0022-538X/11/$12.00 doi:10.1128/JVI.00936-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Genetic and Phylogenetic Analyses of Influenza A H1N1pdm
Virus in Buenos Aires, Argentina
䌤
†
P. R. Barrero,* M. Viegas, L. E. Valinotto, and A. S. Mistchenko
Laboratorio de Virología Hospital de Nin˜os Dr. Ricardo Gutie´rrez, Buenos Aires, Argentina, Consejo Nacional de
Investigaciones Científicas y Te´cnicas (CONICET), and Comisio´n de Investigaciones Científicas de
la Provincia de Buenos Aires (CIC), Buenos Aires, Argentina
Received 30 April 2010/Accepted 18 October 2010
An influenza pandemic caused by swine-origin influenza virus A/H1N1 (H1N1pdm) spread worldwide in 2009, with 12,080 confirmed cases and 626 deaths occurring in Argentina. A total of 330 H1N1pdm viruses were detected from May to August 2009, and phylogenetic and genetic analyses of 21 complete genome sequences from both mild and fatal cases were achieved with reference to concatenated whole genomes. In addition, the analysis of another 16 hemagglutinin (HA), neuraminidase (NA), and matrix (M) gene sequences of Argentinean isolates was performed. The microevolution timeline was assessed and resis-tance monitoring of an NA fragment from 228 samples throughout the 2009 pandemic peak was performed by sequencing and pyrosequencing. We also assessed the viral growth kinetics for samples with replace-ments at the genomic level or special clinical features. In this study, we found by Bayesian inference that the Argentinean complete genome sequences clustered with globally distributed clade 7 sequences. The HA sequences were related to samples from the northern hemisphere autumn-winter from September to December 2009. The NA of Argentinean sequences belonged to the New York group. The N-4 fragment as well as the hierarchical clustering of samples showed that a consensus sequence prevailed in time but also that different variants, including five H275Y oseltamivir-resistant strains, arose from May to August 2009. Fatal and oseltamivir-resistant isolates had impaired growth and a small plaque phenotype compared to oseltamivir-sensitive and consensus strains. Although these strains might not be fit enough to spread in the entire population, molecular surveillance proved to be essential to monitor resistance and viral dynamics in our country.
Despite the worldwide countermeasures against the circula-tion of emerging viruses, an influenza pandemic caused by swine-origin influenza virus A/H1N1 started in Mexico City, Mexico, on 18 March 2009 (33). The virus then spread world-wide, affecting more than 213 countries and with 425,650 lab-oratory-confirmed cases and at least 16,813 deaths occurring (34). In Argentina, the virus was first detected on 17 May 2009. Since then, 12,080 confirmed cases and 626 deaths have been reported (21). In Buenos Aires, Argentina, the number of cases of influenza-like illnesses and pneumonia widely ex-ceeded that in the preceding years, and 742 H1N1 cases were reported in 2009 (10).
The molecular signature of the 2009 H1N1 virus (H1N1pdm) has revealed that a reassortant probably arose from the North American H3N2 and H1N2 swine viruses and led to the emer-gence of a new epidemic virus through host switching, transform-ing an underestimated zoonosis into a pandemic threat that rap-idly spread from human to human (7, 9). Phylogenetic analyses have revealed the multiple origins of H1N1pdm, which comprises genes derived from the avian lineage (polymerase basic 2 [PB2] and polymerase A [PA]), the human H3N2 lineage (polymerase
basic 1 [PB1]), and the classical swine lineage (hemagglutinin [HA], nuclear protein [NP], and nonstructural [NS]). In addition, the neuraminidase (NA) and matrix (M) gene segments have their origin in the Eurasian avian-like swine H1N1 lineage (30). Amino acid signatures that were relevant for host speci-ficity and virulence for the three different pandemic H1N1 influenza viruses from 1918, 1977, and 2009 have previously been mapped at the genome level (28). Moreover, prelimi-nary analyses have described the H1N1pdm viruses as sen-sitive to neuraminidase inhibitors and resistant to adaman-tanes (23).
Phylodynamics merges evolutionary analysis methods with the investigation of viral dynamics. Viral genomes constitute an important and independent source of information about epidemiological processes that support and confirm the find-ings of standard surveillance methods (25). A recent study based on concatenated coding regions of available whole-ge-nome H1N1pdm has shown that at least seven different clades of viruses have been circulating globally (22).
In this report, we describe the analysis of the complete genome sequences from 21 Argentinean isolates from both mild and fatal cases and the analysis of another 16 HA, NA, and M gene sequences from Argentinean isolates in order to identify the origin of H1N1pdm in Buenos Aires. We also analyzed the viral growth kinetics in strains with clinical rele-vance. Furthermore, we describe the microevolution timeline and resistance monitoring of an NA fragment from 228 sam-ples throughout the 2009 pandemic peak by direct sequencing and pyrosequencing.
* Corresponding author. Laboratorio de Virología, Hospital de Ni-n
˜os Dr. Ricardo Gutie´rrez, Gallo 1330, 1425 Buenos Aires, Argentina. Phone: 54 11 49643118. Fax: 54 11 49644320. E-mail: paola.barrero @conicet.gov.ar.
† Supplemental material for this article may be found at http://jvi .asm.org/.
䌤Published ahead of print on 3 November 2010.
1058
on November 7, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Samples and viral isolation.Nasopharyngeal aspirates (NPAs) were referred to the Virology Laboratory of the Dr. Ricardo Gutie´rrez Children’s Hospital, Buenos Aires, Argentina, within the first days after the onset of symptoms to ensure viral recovery. Diagnosis of H1N1pdm infection was carried out for all samples by following the CDC real-time quantitative reverse transcription-PCR (qRT-PCR) protocol for detection and characterization of swine influenza virus (materials were kindly provided by the Influenza Branch, CDC) (4).
Viral isolation was performed by inoculating filtered NPAs into the amniotic cavity of pathogen-free embryonated hen eggs (kindly provided by Immuner, Entre Ríos, Argentina). Allantoic fluid was harvested after incubation for 5 days at 37°C, following biosafety level 3 good laboratory practice. Viral RNA was obtained with a PureLink viral RNA/DNA minikit (Invitrogen Life Technolo-gies, Carlsbad, CA).
Molecular characterizations. (i) Polymorphism and phylogenetic analyses. Sequences of the full-length genomes and HA, NA, and M gene segments were obtained from the isolated viruses following the WHO-recommended protocol (23) in an Applied Biosystems 3500 genetic analyzer (Foster City, CA). The sequences were submitted to GenBank. Nucleotide alignments were obtained with the ClustalX program (version 2.012) (15) and manually edited with the BioEdit program (version 7.0.5.3) (11). For further analyses, all sequences avail-able up to February 2010 were downloaded from the NCBI Influenza Virus Resource (1). Because of the vast number of highly similar sequences, we de-termined the optimal data set using a preliminary neighbor-joining analysis with 1,000 bootstraps performed with the MEGA program (version 4.0) (31). The final data sets for genomic analysis included defined strains for the seven clades described by Nelson et al. in 2009 (22).
Nucleotide substitution models were evaluated with the Jmodeltest program (24). Bayesian consensus phylogenetic trees were inferred using Mr Bayes soft-ware (version 3.1; 20,000,000 ngen, 5,000 samplefreq, 4 nchains, 5,000 burnin) (27). Other phylogenetic inferences were estimated with algorithms available at the Mobyle@Pasteur web server (http://mobyle.pasteur.fr). To infer the evolu-tionary relationships and the most recent common ancestor (MRCA) for the HA and NA Argentinean sequences, a Bayesian Markov chain Monte Carlo (MCMC) method was applied using a strict molecular clock, as implemented in the BEAST program (version 1.4.8). Trees were visualized and edited with the FigTree program (version 1.2.3) included in the BEAST software package (6). Amino acid sequences were inferred using the universal code, and polymor-phisms were determined with Seqscape software (version 2.7; Applied Biosys-tems) by comparing the sequences with the sequence of H1N1pdm reference strain California/04. A hierarchical clustering of samples was performed for visualization of the replacement changes.
Nonsynonymous changes were further classified as conservative or nonconser-vative according to their polarity. Theratio was calculated as number of nonsynonymous substitutions versus the number of synonymous substitutions (dN/dS). Recombination and the overall, gene-specific, and site-specific selection pressures acting upon the viruses were determined using the procedures avail-able in the HyPhy package and accessed through the Datamonkey web server (13, 14).
(ii) Oseltamivir resistance monitoring.A 620-bp fragment (N-4) from the NA gene ranging from amino acids 255 to 440 and including the codon positions 275 and 295 codons was obtained directly from NPAs following the WHO-recom-mended sequencing protocol (23). Purified DNA fragments were sequenced using the DYEnamic ET terminator cycle sequencing kit in an automated cap-illary sequencer (MegaBACE 1000; GE Healthcare, Piscataway, NJ).
Pyrosequencing was performed by following the WHO protocol for influenza A virus (H1N1) NA-H275 (4) using the PSQTM96 MA platform (Biotage AB, Uppsala, Sweden). The relative proportions of sensitive and resistant variants were determined with Pyromark ID software (version 1.0), following allele quan-titation analysis.
Susceptibility of influenza viruses to oseltamivir was assessed by chemilumi-nescence using an NA-Star influenza virus neuraminidase inhibitor resistance detection kit (Applied Biosystems), following the manufacturer’s instructions. Prior to the assay, oseltamivir phosphate (50M; kindly provided by Sidus S.A., Buenos Aires, Argentina) was activated by incubation with rat plasma at 37°C for 30 min and then diluted in half-log series (range, 0.03 to 1,000 nM) (16). The tests were performed directly on NPA dilutions. In cases when the original aspirate was no longer available or suitable for analysis, viral isolates were used. Viral replication kinetics.For assessment of growth properties, MDCK cell monolayers (kindly provided by INEI ANLIS Dr. Malbra´n, Buenos Aires, Ar-gentina) were infected at a multiplicity of infection of 0.001 plaque-forming units (PFU/cell). Supernatants were collected at 12, 24, 36, 48, 60, and 72 h
postin-fection for viral titration by standard plaque assays. Plaque area was measured at 48 h after infection, using Quantity One software (version 4.6.8; Bio-Rad, Her-cules, CA).
Nucleotide sequence accession numbers.The GenBank accession numbers for the viruses obtained in this study are listed in Table S1 in the supplemental material.
RESULTS
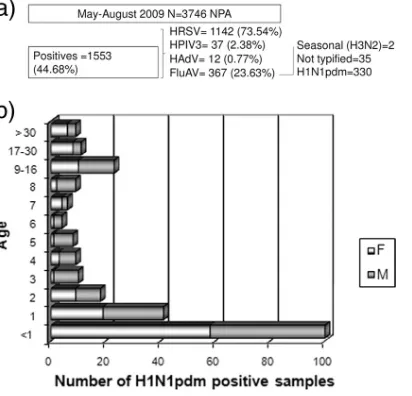
Viral detection. A total of 3,476 NPA specimens were
re-ferred to the Virology Laboratory of the Dr. Ricardo Gutie´rrez
Children’s Hospital between May and August 2009 and tested for human respiratory syncytial virus (HRSV), influenza A and B viruses (FluAV and FluBV, respectively), human parainflu-enza virus (HPIV), and human adenovirus (HAdV). None of the samples tested positive for HPIV type 1 or 2 or FluBV. Three HRSV-FluAV coinfections without major clinical dif-ferences were detected in June. A total of 330 out of the 367 FluAV-positive samples were typed as H1N1pdm by
qRT-PCR. Most cases were males (n⫽190, 63.3%), and the cases
were mainly less than 1 year old (n⫽143, 47.66%). The results
are summarized in Fig. 1.
In our laboratory, H1N1pdm detection began in May (first case, 27 May 2009), had its peak in June (243 cases), decreased in July (84 cases), and ended in August (2 cases). Sixteen patients did not survive (4.84%); among them, the viruses from 11 patients were isolated and studied.
We also included in the analysis virus from a sporadic fatal case from January 2010.
Genetic analyses.A total of 105 viruses were inoculated, and 62 were isolated after the first passage in the allantoic fluid of embryonated hen eggs. From the 62 isolates, 37 HA, NA, and M gene sequences were selected for genetic analyses. The complete genome sequence was accomplished for 21 of those isolates, including 12 viruses from mild cases and 9 viruses from fatal cases. Details of the samples and GenBank acces-sion numbers are listed in Table S1 in the supplemental ma-terial.
Most of the amino acid signatures for H1N1pdm were
con-FIG. 1. Summary of findings. (a) Total number of NPAs and viral etiology; (b) sex and age distributions of H1N1pdm cases.
VOL. 85, 2011 H1N1pdm VIRUSES IN BUENOS AIRES, ARGENTINA 1059
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.317.523.70.268.2]served in all Argentinean samples. The exceptions were the nonconservative change in PB2 S199A and the H1N1 (1918) marker NP I100 (28). All the isolates presented a nonfunc-tional PB1-F2 protein due to stop codons, as in all H1N1pdm viruses described.
Replacements in HA (S220T), NP (V100I), NS1 (I123V), and NA (V106I and N248D), known as specific gene markers for clade 7, were present in all analyzed sequences (22). Ac-cording to the first reports, three NA variants were identified among the H1N1pdm viruses: the A/California/04/2009 group with variants V106 and N248, the A/Osaka/164/2009 group with I106 and N248, and the A/New York/18/2009 group with I106 and D248. All the samples analyzed belonged to the last group (9, 12).
The 37 HA, NA, and M polymorphisms as well as compar-isons of the 21 complete genomes are shown in Fig. 2.
The hierarchical clustering showed that there was a common genetic signature in all samples, apart from the ones that be-longed to clade 7 and H1N1pdm that could represent the consensus sequence or genetic backbone for our data set (HA, P100S, T214A, and I338V; NA, V106I; PA, P224S) and many replacements that arose during the period but that did not persist.
Unique replacements in HA, NA, and M were present in viruses from severe and fatal cases without a common pattern. Two isolates carried the NAH275Y oseltamivir resistance mu-tation (HNRG23 and HNRG84), and the alignment of the 37 Argentinean M genes showed that all samples were adaman-tane resistant (S31N, I43T).
[image:3.585.46.547.85.507.2]In summary, the replacements found in the HA and NA proteins (11/15 and 7/14, respectively), as well as in the NS (2/3) and PA proteins (2/4), were mostly nonconservative,
FIG. 2. Hierarchical clustering of samples. Distances were calculated for Argentinean samples; and replacement changes were plotted for the HA, NA, and M protein sequences (a) and whole-genome sequences (b).
on November 7, 2019 by guest
http://jvi.asm.org/
whereas those in the M1, M2, NP, PB1, and PB2 proteins were more conservative.
The sequences derived from NPAs and chicken embryo iso-lates were 100% identical for the N-4 fragment. Most of the mutations described for adaptation in embryonated hen eggs were absent (8).
Selection analysis. For selection analysis, nucleotide sites were explored using the HKY85 model of selection (1668, 1404, and 498 for HA, NA, and N-4, respectively). Duplicated sequences (21/181, 36/213, and 151/180 for HA, NA, and N-4, respectively) were excluded from this analysis.
According to at least one of the assay methods used (SLAC [single likelihood ancestor counting], FEL [fixed effects likeli-hood], IFEL [internal fixed effects likelilikeli-hood], and REL [ran-dom effects likelihood]), at the specified significance levels
(P⫽0.1 and Bayes factor⫽50), the per gene per sitedN/dS
analyses revealed that two NA sites were under positive selec-tion and 75 were under purifying selecselec-tion (negatively
se-lected), with an overall dN/dS of 0.18, whereas for the N-4
fragment, 11 negatively selected codons with an overalldN/dS
of 0.24 were found.
A total of 73 nonneutral codons, 6 positively selected and 67
negatively selected, with an overalldN/dSof 0.3 were found in
HA (see Table S2 in the supplemental material).
No evidence for recombination was detected when the se-quences were assessed by GARD (generic algorithm recombi-nation detection) and SBP (single breakpoint recombirecombi-nation) from the HyPhy package.
Phylogenetic analyses.Whole genomes of viruses from 12 mild and 9 fatal Argentinean cases were associated with clade 7 by Bayesian inference with high posterior probability (PP) values (Fig. 3). Noteworthy, oseltamivir-sensitive and -resistant isolates taken from the same patient a week apart clustered together (isolates HNRG83 and HNRG84 and isolates HNRG15 and HNRG23). The isolates from one of the fatal cases of the outbreak (HNRG45) and the 2010 fatal sporadic case (HNRG102) clustered alone, whereas the others (HNRG isolates 3, 5, 21, 104, 105, 106, and 107) merged with the ones from mild cases.
[image:4.585.44.540.69.422.2]Similar topologies and statistical support were obtained by neighbor-joining, maximum-parsimony, and maximum-likeli-hood analyses (data not shown).
FIG. 3. Phylogenetic tree of concatenated complete genomes of Argentinean samples. Phylogeny was inferred with representative sequences retrieved from GenBank using Mr Bayes software (version 3.1; settings, 20,000,000 ngen, 5,000 samplefreq, 4 nchains, 5,000 burnin). Clades are indicated as 1 to 7, according to Nelson et al. (22). Argentinean strains are clustered in clade 7. The tree was rooted with H1N1pdm reference strain California/04 (clade 1).
VOL. 85, 2011 H1N1pdm VIRUSES IN BUENOS AIRES, ARGENTINA 1061
on November 7, 2019 by guest
http://jvi.asm.org/
Phylogenetic analysis of the HA gene revealed that Argen-tinean strains were further spread among four subclades in clade seven (see Fig. S1 in the supplemental material). The first subclade was composed of 18 identical sequences from May to July 2009 (HNRG isolates 8, 13, 14, 17, 18, 20, 30, 31, 33, 41, 45, 48, 72, 83, 84, 91, 105, and 107). Only full-length HA sequences were included in the tree. These strains were related to previous isolates from the northern hemisphere, New York/ 3209 from April 2009 and Stockholm/33 from May 2009, and to subsequent strains, Norway/3036 from August 2009 and Mex-ico City/WR1312N from September 2009. The second subclade
was composed of isolates HNRG39, -42, and -44 (PP⫽0.73)
and was 99.8% identical to subclade 1. The third subclade was
composed of isolates HNRG isolates 15, 82, and 23 (PP ⫽
0.76), taken from the same patient 2 and 7 days after the first sample. In this patient we observed the intratreatment emer-gence of oseltamivir resistance by pyrosequencing, showing the transition of the viral population from sensitive to resistant (32). The last subclade was composed of the isolate from the sporadic case, HNRG102, from January 2010, which was closely related to Taiwan/143 from September 2009 and
Nor-way/3797 from October 2009 (PP⫽0.63), as well as to New
York/6806 from December 2009 (not included in the tree). Phylogenetic analysis of the NA gene showed that the Ar-gentinean samples were distributed in the same clade repre-senting the A/New York/18/2009 group with I106 and D248 (data not shown).
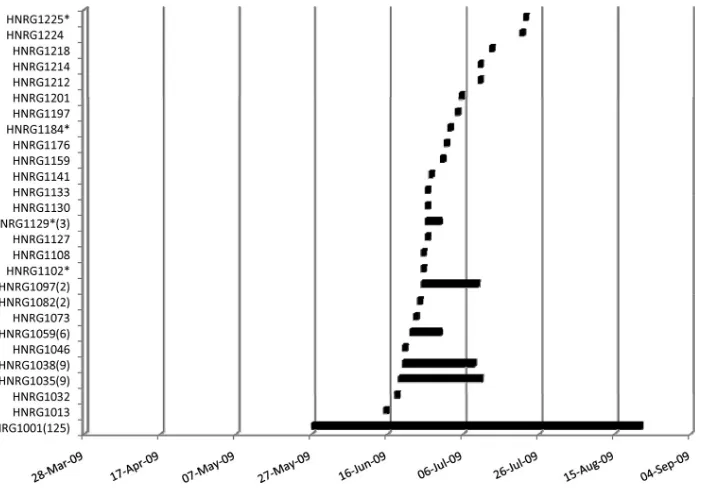
Phylogenetic analysis of the N-4 fragments showed that there were 27 different nucleotide variants circulating during the period analyzed and that they were represented by the oldest sequence in the group (data not shown). The main group was represented by isolate HNRG1001 from 27 May
2009 and had 125 identical sequences, and variants from this group were still detected 87 days after the end of the outbreak (27 August 2009). The second group had 10 identical se-quences, and variants from this group were detected from 19 June 2009 to 10 July 2009 (22 days); the third group had 9 identical sequences, and variants from this group were de-tected from 20 June 2009 to 8 July 2009 (19 days); the fourth group had 6 identical sequences, and variants from this group were detected from 22 June 2009 to 30 June 2009 (8 days); the fifth group had 2 identical sequences, and variants from this group were detected only on 24 June 24 2009; the sixth group had 2 identical sequences, and variants from this group were detected from 25 June 2009 to 10 July 2009 (10 days); and the seventh group had 3 identical sequences, and variants from this group were detected from 26 June 2009 to 30 June 2009 (4 days). There were 19 other unique variants that were detected once during the peak of the outbreak (June and July). Among these, the virus in five samples showed oseltamivir resistance (Fig. 4).
The MRCA of Buenos Aires H1N1pdm, calculated from the HA full-length sequences, was detected from 9 May to 26 May 2009 (95% highest probability density [HPD]), 2 weeks before our first local detection of isolate HNRG14 (27 May 2009), and
had a mean nucleotide substitution rate of 3.58⫻10⫺5
nucle-otides (nt)/site/day. When the same analysis was performed with 174 sequences of the N-4 segment from the NA gene, the MRCA was detected from 17 May to 9 June 2009 (95% HPD). These results suggest that the virus was circulating in our country only 1 or 2 weeks prior to our first detection. In addition, the mean substitution rate of the last region was
4.35⫻ 10⫺5nt/site/day, on the same order as that for the HA
gene.
The monophyletic clustering of HA, NA, and N-4 data
con-FIG. 4. Timeline distribution of nucleotide variants of N-4 fragment. The 27 different nucleotide variants that circulated from 27 May 2009 to 27 August 2009 were represented by the oldest sequence in the group. The number of sequences in each group is indicated in parentheses. Oseltamivir-resistant strains are indicated with an asterisk.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.117.474.66.312.2]firmed their common ancestry, being detected from 9 May to 9 June 2009 (95% HPD).
NA H275Y oseltamivir resistance screening. A total of 291 out of the 330 H1N1pdm-positive samples were suitable for re-sistance screening by Sanger sequencing, pyrosequencing, or both.
Oseltamivir-resistant variant H275Y was found in five pa-tients. The 50% inhibitory concentration values for these re-sistant samples were measured, rendering mean values of
61.54⫾32.89 nM (range, 13.82 to 97.93 nM), while sensitive
samples showed values of 0.27⫾0.04 nM.
Mutation N295S, also known to alter oseltamivir suscepti-bility, was found in the direct sequencing of N-4 from sample HNRG1202 (5).
Viral replication kinetics.On the basis of the whole-genome-sequence polymorphism analysis detailed above, we analyzed if there were phenotypic differences between oseltamivir-resistant and oseltamivir-sensitive strains, as well as between strains from mild and fatal cases, by assessing the viral replication kinetics.
As regards the growth of oseltamivir-sensitive and oseltami-vir-resistant strains, we observed that the titer of the resistant strain (HNRG23) was 100-fold lower than that of the sensitive one (HNRG15) in the first 24 h postinfection. Nevertheless, after 36 h both growth curves reached the same titers. With regard to this point, we verified that the resistant strain main-tained the H275Y replacement at 72 h postinfection in MDCK cells. Moreover, the plaque area of the resistant strain was smaller than that of the sensitive one (Fig. 5).
In the case of mild versus fatal cases, we compared four strains: three from fatal cases (HNRG5, HNRG45, and HNRG102) and one from a mild case (HNRG14). Strains
HNRG14 and HNRG5 showed a pattern of increased growth compared to that of strains HNRG45 and HNRG102. Whole-genome analysis of the strains from fatal cases showed major changes in PA, PB2, PB1, and NEP (nuclear export protein [NS2]). Besides, the plaque area of the strains from fatal cases was smaller than that of the strain from the mild one (Fig. 5). In addition, we performed a viral growth assessment of strains which represent the outbreak timeline (HNRG isolates 14, 42, 49, 66, and 72) without major clinical distinctive char-acteristics. Although minor genetic differences were found among them (Fig. 2), all the strains had a similar increased viral growth pattern in relation to the pattern from strains from fatal cases, which showed impaired growth and a small plaque phenotype (data not shown). Some of these, including the resistant strains, arose once and did not persist over time in the N-4 timeline distribution (Fig. 4).
DISCUSSION
[image:6.585.70.511.70.331.2]Given the time course of the pandemic and due to the high seasonality and transmissibility of influenza viruses, Argentina might have played an important role in raising and spreading strains. As the emergence of H1N1pdm overlapped with the southern hemisphere annual peak of respiratory virus infec-tions, climate and social conditions might have been an optimal source to generate new variants without restraints. In fact, in our hospital, H1N1pdm predominated over other viruses that regularly occur in winter, such as HPIV and HAdV. Neverthe-less, in children under 5 years of age, bronchiolitis caused by HRSV remained predominant both in our setting and in the entire country (73.54% for HRSV versus 21.25% for other
FIG. 5.In vitro viral replication kinetics. Viral titrations in the time series were plotted for wild-type (HNRG15) and H275Y resistant (HNRG23) Argentinean strains (a) and isolates from mild cases (HNRG14) and fatal cases (HNRG5, HNRG45, and HNRG102) (b) in MDCK cells. Corresponding plaque characteristics are shown below.
VOL. 85, 2011 H1N1pdm VIRUSES IN BUENOS AIRES, ARGENTINA 1063
on November 7, 2019 by guest
http://jvi.asm.org/
viruses, respectively, and 66.24% for HRSV versus 22.13% for other viruses, respectively) (21).
Our study mainly focused on pediatric cases with different outcomes from the most populated city in Argentina and one of the hospitals in Buenos Aires where patients are most often referred. This is relevant, considering that children less than 5 years of age were the age group most affected by fatal or severe H1N1pdm infection in Argentina (rate, 76.41/100,000 citizens) (21). We found that 16/330 patients with confirmed H1N1pdm infection died, regardless of their underlying condition. Other research groups in our country have analyzed the situation in pediatric intensive care units and reported mortality rates ranging from 5 to 50% (3, 17). Considering our previous records for seasonal FluAV infection in the last 5 years, there was a marked increase in the rate of mortality from H1N1pdm infection in the 2009 season (P. R. Barrero et al., unpublished data).
We analyzed three main components of the viral particle, namely, the HA, NA, and M genes, in 37 isolates. This com-bination was found to be essential when an attempt was made to evaluate multiple sources of diversity. We detected some discordance in diagnostic procedures that might be relevant in the viral landscape. A total of 14 discordances were found at the qRT-PCR level by evaluating the NP gene for universal swine virus and the HA gene of swine H1 virus. Although these changes might have affected the NP or HA gene, other com-pensatory mutations cannot be ruled out. As an example, we found a nonconservative change in the polymorphic site D239 in an isolate with a nonconclusive result by qRT-PCR (HNRG8). These findings have to be monitored closely in order to have up-to date diagnostic tools and to rapidly eval-uate newly emerging variants.
The complete-genome phylogenetic analysis of the 21 Ar-gentinean H1N1pdm isolates revealed that they were clustered together and had sequences that defined them to be members of clade 7, a globally distributed group of isolates. Therefore, the possibility of multiple introductions before local dissemi-nation cannot be ruled out. In addition, two coupled oseltami-vir-sensitive and -resistant strains clustered together, and two strains from fatal cases clustered alone, while the rest of the strains from fatal cases were mixed into the data set.
The phylogenetic analyses of the full length of the HA and NA genes also related Argentinean sequences to clade 7 se-quences from the rest of the world. The HA analysis revealed that the Argentinean strains, isolated mostly in July 2009, clus-tered with samples from the northern hemisphere autumn-winter from September to December 2009, thus suggesting evolutionary convergence or exportation of viruses and show-ing that viruses might be circulatshow-ing in waves chasshow-ing winters, as suggested by Rambaut et al. in 2008 (26). Moreover, we found that strain HNRG102 from a sporadic case from January 2010 was also related to similar strains, probably suggesting a new introduction of the virus in our country without evidence of local spread.
Genetic and phylogenetic analyses of the N-4 fragment showed that a major consensus sequence dominated the viral landscape from the beginning to the end of the outbreak, probably as a result of previous selection phenomena. Never-theless, many other attempts to generate new variants have been detected, but they lasted for only a short period and were
discarded by purifying selection. That was the case for the oseltamivir-resistant variants that arose at least three times in different locations but that may have not been fit enough to spread in the entire population. In this study, we showed that there were fixed replacements that appeared in all samples as well as diversity attempts that did not persist by means of hierarchical sample clustering and direct sequencing of N-4 from NPAs.
The MCRAs for HA and N-4 were dated 6 and 17 May 2009, respectively, and clearly overlapped the date when local circu-lation was first reported, on 17 May 2009 (20). As far as we were able to test, the HA and NA genes seem to have highly similar mutation rates and might be coevolving, thus minimiz-ing the possibility of emergence of new reassortants.
The switch from low-circulation imported cases to high-circulation local cases resulted in a change in palliative regu-lations and practices on 24 June 2009, at the peak of the epidemic. At that time, empirical treatment with oseltamivir was indicated for every hospitalized child with influenza-like symptoms, groups at higher risk, and close contacts of con-firmed cases (20). Nevertheless, it seems that the massive an-tiviral therapy application leads to the advent of resistant strains in both at-risk and previously healthy patients. It is noteworthy that we found that five patients presented with oseltamivir-resistant variant H275Y and that one patient pre-sented with the N295S mutation. These mutants might have been favored by the high degree of selective pressure and thus resulted in virus more fit to survive in this modified environ-ment over a short period (32). However, to the best of our knowledge, these resistant strains did not show person-to-per-son transmission; therefore, compensating mutations occurring elsewhere in their genomes that might improve their fitness and transmissibility need further study.
The growth differences between the oseltamivir-sensitive and oseltamivir-resistant strains in cell culture were mainly found in the first 24 h, when the initial setting of the natural infection occurs, and might show that sensitive strains have a clear advantage over resistant ones, as the former are the viruses occurring most frequently in the natural environment. This might explain the lack of spread of resistance to the whole population so far. Furthermore, we showed that the resistance was maintained in cell culture in the absence of oseltamivir pressure, as is known to occur for seasonal H1N1 influenza virus strains with an adequate genetic backbone (2). In addi-tion, the smaller plaque phenotype of the resistant strain might show that it has an impaired neuraminidase activity that dis-abled the normal spread of the resistant viruses to the vicinity. Our study shows that two different strains that were isolated from the same patient (namely, oseltamivir-sensitive and osel-tamivir-resistant strains) had markedly different growth kinet-ics and plaque phenotypes. This might be supported by re-placements at the whole-genome level that improve and/or impair viral growth or function as compensative changes to restore or reinforce their fitness. In our example, the sensitive strain had HA A232S and PB1 H47N, whereas the resistant strain had only HA S457L at HA2 position 108.
Except for strain HNRG5, strains from fatal cases also showed impaired growth and the small plaque phenotype. In particular, HNRG5 showed the unique NEP replacement N92S. Changes in this residue have been shown to make H1N1
on November 7, 2019 by guest
http://jvi.asm.org/
human virus highly virulent in pigs (29) and have been associ-ated with increased resistance to interferon (18). The isolate from a sporadic fatal case, HNRG102, had the highly virulent K22R mutation in PA in association with HA D239G and E391K. The former HA replacement has been related to se-vere outcomes in Europe and Asia, whereas the latter has been associated with increased spread and H1N1pdm vaccination failure, as it is located in HA2 position 47, known to strength the interaction between the HA monomers and to bear a highly conserved epitope recognized by antibodies that neu-tralize the closely related 1918 H1N1 virus (19).
As the first detected case of local spread, the nonfatal HNRG14 virus had the unique PA R531K replacement that was not conserved over time. For HNRG45, there were no additional mutations; therefore, whether the severity of the disease was related to host immune failure has to be further evaluated.
The recent H1N1 pandemic was not only a threat but also a challenge both for the scientists and for the community, in terms of preparedness of human resources, detection tools, and awareness to investigate and fight emerging viruses appro-priately. In Argentina, H1N1pdm circulation decreased and ceased in spring and summer, and the molecular surveillance system is prepared to monitor resistance and viral dynamics in the following winter.
ACKNOWLEDGMENTS
We acknowledge M. E. Acevedo, M. C. Alvarez Lo´pez, M. Campal, M. Gonza´lez, O. A. Jacquez, O. A. Luna, M. A. Ma´rques, P. M. Riveiro, and P. F. Scirica from the laboratory staff and all the physi-cians and authorities of Dr. Ricardo Gutie´rrez Children’s Hospital for their invaluable work in the 2009 H1N1 pandemic. We appreciate the excellent technical support of S. B. Lusso and the comments of M. A. Abba. We thank D. Posik, M. V. Ripoli, A. Rogberg Mun˜oz, G. Giovambattista, and P. Peral García from IGEVET, UNLP, for dis-cussions and assistance with pyrosequencing; J. Molina from CDM for allowing us to use CDM equipment; and R. A. Diez for continuous support.
This work was supported by ANPCYT, Ministerio de Ciencia, Tec-nología e Innovacio´n Productiva de la Nacio´n, Argentina (grant PICT1624/07).
REFERENCES
1.Bao, Y., P. Bolotov, D. Dernovoy, B. Kiryutin, L. Zaslavsky, T. Tatusova, J. Ostell, and D. Lipman.2008. The Influenza Virus Resource at the National Center for Biotechnology Information. J. Virol.82:596–601.
2.Bloom, J. D., L. I. Gong, and D. Baltimore. 2010. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328:1272–1275.
3.Caprotta, G., P. Gonza´lez Crotti, Y. Primucci, H. Alesio, and A. Esen.2010. Influenza A H1N1 respiratory infection in an intensive care unit in Argen-tina. An. Pediatr. (Barc.)72:62–66.
4.Centers for Disease Control and Prevention.2009. H1N1 flu (swine flu): resources for laboratories. Centers for Disease Control and Prevention, Atlanta, GA. http://www.who.int/csr/resources/publications/swineflu/NA _DetailedPyrosequencing_20090513.pdf. Accessed 20 March 2010. 5.Deyde, V. M., T. G. Sheu, A. A. Trujillo, M. Okomo-Adhiambo, R. Garten,
A. I. Klimov, and L. V. Gubareva.2010. Detection of molecular markers of drug resistance in 2009 pandemic influenza A (H1N1) viruses by pyrose-quencing. Antimicrob. Agents Chemother.54:1102–1110.
6.Drummond, A. J., and A. Rambaut.2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol.7:214.
7.Fraser, C., C. A. Donnelly, S. Cauchemez, W. P. Hanage, M. D. Van Kerk-hove, T. D. Hollingsworth, J. Griffin, R. F. Baggaley, H. E. Jenkins, E. J. Lyons, T. Jombart, W. R. Hinsley, N. C. Grassly, F. Balloux, A. C. Ghani, N. M. Ferguson, A. Rambaut, O.G Pybus, H. Lopez-Gatell, C. M. Alpuche-Aranda, I. B. Chapela, E. P. Zavala, D. M. Guevara, F. Checchi, E. Garcia, S. Hugonnet, C. Roth, and WHO Rapid Pandemic Assessment Collabora-tion.2009. Pandemic potential of a strain of influenza A (H1N1): early findings. Science324:1557–1561.
8.Gambaryan, A. S., J. S. Robertson, and M. N. Matrosovich.1999. Effects of egg-adaptation on the receptor-binding properties of human influenza A and B viruses. Virology258:232–239.
9.Garten, R. J., C. T. Davis, C. A. Russell, B. Shu, S. Lindstrom, A. Balish, W. M. Sessions, X. Xu, E. Skepner, V. Deyde, M. Okomo-Adhiambo, L. Gubareva, J. Barnes, C. B. Smith, S. L. Emery, M. J. Hillman, P. Rivailler, J. Smagala, M. de Graaf, D. F. Burke, R. A. Fouchier, C. Pappas, C. M. Alpuche-Aranda, H. Lo´pez-Gatell, H. Olivera, I. Lo´pez, C. A. Myers, D. Faix, P. J. Blair, C. Yu, K. M. Keene, P. D. Dotson, Jr., D. Boxrud, A. R. Sambol, S. H. Abid, K. St. George, T. Bannerman, A. L. Moore, D. J. Stringer, P. Blevins, G. J. Demmler-Harrison, M. Ginsberg, P. Kriner, S. Waterman, S. Smole, H. F. Guevara, E. A. Belongia, P. A. Clark, S. T. Beatrice, R. Donis, J. Katz, L. Finelli, C. B. Bridges, M. Shaw, D. B. Jernigan, T. M. Uyeki, D. J. Smith, A. I. Klimov, and N. J. Cox.2009. Antigenic and genetic character-istics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science325:197–201.
10.Gobierno de la Ciudad de Buenos Aires, Argentina, Ministerio de Salud, Direccio´n de Epidemiología.15 March 2010. Infecciones respiratorias agu-das: sala de situacion 2009. http://estatico.buenosaires.gov.ar/areas/salud /archivos/sala_ira_semana_25_2009.pdf. Accessed 20 March 2010. 11.Hall, T. A.1999. BioEdit: a user-friendly biological sequence alignment
editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser.41:95–98.
12.Itoh, Y., K. Shinya, M. Kiso, T. Watanabe, Y. Sakoda, M. Hatta, Y. Mu-ramoto, D. Tamura, Y. Sakai-Tagawa, T. Noda, S. Sakabe, M. Imai, Y. Hatta, S. Watanabe, C. Li, S. Yamada, K. Fujii, S. Murakami, H. Imai, S. Kakugawa, M. Ito, R. Takano, K. Iwatsuki-Horimoto, M. Shimojima, T. Horimoto, H. Goto, K. Takahashi, A. Makino, H. Ishigaki, M. Nakayama, M. Okamatsu, K. Takahashi, D. Warshauer, P. A. Shult, R. Saito, H. Suzuki, Y. Furuta, M. Yamashita, K. Mitamura, K. Nakano, M. Nakamura, R. Brock-man-Schneider, H. Mitamura, M. Yamazaki, N. Sugaya, M. Suresh, M. Ozawa, G. Neumann, J. Gern, H. Kida, K. Ogasawara, and Y. Kawaoka. 2009. In vitro and in vivo characterization of new swine-origin H1N1 influ-enza viruses. Nature460:1021–1025.
13.Kosakovsky Pond, S. L., and S. D. W. Frost.2005. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioin-formatics21:2531–2533.
14.Kosakovsky Pond, S. L., and S. D. W. Frost.2005. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol.22:1208–1222.
15.Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thomp-son, T. J. GibThomp-son, and D. G. Higgins.2007. Clustal W and Clustal X, version 2.0. Bioinformatics23:2947–2948.
16.Li, W., P. A. Escarpe, E. J. Eisenberg, K. C. Cundy, C. Sweet, K. J. Jakeman, J. Merson, W. Lew, M. Williams, L. Zhang, C. U. Kim, N. Bischofberger, M. S. Chen, and D. B. Mendel.1998. Identification of GS 4104 as an orally bioavailable prodrug of the influenza virus neuraminidase inhibitor GS 4071. Antimicrob. Agents Chemother.42:647–653.
17.Libster, R., J. Bugna, S. Coviello, D. R. Hijano, M. Dunaiewsky, N. Reynoso, M. L. Cavalieri, M. C. Guglielmo, M. S. Areso, T. Gilligan, F. Santucho, G. Cabral, G. L. Gregorio, R. Moreno, M. I. Lutz, A. L. Panigasi, L. Saligari, M. T. Caballero, R. M. Egu¨es Almeida, M. E. Gutierrez Meyer, M. D. Neder, M. C. Davenport, M. P. Del Valle, V. S. Santidrian, G. Mosca, M. Garcia Domínguez, L. Alvarez, P. Landa, A. Pota, N. Bolon˜ati, R. Dalamon, V. I. Sanchez Mercol, M. Espinoza, J. C. Peuchot, A. Karolinski, M. Bruno, A. Borsa, F. Ferrero, A. Bonina, M. Ramonet, L. C. Albano, N. Luedicke, E. Alterman, V. Savy, E. Baumeister, J. D. Chappell, K. M. Edwards, G. A. Melendi, and F. P. Polack. 2010. Pediatric hospitalizations associated with 2009 pandemic influenza A (H1N1) in Argentina. N. Engl. J. Med. 362:45–55.
18.Lipatov, A. S., S. Andreansky, R. J. Webby, D. J. Hulse, J. E. Rehg, S. Krauss, D. R. Perez, P. C. Doherty, R. G. Webster, and M. Y. Sangster.2005. Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: the role of cytokines and B- and T-cell responses. J. Gen. Virol. 86:1121–1130.
19.Maurer-Stroh, S., L. Tze Chuen, F. Eisenhaber, C. Lin, P. P. Shiau, and R. T. P. Lin.1 June 2010. A new common mutation in the hemagglutinin of the 2009 (H1N1) influenza A virus. PLoS Curr. RRN1162.
20.Ministerio de Salud, Presidencia de la Nacio´n, Argentina. 17 June.2009. Inicio de la pandemia de influenza 2009: cambio de fase 5 a fase 6. Ministerio de Salud, Presidencia de la Nacio´n, Buenos Aires, Argentina. http://www .msal.gov.ar/htm/site/pdf/alerta-epidemiologico-8.pdf. Accessed 20 March 2010.
21.Ministerio de Salud, Presidencia de la Nacio´n, Argentina.26 February 2010. Influenza pande´mica (H1N1) 2009/2010. Ministerio de Salud, Presidencia de la Nacio´n, Buenos Aires, Argentina. http://www.msal.gov.ar/archivos /Informe%20Influenza%20SE%207%2026%20feb.pdf. 20 March 2010. 22.Nelson, M., D. Spiro, D. Wentworth, J. Fan, K. St. George, E. Ghedin, R.
Halpin, J. Bera, E. Hine, K. Proudfoo, T. Stockwell, L. Xudong, S. Griese-mer, M. Bose, L. Jurgens, S. Kumar, C. Viboud, E. Holmes, and K.
Hen-VOL. 85, 2011 H1N1pdm VIRUSES IN BUENOS AIRES, ARGENTINA 1065
on November 7, 2019 by guest
http://jvi.asm.org/
drickson. 3 November 2009. The early diversification of influenza A/H1N1pdm. PLoS Curr. RRN1126.
23.Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team.2009. Emergence of a novel swine origin influenza A (H1N1) virus in humans. N. Engl. J. Med.360:2605–2615.
24.Posada, D.2008. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25:1253–1256.
25.Pybus, O. G., and A. Rambaut.2009. Evolutionary analysis of the dynamics of viral infectious disease Nat. Rev. Genet.10:540–550.
26.Rambaut, A., O. G. Pybus, M. I. Nelson, C. Viboud, J. K. Taubenberger, and E. C. Holmes.2008. The genomic and epidemiological dynamics of human influenza A virus. Nature453:615–619.
27.Ronquist, F., and J. P. Huelsenbeck.2003. MRBAYES 3: Bayesian phylo-genetic inference under mixed models. Bioinformatics19:1572–1574. 28.Schnitzler, S. U., and P. Schnitzler.2009. An update on swine-origin
influ-enza virus A/H1N1: a review. Virus Genes39:279–292.
29.Seo, S. H., E. Hoffmann, and R. G. Webster.2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med.8:950–954.
30.Smith, G. J. D., D. Vijaykrishna, J. Bahl, S. J. Lycett, M. Worobey, O. G. Pybus, S. K. Ma, C. L. Cheung, J. Raghwani, S. Bhatt, J. S. M. Peiris, Y. Guan, and A. Rambaut.2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature459:1122–1125. 31.Tamura, K., J. Dudley, M. Nei, and S. Kumar.2007. MEGA4: Molecular
Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol.24:1596–1599.
32.Valinotto, L. E., R. A. Diez, P. R. Barrero, J. Farias, E. Lo´pez, and A. S. Mistchenko.2010. Emergence of intra-treatment resistance to oseltamivir in pandemic influenza A H1N1 2009 virus. Antivir. Ther.15:923–927. 33.World Health Organization.24 April 2009. Influenza-like illness in the
United States and Mexico. World Health Organization, Geneva, Switzer-land. http://www.who.int/csr/don/2009_04_24/en/index.html. Accessed 20 March 2010.
34.World Health Organization.19 March 2010. Pandemic (H1N1) 2009—up-date 92. World Health Organization, Geneva, Switzerland. http://www.who .int/csr/don/2010_03_19/en/index.html. Accessed 20 March 2010.