
DON SENS, DON ESHENBAUGH, AND ERIC JAMES*
DepartmentofChemistry, University of South Carolina, Columbia, South Carolina 29208 Received forpublication 14February 1975
The
DNA strands of lambdoidphages
with deletions orsubstitutions of
theguanine plus
cytosine-rich region
in theleftarmarenotresolvableby complexing
with
poly UG
followedby
centrifugation
in CsCl. This work describes acompletely general procedure
for thestrand
resolution
of these
phages by
hybridization
withfragments
ofseparated
strands of the parentphage.
Inparticular,
resolution of the DNA strands of thespecialized transducing phage
XhBOC,857dargF
is described, and
evidence is presented
which indicates thatargF
istranscribed from the rstrand.Genetic information
is,
with
fewexceptions,
encoded
by double-stranded
DNA with
onestrand
complementary to the other as first
pro-posed
by
Watson
and Crick
(21).
The
specific
information for any given gene is defined
by
oneof
the
two
strands. All
senseinformation may be
encoded
by
onestrand
asin
the
caseof
bacterio-phage T3 and T7 (17, 18); however, in
general
some
genes
aretranscribed
from
onestrand and
other genes
areread
inthe
opposite direction
from
the other
strand.
This has
been
clearly
demonstrated for the
coliphages
Xand
T4(5,
19), and in
some casesparticular
regions
aretranscribed from both DNA strands
(1). The
direction of transcription of
anumber
ofgenes
in
Escherichia coli has been determined (20),
and of particular
interestis
the
divergent
tran-scription
of
the
argECBH
genes of the
arginine
regulon.
It
has been shown that argE is
tran-scribed
anticlockwise from
acontrol region
between
argE
and
argC
and that the
argCBH
genes
aretranscribed
in
the
opposite direction
from a
closely linked control region (7, 11, 16).
The determination of the orientation of
tran-scription
of
various
genes and the detailed study
of
their
regulation in vitro have been made
possible by
the
availability of
techniques for the
resolution of the DNA strands of various
bacte-riophages.
The method of choice for resolving the DNA
strands ofX,
k80,
and T7 is
to
complex the DNA
with aribopolymer, poly UG, followed by
cen-trifugation to equilibrium in a CsCl gradient as
described by
Hradecna and Szybalski (10). In
the case of
Xthis
procedure takes
advantage of a
guanine plus cytosine-rich region on the left
arm; however,
Xmutants with
deletions in this
region and
specialized transducing
bacterio-phages
(Xh8OC1857dara
[9],
Xh8OC1857dhisgnd
8g
[22
],
Ah80C 1857dargF1
[E.
James and L. Gorini,
Fed. Proc.
31:3710, 1972 ]) which have this
region
replaced by
bacterial genes have
DNAwhich
is
notamenable
tostrand
separation
by
established
procedures.
We
require
resolved strands of Xh8OC
i-857dargF
for
studying
the
regulation
of
gene
expression
inthe
arginine
biosynthetic
path-way. A
facile technique for
resolving the
DNA
strands of any X80
or Xphage is
described,
and
inparticular
wereport that the
rstrand
ofXh80C1857dargF
carries the sense
information
for
argF.
MATERIALS AND METHODS
Materials. Ready-Solv IV and VI scintillation fluid and scintillation vials were obtained from Beckman Instruments, Atlanta, Ga. All radioisotopes were purchasedfromNewEnglandNuclearCorp., Boston, Mass.ElectrophoreticallypurifiedDNase and RNase wereobtainedfromWorthingtonBiochemicalsCorp., Freehold, N.J. Selectron B6 filters were obtained from Arthur Thomas. Media was purchased from Difco Laboratories, Detroit, Mich. Cesium chloride was obtained from Columbia Organic Chemicals, Columbia, S.C. Poly UG was purchased for Bio-polymers,Inc. andhad a U-G ratio of 1.9:1.
Media.
Bacteria
used tor preparing phage stocks weregrown in L liquid medium (13) or L agar plates except for the preparation of32P-labeled
phage when bacteriaweregrowninS medium (Table 1). Bacterio-phageweretitered on H plates in H top agar (9). F top agar wasusedforplating cells on minimal medium (14). Selection plates contained medium A (6) and supplemental growth factors as required, 2% agar, and 0.5% glucose as carbon source. Supplements were usedatthe concentrations previously described (8).Bacteria and bacteriophage. Bacterial strains and bacteriophage used in this work are listed in Table2.
Propagation of bacteriophage. XCb2was
on November 10, 2019 by guest
http://jvi.asm.org/
86 SENS, ESHENBAUGH, AND JAMES TABLE 1. Composition of Sl mediuma
Contents Concn
Tris-hydrochloride (pH 7.4) 0.1M
NaCl 7mM
NH4Cl 10mM
KH2PO4 3mM
MgSO4 2mM
CaCl2 0.2mM
VitaminB1 4mg/liter
20%Glucose 20mi/liter
a
Si
andS2 medium also contained all amino acids [image:2.498.72.262.84.185.2]and nucleic acidbasesatthe concentrationsdescribed (8). S2 medium has thesamecomposition except for the omission ofphosphate.
TABLE 2. Listofstrains
Strain Source
CA 8000 Hfr thi J. Beckwith
EJ 81thi thr leu argFargl Our collection gal lac
EJ 83.1thi thr leu argFargl Our collection gal lac strr
(Gh8OC1857)
(Xh8OC1857dargF)
EJ107thi argI
argR,5
spcr Our collectionACb2 W.Szybalski
Xh80C1857 Our collection
Xh80C1857dargF Our collection
XC1857S7 N. Kelker
gated by the plate procedure using the sensitive bacterial strain CA8000; Xh8OC1857dargF and XC1857S7were prepared from appropriate lysogenic strains as described by E. James and L. Gorini
(manuscript inpreparation).
Bacteriophage labeled with 32Pwerepreparedfrom lysogens grown in S1 (Table 1) medium with 2 mM KH2PO4.Cellsweregrowntoanopticaldensityat575
nm
(OD.7.)
of 0.40, at which time they weresedi-mentedby centrifugationat8,000rpmfor4min;the resulting pelletwasrapidlyresuspended in S2(Table
1) medium, grownat42C for15min, transferred to
37C, and grown until confluent lysishad occurred. PhageDNA withaspecific activityof 1.0 x105to7.0
x 106counts permin/gg wasprepared asdescribed
except the Casamino Acids solution used inboth Si and S2 media was treated with 1.90 ml of 1.0 M
calcium chloride per25 ml of 20% Casamino Acids, and precipitated calciumphosphate wasremovedby
centrifugationat6,000xgfor 10min.
Purification ofbacteriophage.Phage lysateswere
clarified by centrifugation at 8,000rpm for 15 min, and phage was sedimented for 12 to 16 h in the
presence of 10% polyethylene glycol 6000 (Arthur Thomas)and 0.5 MNaClasdescribedbyYamamoto
etal. (24).The sedimentwascollected by
centrifuga-tion at 5,000 rpm for 5 min and the pellet was
carefully resuspended in l/5o of the original lysate
volume of T1buffer (6 x 10-4MMgSO4,5 x 10-4M CaCla, 6 x 10- M Tris-hydrochloride, p-H 7.3, 0.1% [wt/vol] gelatin). The concentrated phage was sedi-mentedfor2h at 30,000 rpm into acushion compris-ing 2ml of1.4-density CsCl above 2 ml of1.6-density CsClusinga 50.2Tirotorin the Beckman ultracentri-fuge.Phagewerefurtherpurified byrepeated centrif-ugation in a cesium chloridedensity gradient using 1.4, 1.5, 1.6block gradients for a period of4to 6h, 1.4, 1.6 block gradients for 12 to 16 h, or running to equilibrium in 1.5-density CsCl using either the 50.2 Ti, 50Ti, or 75 Ti angle rotors. Aftercentrifugation, gradients were harvested by upward displacementby fluorientFC 40(Minnesota Mining and Manufactur-ing Co., St. Paul, Minn.) with an ISCO gradient fractionator coupled to a Chromatronix dual-wave-length absorbance monitor with an 0.5-mm path length flow cell.
Resolutionof phage DNA strands by complexing with poly UG. Strand resolution of phage DNAby complexing with poly UGwasperformedasdescribed by Hradecna and Szybalski (10) and Szybalski (per-sonalcommunication).Cesiumchloride was added to high puritywatertogiveasaturated solution at 60C, and thewarmsolutionwasrapidly filtered through a
0.45-jum
membrane filter (Millipore).Saturated CsCl solutions hadanOD2,0
ofless than 0.010.Polyallomer centrifuge tubes (% by3 inch; ca. 1.7by7.6cm)were placedin 1literof0.1MEDTA, pH10.0, and heated to 100C for15min; thetubeswerewashed withglass distilled water and stored at room temperature. A quantity of phage containing 350 to 450 jtg of DNA, freshly purified in a CsCl gradient, with anOD2,0
greaterthan60,wasplacedintoadialysis bag,both endswereclosed withhemostats,and thephage wasdialyzed for2 hagainst 1 liter ofphage dialysis bufferat 4C. Thephageweretransferredto a testtube (16by125mm), and the volumewasadjustedto1.83 ml withdialysis buffer;0.50ml ofanaqueous solution ofpoly UG (1 mg/ml)and0.015 ml of 10%Sarkosyl NL97wereadded,and the mixturewasheated for2.5 minat95Cin ahotwaterbath withgentleswirlingfor the first 30 s.The mixturewasrapidlycooledto 5C followed by the addition of 0.45 ml of 0.5 M Tris-hydrochloride (pH 7.6) and 10.9 ml of saturated CsCl. The density was adjusted to 1.725 (refractive index 1.4022), and the mixture was centrifugedina 75 Ti or 50Tirotor at35,000 rpmfor67 to 72hat 17C. At the termination of the run each preparation was frac-tionatedasdescribed for the isolation of phage.
The separated strands were dialyzed overnight against140mMphosphate buffer (pH 7.6),adjustedto 0.3 NKOH, and incubatedfor 12hat 37C toremove polyUG. Thereactionmixture was neutralized with 3 NHCl, annealed at 68Cfor 3 handpassed througha 5-mlhydroxyapatite column at 60C to remove dou-ble-stranded DNA. Single-stranded DNA was dialyzed against 1mM EDTA(pH 8.0) and storedat 4C over adropofchloroform.
Isolation of DNA from the phage
Xh8OC1857dargF.
Bacteriophage was purifiedimme-diately priortoisolationofDNAby centrifugationto equilibrium in1.5-density cesiumchloride, and puri-fied phage was adjusted with 1.5-density cesium J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.498.69.265.246.387.2]STRAND SEPARATION OF Xh8OC1857dargF 87 chloride toyieldaconcentration correspondingto an
OD2,,0
of 13.The phage solution was dialyzed against 1xSSC(0.15 MNaClplus0.015M sodiumcitrate, pH 7.4) for 3 h without stirring, and the phage was extractedthree times with an equal volume of redis-tilled phenol saturated with 1 x SSC. For the first extraction 15
Al
of a 25% solution of sodium dodecyl sulfate wasadded per ml. The third phenol extraction was followed by extraction with chloroform-isoamyl alcohol (24:1, vol/vol); the aqueous phase was ad-justed to 25Ag/ml
inPronase andincubated for2h at 37C. Two additional phenol extractions were per-formedfollowed by a final chloroform-isoamyl alcohol extraction. The aqueous layer was dialyzed exhaus-tively against 10 mMEDTA, pH 8.0, and stored at 4Coveradropofchloroform.Preparation of 4 to 5S siingle-stranded DNA fragments. A quantity ofsingle-strandedDNA, usu-ally 800Mg, in 13ml of 1 mM EDTA was sonically treated with a Bronson Heat Systems sonifier at a power output of 85 W using four pulses of 30-s duration with 5-min cooling at 0C between each pulse. The fragments wereconcentrated to avolume of 3 ml by centrifugal lyophilization in a silanized corextube, and 0.5-mlaliquotswerelayeredon a 5 to 30%sucrosegradient (0.5 MNaCl-1mMEDTA)with a0.5-mlcushion of 50% sucrose and centrifuged in an SW50.1rotorat45,000 rpmto anw2tvalue of 55,000. Thegradientswerefractionated from thebottom;the upper 2.0 mlcontained4 to5S fragments whichwere pooled,concentratedby centrifugal lyophilizationto a
volume of 5 ml, and desalted by gel filtration on a Sephadex G15 column equilibrated with 1 mM EDTA. DNA larger than4 to5S,recovered from the bottom of the gradient, was used in subsequent solification experiments.
Hydroxyapatite chromatography.Denatured and native DNA were separated by differential ad-sorption to and elution from hydroxyapatite (Bio-Rad, Richmond, Calif., Bio Gel control no. 11704) according to Kohne and Britten (12). Columns of hydroxyapatite were poured in disposable plastic syringebarrels ina60C water bath (J. F.Sambrook, personal communication). Mixtures of native and renatured DNAwere applied in 140 mM phosphate buffer(pH 7.0).Single-strandedDNApassedthrough the column, which was washed with eight column volumes of 140 mM phosphate buffer. Duplex DNA was eluted with six column volumes of 400 mM phosphate buffer.
Determination of radioactivity. Bovine serum albumin was addedascarrier forsampleswhichwere precipitated with 10% trichloroacetic acid. The pre-cipitates were collectedon glass fiber filters (What-man), dried, andcounted inReady-SolvIV scintilla-tion fluid. Samples containing 32p were counted without fluorby Cerenkov radiationinaBeckmanLS 230scintillation spectrometer.
Hybridization conditions. Allhybridizationswere
performed at68C in 140mMphosphate buffer(pH 7.6)-0.4% sodium dodecyl sulfate. Native DNA was denatured at low salt concentration by heating to 97C for 4 min, quick cooled, adjusted to 140 mM phosphate, and passed through a hydroxyapatite
column to remove any tangled DNA. In experiments to determine the kinetics of reassociation, samples were withdrawn from the annealing mixture at appropriate times,diluted 10-fold into 140 mM phos-phate buffer, and storedat 4 C until the proportion of single- anddouble-stranded DNA was determined by chromatography on hydroxyapatite.
Preparation of argF mRNA. Strain EJ107 was grown at 37 C in S1 medium without uridine to a concentration ofapproximately 5 x 108cells/ml, at which time the culture was pulsed for 50 s by the addition of 4uCi of [3HJuridineper ml. At the end of the pulse period a freshly prepared solution of sodium azide was added to 0.02 M final concentration, and the culture was immediately poured on to the same volume of frozen crushed buffer containing 20 mM Tris-hydrochloride (pH 7.3)-5 mM MgCl2. The cells were allowed to thaw,concentrated by centrifugation for 10 min at 8,000 rpm, and resuspended in a 0.1 volume of 0.1 x SSC(pH 7.0). The cells were lysed by a single passage through a French press at 8,000 lb/in2.Bentonite was immediately added to the lysate to afinal concentration of 0.01 mg/ml, the mixture wasstirred gently at 4 C for 5 to 10 min,and bentonite was removed by centrifugation for 10min at 13,000 rpm at4C.
DNase(RNase free) was added to the supernatant liquid at a concentration of 20Mg/ml,and the mixture wasincubated at room temperature for 15 min. The solution of RNAwasthenextracted three times with phenol saturated with 0.1 xSSC. The first extraction was performed in the presence of 0.4% SDS. After each extraction the mixture was centrifuged at 6,000 rpm for 5 min to facilitatephase separation. After the last phenol extraction, residual phenol was removed from the aqueous phase by extraction with chloro-form-isoamyl alcohol (24:1, vol/vol). The aqueous phase was heated in aboilingwaterbath for 4 min and quick chilled, and 20 x SSC was added to give an overall concentration of 2 x SSC. The cold solution was passed through a double layer of nitrocellulose filters. To the filtered solution a0.1 volume of 20% sodium acetate (pH 5.2) and2volumes of ethanol (at -20C)wereadded, and precipitationwasallowedto occurovernight at -20 C.
TheRNA precipitate wascollected by centrifuga-tion and resuspended in 0.1 x SSC and passed throughaSephadexG-50(fine) column (0.9by30cm, equilibratedwith0.1 x SSC).RNA elutedinthe void volume and was concentrated by ethanol precipita-tion. RNA was resuspended in 2 x SSC, saturated with phenol, and stored at4C.
RESULTS
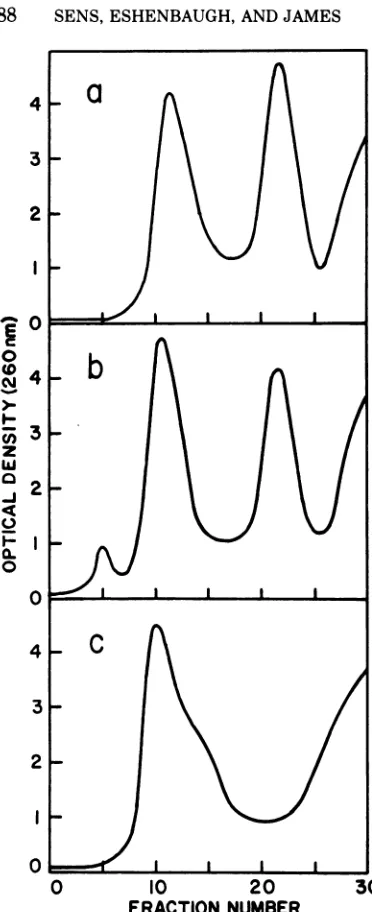
The resolution of the DNA strands of
bacte-riophage
Xand
080
isreadily affected by
com-plexing
withpoly UG
followedby centrifugation
to
equilibrium
in a1.725-density
cesiumchlo-ride
gradient
as shown inFig.
la and b forXC1857S7 and
XCb2,
respectively. Separations
such
as those shown inFig.
laand b
are efficacious due to the specific complexing of VOL.16,1975on November 10, 2019 by guest
http://jvi.asm.org/
88 SENS,ESHENBAUGH, AND JAMES
4 3 2
9 (0
I-a 2
0
I
0L
0
0
4
3
2
0 10 20 30
FRACTION NUMBER
FIG. 1. Comparison of efficacy of complexing with poly UGfollowed by centrifugationtoequilibrium in 1.710-density cesium choride for the resolution of the DNA strands ofvarious bacteriophage. (a) XCb2; (b)
Xh8OC1857; (c) Wh8OC1857dargF.
poly UG with the guanine plus cytosine-rich region ofthe left arm ofthe
bacteriophage.
Inthosecases inwhich this guanineplus
cytosine-rich region is removed by a deletion or
substi-tution, as in the case of
080dargF
andXh8OC1857dargF,
the guanineplus cytosine-richregion is no
longer
available to complex poly UG.The DNAisolated from suchphages isnot amenable to strand separation using poly UG(Fig.
lc). The
work ofWestphal
(22)
and of Sambrook et al.(16)
with simian virus 40 andthe
following
considerationssuggest
analter-nate
procedure
forresolving
the DNAstrands
ofAh8OC,857dargF.
(i)
DNA isolated fromXh8OC1857
and080
may
bereadily
resolvedusing
poly
UG.(ii)
Single-stranded Xl fragments
shouldcom-pete
for theXh8OCidargFr
strand in areas-sociation
experiment
withdenatured
Xh8OC1857dargF.
(iii)
The rate ofreassociation
ofhigh
concen-trations
of Xlfragments with the
complemen-tary
strand
ofXh8OC1857dargF should be
fasterthan that of the
corresponding
strand ofXh8OC1857dargF,
indirect proportion
to theincreased concentration of
fragments relative
toAh8OC1857dargF.
(iv) The
unitlength-fragment DNA hybrid
duplex
maybe
separated from single-stranded
DNA
by chromatography
onhydroxyapatite.
(v) Single-stranded fragments
of 4 to5S canbe
readily
separated
from unitlength
Xh8OC1857dargF DNA by
centrifugation in
asucrose
density gradient.
If
unit
length-denatured Xh8OC1857dargF
DNA
washybridized
toeither
Xh8OC1857
rorI
strand
fragments
of 4 to5S,
only
oneof the
DNA strands
ofthe specialized
phage should
hybridize
tothe
4 to5S-resolved DNA. By
performing the
reactionwith
alarge
excessof
4to
5S
fragments, the
rateof
hybridization
of
fragments
totheir
complement
should
be
con-siderably
faster thanthat
ofthe
homologous
Xh8OC1857dargF
r or Istrand;
excesssingle-stranded
fragments
together with the
samestrand
ofXh8OC 1857dargF should
notbe
re-tained when
chromatographed
onhydroxy-apatite at 140
mM
phosphate, whereas unit
length-fragment hybrid DNA would be retained
and
eluted
at 400mMphosphate.
Data
arepresented
inFig.
2which
summarizethe
results
of anumber
ofreassociation
experi-ments in
which
3H-labeled
unitlength
Xh8OC
,dargF
DNA
wasdenatured
andper-mitted toreassociate in thepresence of various concentrations of 4 to
5S fragmented
rstrand
DNA isolated
fromXCb2. The fraction ofradio-active
single-stranded
DNA present afterhy-bridization had been permitted
toproceed
forvarying
times was ascertainedby
chromatog-raphy
onhydroxyapatite. Increasing
concentra-tionsofXCb2fragments lead toadecreaseinthe percentage of
labeled
duplex DNA, but
even with a 30-fold excess offragments less than 50%radioactive-labeled
single-stranded DNA waspresent;
however,
the use ofXh80C1857
frag-ments ora mixture offragments fromXCb2
and J.VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.498.75.261.49.505.2]STRAND Xh8QC,57dargF 89
and
the solution was rapidly cooled
to 0C and
applied to a 3-ml hydroxyapatite column
main-tained at 60
C, with occasional
stirring of
40
p:)
the
hydroxyapatite
bed. The fraction
passing
40
through the column contained
more
than 90%
of the single-stranded DNA;
the
remaining
49
/single-stranded
DNA
eluted with
10ml of
140cz
30
X 30-0~~~~~~~~~~~~~~~~~10
*C 20 -
75-.0
0 . * a 2
0 5 10 IS 20
ACb2
(pg/mi)
FIG. 2. Relationship between
'H-labeled
single- 01 10 10 100stranded
Ah8OC,857dargF
present and concentration Tira. (hr)of added single-stranded XCb2 fragments. Xh8OC1- FIG. 3. Rate of conversion of single-stranded
857dargF
DNA (1.0ug)
wasdenatured in 0.5 ml of 1Xh8OC1857dargF
DNA into duplex DNA in the pres-mM EDTA by heating at 97 C for 5min
and adjusted ence of a30-fold
excess ofsingle-strandedXh8OC,8574 to a total volume of 2ml
in 140 mMphosphate buffer to 5Sfragments.Xh8OC1857dargF
DNA (2.0Mg)
was containing various concentrations of 4 to 5S XCb2 denatured in I ml of 1 mMEDTAbyheatingat97C single-strandedDNA. The mixture was incubated at for 5min and adjusted to a totalvolumeof 4 mlin 140 68 C for 60 h, at which time 1 ml was removed, diluted mM phosphate buffercontaininga30-fold excessof 4 with 9 ml of 140 mM phosphate buffer-0.4% sodium to 5SXh8OC1857,
strand DNA. The mixture was dodecyt sulfate, and stored at 4 C until assayed for incubated at 68 C. Samples(0.5 ml) wereremovedat single-stranded'H-labeled
DNA by chromatography intervals, diluted with 3.5 mlof 140 mMphosphate on hydroxyapaptite. buffer-0.4%sodiumdodecylsulfate, andstoredat 4 C until assayed forsingle-stranded'H-labeled DNA by480 led to the rapid reassociation of 50% of the
chromatography
on
hydroxyapatite.
labeled DNA to the duplex form (Fig. 3 and 4)
with 50% of the radioactive-labeled single-
100stranded DNA present. These data clearly
indi-cate that annealing denatured DNA, isolated
from a specialized transducing bacteriophage,
It
75-with a 30-fold excess of single-stranded frag-
a
ments from an appropriate phage, should per-
Cmit separation of the strands of the specialized
50-phage.
Preparation
of separated
strands
of
,,
Xh8OC1857dargF.
The strands of
Xh80C1857-
2-dargF
are
separated routinely by using the
following protocol: 5
ug
of
Xh8OC1857dargF
DNA was diluted to 4.0 ml with sterile
water,
heated at 97C for 5 min, and immediately
°01
lo
10 100cooled to
0 C.
Purifieca
4 to
5S fragmented
Time
(hr)
DNA(150
M&g)
isolated fromAh80C1857
1strand FIG. 4. Rate of conversion of single-strandedwas
added,
and the mixture was adjustedh0C0857dargF
DNAinto duplex DNA
inthe
pres-to 140 mM phosphate (pH 7.6)-0.4% sodium ence of a20-fold
excessofsingle-stranded
Xh8OC,8574
dodecyl sulfate in a total volume of 10 ml. to 5S fragmentsanda10-fold
excess
of
XCb2.
Experi-The mixture was incubated at 68 C for 1.5 h, mentalconditionsas
described in
the
legend
to
Fig. 3.
VOL.16,1975on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.498.39.222.63.307.2] [image:5.498.237.431.167.323.2] [image:5.498.240.429.463.615.2]90
SENS,ESHENBAUGH, ANDJAMES mM phosphate buffer. Double-stranded DNA waseluted from the hydroxyapatite column with 6ml
of 400mM phosphate buffer
(pH 7.6) with 0.4% sodium dodecyl sulfate; an addi-tional 8 ml of the same buffer removed morethan
95% ofthe double-stranded
DNA.Radioactive DNA was equally* divided be-tween
the
fractionseluting
at 140 mMand
400mM
phosphate, and the
totalyield
wasgreaterthan
95%.The fraction eluting
at 140 mMphosphate was concentrated to dryness
by
cen-trifugal lyophilization,resuspended
in 3 ml of sterile glass distilled water,and
desalted on acolumn
(1.5by
20cm) ofSephadex G15
equili-brated
with 1 mM EDTA (pH 8.0). DNA waseluted
inthe void volume and concentrated
to avolume of 2 ml
by
centrifugal lyophilization.Aliquots
of 0.5 ml werelayered
on 5 to 30%sucrose
gradients with
a0.25-ml
cushion
of 50% sucrose andcentrifuged
at42,000 rpm in anSW50.1
rotor to an w2tvalue
of 55,000.The
sucrose
gradients
werefractionated
fromthe
bottom
intothree
fractions;the lower
2.5 mlcontained Xh8OC 1857dargF light strand, the
next 0.5 mlcontained
no DNA, and the upper2.5
ml
ofgradient
contained
Xh8OC,857
Istrand
4 to
5S fragments.
Fractionscontaining
Xh80C1857dargF light strand
werepooled,
250,ug
of
poly(A) carrier
wasadded,
and the solutionwas
concentrated
to avolume
of 7ml
by
centrifugal lyophilization, desalted
asde-scribed, and again concentrated
to avolume
of 3ml.
The sample
wasstored
at 4C
over adrop
ofchloroform with
afinal
yield
of77%.Fractions
from
the
upper 40% ofthe gradients
werepooled, concentrated by centrifugal
lyophiliza-tion,
desalted, and reconcentrated
asdescribed
to
yield
88% ofthe
fragments
originally used
inthe
experiment.The
fractioneluting
in400mM
phosphate
wasconcentrated, desalted, and
con-centrated
asdescribed. Aliquots
of 0.5 ml werelayered
on a 5 to 30%alkaline
sucrosegradient
(0.5 M
NaCl,
0.2MNaOH,
1mM
EDTA) with
a
0.25-ml cushion of
50%
sucroseand
cen-trifuged at 42,000 rpm at 4 C in an SW50.1 rotor to an
w2t value
of55,000. The lower 2.5 ml of the gradients was pooled and neutralized with 1.0 NHCl,
and 250 ug ofpoly
A carrier wasadded,
and the mixture was concentrated,desalted,
and concentrated once more as described to give a final recovery of 82% ofXh8OC,857dargF
r strand. The individual resolved strands wereunit
length
asdemonstrated
inFig.
5 inwhichthe
sedimentation
profile
of Xh8OC,857dargF
freshly prepared
by phenol
extraction(Fig. 5a),
after resolution of the I strand from the 140mM fraction
(Fig.
5b),
and after resolution of the r strand from the 400 mM fraction(Fig. 5c)
arero
0
E
0. 3o
3
2
0 5 10 15 ZO 0 5
FRACTION NUMBER
FIG. 5. Sedimentation of XhOC,857dargF single-stranded DNA through alkaline sucrose gradients.
Samples of 32P-labeledIandrstrandXh8OC,857dargF
DNA were sedimented through a5 to 30% alkaline
sucrosegradient at42,000rpm at4CinanSW50.1
rotor toanW2t valueof55,000.Fractions(0.2ml)were
collected, and the 32Pwas determined by measuring
Cerenkov radiation. (a) Xh80C,857dargF freshly
pre-pared by phenol extraction; (b)Istrand obtainedfrom 140 mM phosphate fraction; (c) r strand obtained
from 400 mM phosphate fraction; (d) I strand
Xh8OC1857dargF after self-annealing for 16hat68C in 0.2 MNaCl.
compared by sedimentation through a 5to30% alkaline sucrose gradient (0.5 M NaCl-0.2 M
NaOH).
Each of the resolved fractions was
self-annealed for 16 h at 68 C in 0.2 M NaCl to permit any contaminating complementary strandtoreanneal; the phosphate concentration
wasadjusted to 140mM, and each fractionwas
chromatographed on hydroxyapatite at 60C. More than 98% ofthe DNApassed through the column, indicating that therewas insignificant
cross-contamination with the complementary strand. A sample of the single-stranded DNA self-annealed as described was sedimented
through an alkaline sucrose gradient as
de-scribed (Fig. 5d). In view of the considerable thermal shearing obtained after self-annealing at68C andbecause ofnegligible cross-contami-nation of one resolved strand with the other,
this step of the preparation procedure was
usually omitted.
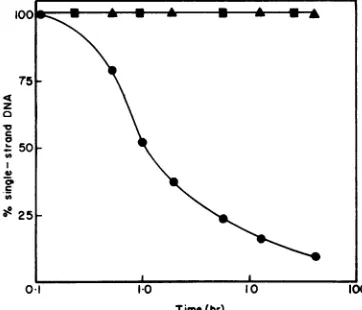
We have performed twoexperiments to
con-firm that the 140and400mMfractions indeed constitute resolved strands of
Xh8OC1857dargF.
(i) Self- and cross-hybridization. Whend
c
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.498.281.467.76.282.2]STRAND SEPARATION OF
Xh8OC,857dargF
91Xh8OC1857dargF
Iand
rstrand DNAwereincu-bated separately at 68 C in 140 mM phosphate bufferat aconcentration of 0.5 Mg ofDNA/ml,
there was no conversion to a double-stranded
form detectable bychromatographyon
hydroxy-apatite; however, a mixture containing equal
proportions of both strands renatured with second-order kinetics (Fig. 6).
(ii) Hybridization of argF mRNA to
Xh-80C1857dargFrand
I
strands. Purified mRNA wasprepared fromastrain making constitutivelevels of argF message and ornithine transcar-bamylase asdescribed. Six aliquots, each
com-prising 3.05 x 106 counts/min of [3H
]uridine-labeled RNA, were
hybridized
to 5 ,gofdena-tured Xh8OC1857dargF DNA immobilized
on anitrocellulose filter. One filterwaswashed, and
boundradioactivity wascounted in Ready-Solv
IV; RNA was eluted from the other filters as
describedby B0vre and Szybalski (2). Approxi-mately 0.2% of the RNA input was recovered
from specific hybridization todenatured DNA
isolated from
Xh8OC1857dargF (Table 3). In
100
75
z
C
50
25
0o1 10 10 100
Time(hr)
FIG. 6. Reassociation kinetics of I and r strand
Xh8OC,dargF DNA. Hybridization solutions
con-tainedatotalof2.5 Mgof strand DNA(U),rstrand DNA (A),or amixtureof equal proportion of andr
strand DNA (0) in 2.5 ml of 140 mMphosphate buffer-0.4% soldium dodecyl sulfate. After various timesof incubationat68C, samples (0.250 ml)were
diluted into2.5 mlof140mMphosphate buffer-0.4% sodium dodecyl sulfate, and thequantity of single-stranded and nativeDNAwasdeterminedby chroma-tographyonhydroxyapaptite.
constrast to
these
data, when mRNA
waspre-pared under conditions of physiological
repres-sion
from
CA8000,
astrain
carrying
the argR+
allele, 0.06% of the RNA input was found to
hybridize
specifically to DNA isolated fromXh8OC1857dargF (Table
4). The purity of argFmRNA isolated under
conditons of constitutivesynthesis
wasdemonstrated
by rehybridizationto
denatured
Xh8OC,857dargF
DNA and to
'80
DNA
immobilized
on nitrocellulose filters. Itwas
found that
93% ofthis
input radioactivitybound specifically
toargF-containing DNA,
whereas
only
2%bound
to080
DNA.
Hybridiza-tion of
1,400counts/min
of this
purified argF
mRNA to 0.33
,ug
of
Xh8OC1857dargF
r strand
and
lstrand, isolated
asdescribed, resulted
in76%
of the
inputradioactivity
hybridizing
spe-cifically
tothe
rstrand and
9% tothe
lstrand
(Table
5).
DISCUSSION
The
resultspresented
in this work showthat
it is
possible
to separate the strands of thespecialized
transducing
bacteriophage
Ah8OC 1857dargF utilizing
a procedurewhich
takes
no cognizance ofthe specific parts ofthe
molecules which
areeither
present ordeleted. It
is,
therefore, possible
toresolve the strands of [image:7.498.43.224.327.482.2]any
deletion
orsubstitution
mutantof
X or080
TABLE 3. Purification ofargFmRNAisolatedfrom EJ107
counts/min counts/min
TotarRNA
bound to: % bound to:input('H
counts/min) argF 080DNA
argF
480DNADNA DNA
3,054,800 6,210
1227
10.2 10.007
TABLE 4. Isolationof argFmRNAfrom CA8000 grown in the presenceof 100
Mg
ofarginineper ml'Hcounts/min 'Hcounts/min
bound to: %bound to: Total RNA
input
argF
4)80DNAargF
O8DNA
DNA DNA
901,000 564 49 0.06 0.005
TABLE 5. Hybridization of purified argFmRNAtoargFand
480
DNAPurifiedargF 'Hcounts/minbound to: %'Hcounts/minbound to: mRNA'H
counts/min
input argFDNA 480 DNA argFr argF I argF DNA O80DNA argFr argF I
1,400 1,290 330 | 1,060 | 130 93 | 2 | 76 99
VOL.16, 1975
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.498.243.434.390.495.2]92 SENS,ESHENBAUGH, AND JAMES
by
use ofthe techniquedescribed
usingsingle-stranded
fragmentswith
anS value
ofbetween
4
and
5derived
from one or morelambdoid
phages to
provide
the maximumhomology
be-tween the fragmentsand
the genome tobe
resolved. The resultsobtained
usingXCb2
frag-ments toresolve
aXh8O
hybrid phage indicated
that the choice
of fragmentsisolated
fromanother source
should be
more efficacious. Theuse of
resolved
fragments ofXh8OC,857
or amixture of
XCb2 and
080
fragments wasfound
to provide a statisfactory solution to the prob-lem. An important aspect of this work is theapplicability
ofthe
procedure
we havede-scribed
for theresolution
of the DNAstrands
ofspecific DNA species
obtained
afterdigestion
with theendonuclease
EcoRI
of DNAisolated
from a specialized transducingbacteriophage.
The onlymodificationrequired
forthis
purpose isthe sucrose gradient centrifugation stepused
forthe
separation ofthe
resolvedrestricted
DNA
strand
from excesssingle-stranded
frag-ments.
It is
possible
thatduplex
DNAcomprising
unit
length Xh8OC 1857dargF
rstrand and
Xh8OC,857
Istrand
fragments could be
sepa-rated
from excesssingle-stranded
fragmentsand
Xh8OC1857dargF
Istrand
by centrifugation
to
equilibrium
in a1.7250-density cesium
chlo-ride density
gradient; however,the
use ofchro-matography
onhydroxyapatite
ismuch
morerapid,
considerably
less expensive,and
un-doubtedly leads
to acleaner
separation.
The
data
presented
inTable
4clearly
indi-cate that
control
ofthe
argF
operon inthe
arginine
biosynthetic regulon
iseffected,
atleast in part, at
the
transcriptional level. Thisis
consistent with
data presented by
Pouwelsetal.
(15)
forthe
argECBH
genes ofthe arginine
regulon.
These workers
demonstrated
that
argECBH
mRNA
levels
werebetween
0.02 tc0.036%
under conditions
ofphysiological
re-pression
compared
toaconstitutive
level of
0.23to 0.42% of
the
total RNA
synthesized
in a 50-s pulse of[3Hluridine
asjudged by studiesof mRNA
hybridized
to DNA isolated from a080
transducingphage
carrying the genes ppcand
argECBH.
It isclear
that synthesis ofmRNA from
bacterial operons neighboring argF
is
observed
under conditions ofphysiological
derepression of the arginine biosynthetic
regu-lon;
however,
the data shown inTable
4indicatethat at least 70% of the mRNA,
synthesized
inastraincarrying the argR- allele, which bindsspecifically
to DNA isolated from thephage
Xh8OC
,857dargF
is indeed argF mRNA. Thisis
entirely
consistent with the observation(Table
5) that76%
ofpurified
argFmRNA
binds specifically with
XhSOC1857dargF
rstrand
DNA
and
clearlyindicates
thatthe
Xh8OC
,-857dargF
rstrand
carries sense informationfor the
argF
gene.ACKNOWLEDGMENTS
We thank JoeSambrookforintroducing usto hydroxyapa-titefor thechromatographyofDNA andMichael Vodkinfor
numerousdiscussions. We also thank Dianne Winburn and Philip James for excellenttechnical assistance. Weare most
grateful to the 3M Company for the gift of fluorinertFC40. This work was supported by the Research Corporation Brown-Hazen BH-853 grant and American Cancer Society grantVC 131 provided to Eric James.
LITERATURE CITED
1. Bovre, K., and W. Szybalski. 1969. Patterns of conver-gentandoverlapping transcription within the b2 region ofcoliphageA.Virology 38:614-626.
2. Bovre,K.,and W.Szybalski.1971.Multistep DNA-RNA hybridization techniques, p.350-383.In L. Grossman
and K. Moldave(ed.), Methodsinenzymology, vol.21.
Academic Press Inc., New York.
3. Campbell,A. 1963.Segregantsfromlysogenic
heterogen-otescarrying recombinant lambda prophages. Virology 20:344-356.
4. Campbell, A.1964. Genetic recombination betweenXdg
phage.Virology 23:234-251.
5. Cohen,S. N., and J. Hurwitz. 1968. Genetic transcription
in bacteriophage A: studies of A mRNAsynthesis in vivo. J. Mol. Biol. 37:387-406.
6. Davis, D. D. 1950.Mutants ofEscherichia colirequiring
methionineorvitamin B12.J. Bacteriol. 60:17-28.
7. Elseviers, D., R., Cunin, N. Glansdorff, S. Baumberg, and E. Ashcroft.1972.Control regions within the arg-ECBH gene cluster of Escherichia coli K12. Mol.Gen.
Genet. 117:349-366.
8. Eshenbaugh, D., D. Sens, and E.James. 1974.A solid phase radioimmune assayforornithine
transcarbamyl-ase, p. 61-73. In R. B. Dunlap (ed.), Advances in experimental medicine and biology, vol. 42. Plenum Press, New York.
9. Gottesman, S., and J. R. Beckwith. 1969. Directed transposition of the arabinose operon: atechniquefor the isolation ofspecialized transducingbacteriophages.
J. Mol. Biol. 44:117-127.
10. Hradecna, A., and W. Szybalski. 1967. Fractionation of thecomplementarystrands ofcoliphageADNAbased ontheasymmetricdistributionofthepolyIGbinding
sites.Virology32:633-643.
11. Jacoby, G. A. 1972. Control of the argECBHcluster in Escherichia coli.Mol.Gen. Genet. 117:337-348. 12. Kohne, D. E., and R. J. Britten. 1971. Hydroxyapatite
techniquesfornucleic acid reassociation. p. 500-512. In G. L. Contoni and D. R. Davies (ed.), Proceduresin
nucleic acidresearch, vol.2.Harper& Row, New York.
13. Lennox, E. S. 1955. Transduction of linked genetic charactersofthe hostby bacteriophageP 1. Virology
1:190-206.
14. Miller,J.H., K.Ippen, J. G. Scaife, and J. R. Beckwith.
1968. Thepromoter-operator regionofthe lacoperonof
Escherichia coli.J.Mol. Biol.38:413-420.
15. Pouwels, P. H., R. Cunin, and N. Glansdorff. 1974. Divergent transcription in the argECBH cluster of
genesinEscherichia coliK12. J.Mol. Biol.83:421-424.
16. Sambrook, J. F., P. A. Sharp, and W. Keller. 1972.
Separationof thestrandsofSV40DNAand
hybridiza-J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
tion of theseparated strandstoRNA extractedfrom lytically infected and transformed cells. J. Mol. Biol. 70:57-71.
17. Summers, W. C., and W. Szybalski. 1968. Size, number and distribution ofpolyG binding siteson the
sepa-rated DNA strands of coliphage T7. Biochim. Biophys. Acta 166:371-378.
18. Summers, W. C., and W. Szybalski. 1968. Totally
asym-metric transcription of coliphage T7invivo: correlation withpolyG binding sites. Virology 34:9-16.
19. Taylor, K., Z. Hradecna, and W. Szybalski. 1967. Asym-metric distribution of the transcribing regionsonthe
complementary strands of coliphage A DNA. Proc. Natl. Acad. Sci. U.S.A. 57:1618-1625.
20.Taylor, A. L., and C. D. Trotter. Linkage map of
Escherichiacoli strain K-12. Bacteriol. Rev. 36:504.
21. Watson, J. D., and F. H. C. Crick. 1953. Astructurefor deoxyribose nucleic acid. Nature (London) 171:737-738.
22. Westphal, H. 1970. SV40 DNA strand selection by Escherichia coli RNA polymerase. J. Mol. Biol. 50:407-420.
23. Wolf, R. E., and D. G. Fraenkel. 1974. Isolation of specialized transducing bacteriophages for gluconate 6-phosphatedehydrogenase (gnd) of Escherichia coli. J. Bacteriol. 117:468-476.
24. Yamamoto, K., B. Alberts, R. Benzinger, L. Lawhorne, and G. Preiber. 1970.Rapid bacteriophage sedimenta-tion in the presence of polyethylene glycol and its application to large-scale purification. Virology 40:734-744.