0022-538X/83/020842-13$02.00/0
CopyrightC) 1983, American Society for Microbiology
Porcine Parvovirus: Virus Purification and Structural and
Antigenic Properties of Virion Polypeptides
THOMASW. MOLITOR,1 H.S.
JOO,1
ANDMARC S.COLLETT2*DepartmentofLargeAnimal Clinical Sciences,College of Veterinary Medicine, University of Minnesota, St.
Paul, Minnesota55108,1 and Departmentof Microbiology, University of Minnesota Medical School, Minneapolis, Minnesota554552
Received18October1982/Accepted10 November 1982
Porcine parvovirus
(PPV) was extensivelypurified from infected
swine fetalhomogenates by CaC12 precipitation
followed byCsCl
density centrifugation. Twospecies of particles possessing PPV-specific
hemagglutinating activity wereobserved banding
atdensities of
1.39and
1.30g/ml, representing
full and empty20-nm
virion particles,
respectively. Both classes of particles contained threemajor polypeptides,
A, B, and C, with respective molecular weights of 83,000,64,000,
and
60,000. The amountof
polypeptide
A was similar in bothspecies
(approximately
10%); however, the
Bprotein
wasmostabundant in the1.30-g/ml
particles,
whereas theC
protein
wasthemajor
polypeptide found
in the1.39-g/ml
particles. Antisera generated
toeachsodium
dodecyl
sulfate-polyacrylamide
gel-purified virion
structuralprotein had reactivities
qualitatively
similar to those ofconventional antisera raised against intact PPV in avariety of standard serological assays. The antisera generated
against
the individual sodium dodecylsulfate-denatured
PPVpolypeptides
wereable
to react withnative, intact
PPVvirions
and
werecapable of neutralizing virus
infectivity.
Porcine parvovirus
(PPV) is aubiquitous
in-fectiousagent of pigs. The etiological role ofthis
virus
inreproductive failure
inswine
is wellestablished
(13, 20, 21). Mostoften, disease
caused
by
PPVis manifested
asfetal death andmummification, although infertility,
abortion,
stillbirth, and neonatal death
mayalso
beconse-quences
of
in utero PPVinfection
(13,
21).
Humoral
immunity, either
as aconsequenceof
naturalexposure or
from
vaccination,
willoften
prevent
viremia in
pregnant sows and thuspre-vent
transplacental
virusinfection
andsubse-quent
fetal
death(15, 20).
PPV, like other
parvoviruses,
isasmall,
non-enveloped virus
containing single-stranded
DNA
(23).
PPVappears to beanondefective
orautonomously
replicating parvovirus,
similar inthis respect to the more
extensively
studiedparvoviruses
such as the minute virusof
miceand the Kilham rat
virus,
and in contrast to the defectiveadeno-associated
viruses(35).
Very little is known of the molecular features of PPV. We
began
ourstudy of
PPVby
develop-ing
a scheme forobtaining
the virus inhighly
purified form
frominfected swine
fetal tissueand then
proceeded
with studies on the virionprotein
composition.
We report here our workon the
isolation
andpurification
of PPV frominfected fetuses
and the initialcharacterization
of various
featuresof
the threemajor
virion
polypeptides.
Wepreparedantisera
to eachpuri-fied
viralpolypeptide and
compared the reactivi-ties of these antisera in a variety of tests. Wefound
that the threevirion
proteins
of PPV wereboth
structurally and antigenically quite similar.Finally,
the denatured, gel-purified virionpro-teins
wereable
toelicit
virus-neutralizing
anti-body.
MATERIALS ANDMETHODS
Virus propagation in infected animals. Pregnant sows, at approximately 35 to 40 days of gestation,
were purchased from a local swine producer. The
serological status of these sows was not important because no transplacental immunity has been
ob-served in swine (30). At 40 to 50 days, sows were
laparotimized, and fetuseswereinfected via amniotic fluid inoculation with PPV (0.2ml;1,024 hemaggluti-nating unitsper 50,ul;107 50%tissue cultureinfective doses)aspreviously described (25).Thevirusused for inoculation was PPV strain NADL-8, originally
ob-tainedfrom the National Animal DiseaseLaboratory,
Ames, Iowa. Ten to 15dayslater, the sows were taken to slaughter; the entire uterus of each was collected and transported backtothelaboratory.Pooled
inter-naltissues fromeachfetus(lung, kidneys, liver,heart,
spleen,andintestines)werecollected andmincedin 50 mMTris-hydrochloride-25 mM EDTAbuffer, pH 8.7
(TE buffer). After overnight freezing, minced tissue fluids fromeachfetusweretestedby hemagglutination (HA)ofguinea pig erythrocytes forpresenceof virus (seebelow). Tissues fromfetuses that werehighlyHA 842
on November 10, 2019 by guest
http://jvi.asm.org/
PPV: PURIFICATION AND ANTIGENIC PROPERTIES 843 Infectedfetuses(viscera)
20%homogenate/sonic extractin TE buffer HAassayindividual fetuses
HApositive HAnegative: discard
-12,000 x g, 30min
Supernatant Discard pellet
Adjustto 25 mMCaCl,,4°C, 30min
12,000 x g, 20 min
Discard supernatant
Resuspend in TEbuffer
Clear
Supernatant
- Discontinuous CsClgradient
120,000x g, 24 h
Fractions
K Determine fraction density
Assayfor viralantigen by HA
Poolvirus peak (1.39 g/ml),dialyze againstTEbuffer Repeatdiscontinuous CsCl gradient
Poolvirus peak(1.39g/ml),dialyze againstTEbuffer PurifiedPPV
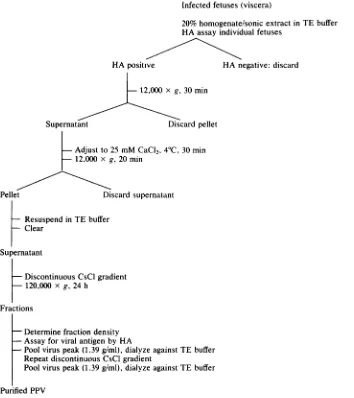
FIG. 1. Scheme forpurificationofPPV.
positive (>1:i,024)werepooled,dilutedtoa20%(wt/ vol) suspension in additional TE buffer, and further homogenizedinaWaringblender. Thesuspensionwas
frozen and thawed twice andwassonicatedat4°Cfor three 1-min intervals beforeviruspurification.
Virus purification. The procedure for the purifica-tion ofPPVfrommummified fetuseswasadaptedfrom theprocedure of Tattersalletal. (33) used for minute virus of mice(Fig. 1).All manipulations werecarried
out at 4°C. Sonicated homogenates of HA-positive fetalviscerawerecleared of cellular debris by
centrif-ugation at 12,000 x g for 30 min. To the resultant supernatant was added CaCl2 dropwise to a final concentration of 25mM, and the viruswasallowedto precipitate for 30 min. The precipitate was collected
by centrifugation at 12,000 x gfor 20 min and was
suspended by gentle sonictreatmentin TEbuffer. Any insoluble material was removed by brief
centrifuga-tion, and the cleared supernatant was adjusted to a final CsCl density of 1.32 g/ml in TE buffer. This solution waslayeredonto anequal volume ofa CsCI solution in TE buffer having adensity of1.40 g/ml. Centrifugationwasperformedat120,000 x gfor18 h at 4°C (Beckman SW41 rotor). Gradients were frac-tionated(approximately25to30fractions),the
refrac-tive index of each fraction was determined, and the presence ofviral antigenwas assayed by HA.
Frac-tionscontaining hemagglutinating activitywerepooled
andsubjectedto asecondcycle ofdiscontinuousCsCl centrifugationsasoutlinedinthelegendtoFig.2.The
pooled HA-positive fractions from the second CsCl gradientswerethendialyzed againstTEbuffer.
Radioiodination of viralproteins. Viralproteinswere labeled with
1251,
using the chloramine T method of Hunter(8). Purified virus (50 to100 ,ug) inTEbuffer (50,il)wasdisrupted by boilingin1% sodium dodecylVOL.45, 1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.496.82.436.78.476.2]sulfate (SDS) for3 min. After coolingto room tem-perature, 100 ,Ci of '25I (carrier free; New England
Nuclear Corp.) was added, followed by 50 ,ug of
chloramineT. After 2min atroom temperature, the
reaction mixture was applied to a Sephadex G-50 column (1 by 12 cm)equilibrated with0.01 Msodium
phosphate (pH 6.8). The excluded peak of
radioactiv-itywas collected and pooled.Thespecific activity of the radiolabeled proteins was approximately 5 x 106 cpm/,ug.
Viralpolypeptide purification. Viral structural
pro-teins, either '25l radiolabeled or unlabeled, were
boiled inSDS-containing sample buffer for2 minand
then were subjected to electrophoresis in SDS-con-taining 10% polyacrylamide gels (17). Proteins were
localized either byautoradiography orby Coomassie
brilliant blue staining ofend sections of thegels. The individual protein bands were excised, and the
pro-tein-containing gel pieces were subjected to another cycleofelectrophoresis on asecond SDS-containing
polyacrylamide gel. After protein localization in the second gel, the excised gel pieces were either used
directlyforone-dimensional partial proteolytic peptide mapping(seebelow)orusedfor theextraction of the protein contained within. Isolation ofviral proteins
frompolyacrylamide gel pieceswaseitherbyrepeated
elution in 0.05 MNH4HCO3-0.1%SDSat37°Corby isotachophoresis inSephadex G-50columns (1). The
leadingbufferwas50 mMTris-hydrochloride (pH 6.8),
andtheterminating bufferwas50 mMTris-glycine (pH
8.8). Bromophenolbluewasusedas atracking dyefor
theleading edgeofprotein.Thedye-containing
Sepha-dexwasremoved from the columns and washed with
watertorecovertheprotein.Bothproceduresresulted
inrecoveries ofproteinof65to90%. Topreparethe
individualvirionpolypeptidesforuseinanimal
immu-nizations, proteinsweretwicegel purified andeluted from the polyacrylamide gel pieces in 0.05 M
NH4HCO3-0.1% SDSasdescribed above. The eluates
werelyophilized, suspendedin1to2ml ofTEbuffer,
andextensively dialyzedover aperiodofseveraldays
againstbuffercontaining10mMpotassium phosphate (pH 6.8), 40 mM NaCl, 1 mM EDTA, 1 mM
2-mercaptoethanol, and 50%glycerol. The method of Lowryetal. (18)wasemployed todetermine protein
concentrations, using bovineserumalbumin (BSA)as astandard.
Peptidemapping.The one-dimensionalpartial prote-olysis mapping procedure of Cleveland et al. was
employedasoriginallydescribed(4). '25I-labeledPPV proteins, purified by two cycles of
SDS-polyacryl-amide gel electrophoresis, were subjectedto
electro-phoresis in SDS-containing 15% polyacrylamide gels
in the presenceof either Staphylococcus aureus V8
protease (Miles Laboratories, Inc.), elastase
(Wor-thington Diagnostics), or chymotrypsin
(Worthing-ton).
Preparation ofantisera. Normal porcine sera were
collected from adult pigs that were free of
PPV-specificantibodiesasdeterminedbyhemagglutination
inhibition(HAI;seebelow) andby
immunoprecipita-tionof radiolabeledPPV-infectedcultured celllysates (datanotshown).Adultpigseracontaining antibodies toPPV, as determined by HAI, werecollected from sows orgilts naturally exposedtoPPV.Fetalpigsera
containing antibodies to PPV were prepared by the
amnioticinjectionof 70-to85-day-oldfetuseswith 0.2 ml ofPPV NADL-8(1,024hemagglutinationunits per
50,u1). Fetuseswerecollected21 dayslaterandbled. Rabbit antisera against intact PPV and gel-purified
viral polypeptides were prepared in the following
manner. Five-pound (2,268-g) New Zealand white rabbitswerebledbytheearveinstoobtainpreimmune (normal)rabbitsera.Thesesera weredemonstratedto
lack antibodies specific forPPVantigensby HAI and
by immunoprecipitation of radiolabeled PPV-infected
cultured cell lysates; they were therefore used for
subsequentimmunizations. Rabbitswereinjected
sub-cutaneously at multiple sites along the back with either 50,ug ofintact,CsCl-purified,fullvirus particles
or50,ug ofgel-purified polypeptidedissolvedin1mlof
TEbufferthathad been emulsifiedinanequalvolume
ofcomplete Freund adjuvant. One rabbitwasused for
each antigen preparation. Rabbits receiving purified viral polypeptides were injected at 3 weeks and 7
weeksaftertheinitial immunizationwith 50jigof the
respective gel-purified protein in incomplete Freund
adjuvant. The rabbit that received intact virus was
boosted once at 3 weeks with 50 jig of virus. All rabbits were maintained in individualcages. Rabbits
injected with live virus did not show demonstrable
virusshedding, and other normalrabbitsmaintainedin the same animal room remained negative for PPV
antibodies throughout thecourseof this experiment.
Table 1 summarizes the various antisera used inthese studies.
HA and HAI. PPV antigen was detected by the
hemagglutination ofguinea pig erythrocytesas
previ-ously described (14). Antibody titers to PPV were
[image:3.496.58.450.565.673.2]measuredby anHAltest(14). Briefly, heat-inactivat-edsera werefirstabsorbed with 25% kaolin in borate-saline solution (pH 9.0)for 20 min and centrifuged.
TABLE 1. Various antiseraused in thepresentstudy
Antiserum
Abbrevia-
Methodofproduction/sourcetion
Normalpigserum NPS HAI-negativeadult animal
AdultpigaPPV PaPPV Naturallyexposedfield animal
FetalpigotPPV FoxPPV Experimentalvirusinfectionbyamnioticinjectionof pregnantsow Normal rabbit serum NRS Pre-immunizationseraof rabbitsused below
Rabbit aintactPPV RaPPV Immunization withCsCl-purifiedintactvirionparticles
RabbitotApolypeptide RaA Immunizationwithgel-purifiedApolypeptide; 10-week post-immu-nizationserum
Rabbit aB polypeptide Ra B Asabove withB polypeptide Rabbit a Cpolypeptide Ra C Asabove withC polypeptide
on November 10, 2019 by guest
http://jvi.asm.org/
PPV: PURIFICATION AND ANTIGENIC PROPERTIES 845 This wasfollowed byincubation for 60 min with50%
guinea pig erythrocytesand centrifugation (500 x gfor 10min). This procedure removed natural nonspecific
hemagglutinins. Sera were then serially diluted with
phosphate-bufferedsaline (PBS) in microtiter plates. A standard amount of cell culture-propagated PPV (8
hemagglutination units per 50 .d) was added to all wells and allowed to incubate for 3 h at 22°C or
overnightat4°C.Then a0.6%suspension of guinea pig
erythrocytes was added to all wells andincubated at 22°C for 2 h. The endpoint is expressed as the recipro-calofthehighest dilution resulting in 50% inhibition of HA.
Other serological assays. An indirect
fluorescent-antibody(IFA) test,previouslydescribed byBommeli
et al. (3), was used to compare the reactivities of various antisera with infected cells in culture. Briefly. PPV-infected ST (swine testicle)cells grown on
Lab-Tek slides were treated with acetone and air dried. Various antisera, diluted 1:8 in PBS, were incubated with the monolayers at 37°C for 60 min, after which
theywerewashedandfurther incubatedwith 0.1 mlof
a 1:50 dilution of a 1-mg/ml solution of protein
A-fluorescein isothiocyanate conjugate (Pharmacia Fine
Chemicals. Inc.). Controls consisted ofknown
PPV-positive and-negative sera, uninfected ST cells, and
pseudorabies virus-infectedcells. Agar gel
immunodif-fusion (AGID)wasperformedaspreviously described (16) to testforthe presenceofprecipitating antibodies.
Protein transfer to nitrocellulose and reaction with antisera. PPVproteins wereelectrophoretically trans-ferredfromSDS-polyacrylamide gels to nitrocellulose paper(Schleicher&SchuellCo.)by the procedureof Bittner et al.(2). After transfer, gelswerestained with Coomassie blue toestimate the efficiency of transfer. Routinely, 70 to95% ofthe proteins weretransferred
tothenitrocellulosesheets. Theprocedureof
Syming-ton etal. (31) wasemployedtoimmunologicallydetect proteins. Briefly, the sheetscontainingthetransferred
proteins were placed in asolution of4% BSA-0.01% sodium azide for 18 h at 22'C. Purified immunoglob-ulinG(100 to 200
p.g;
obtained by ammoniumsulfate precipitation) from various antisera was added. andincubationwascontinuedwithgentle shaking for16to 24 h at22°C.Sheets were then washed five or six times (5 min per wash) with BSA-sodium azide solution.
Protein A (Pharmacia), radioiodinated bythe proce-dureofHunter(8)in BSA-sodiumazide solution(107
cpm; 8x 106
cpm/[.g).
wasadded, andthe sheetswere gently shaken at 22°C for 3 to 4 h. The sheets were again washed five or six times(10 min perwash)withBSA-sodium azide solution, air dried, andexposed to X-rayfilm.
Electron microscopy. CsCl-purified virions,
exten-sively dialyzed against TE buffer, were analyzed by
negativestaining with4% phosphotungsticacid, using standard procedures (7). Grids were examined in a Zeiss 10transmission electron microscope at a plate
magnification of40,000 withan80-kV beam. The ability of the various antisera to form lattices with intactvirion particles was tested by an immuno-electron microscopy procedure(7). Various sera
(di-luted 1:10 in TE buffer) were mixed with equal vol-umes of purified PPV in TE buffer and incubated
overnightat4°C.Themixtures were then diluted 1:100 with water and centrifuged at 30,000x g for 30 min to
pellettheimmunecomplexes.Thepellets were
resus-pended in a small volume ofTE buffer, negatively stained, and analyzed in the electron microscope as described above.
Virusinfectivity neutralization. Standardvirus infec-tivity neutralization assays were performed in
tripli-cate. Sera(1:2or 1:10dilutionsin PBS,total volume 0.3ml)were mixed andincubated with0.3 mlofvirus
(8 HA units per 50
[LI)
for 60 min at 22°C. A 200-,ulvolume of this mixture was then added to Leighton
tubes containing recently trypsinized ST cells
previ-ously washed with PBS. Controls included known positive sera. known negative sera. no serum, andno virus. Theinocula were allowed to absorb for 90 to 120
min. after which the cultureswere washed with PBS andfresh growth mediumwasadded. Fourdaysafter
inoculation,the Leightonslipswereremoved, treated with acetone, and air dried. The slips were then stained directly with fluorescent-antibody conjugate.
washed, and observed under a fluorescent
micro-scope. The fluorescent-antibody conjugate consisted of fluorescent isothiocyanate conjugated to porcine
anti-PPVantibodies producedingnotobioticpigs(21).
Virusinfection of cells wasconsidered positive when
multiple foci offluorescentlylabeled cellswere appar-entonany given Leightonslip.
RESULTS
Purificationof PPV from infected fetuses. The
purification
procedure for PPV frominfected
porcine
fetuses isoutlinedinFig.
1.Asfound forminute virus of mice (33) and H-1 virus (24),
PPV is
significantly
purified
from cellularmate-rial at
high
pH and lowionic
strength
in the presenceof EDTA andisgreatly
enrichedfor by
CaCl.
precipitation.
CaClprecipitation
of thevirus was found to be essential for obtaining
highly
purified
preparations
and wasfar superiorto other methods
of
virusconcentration
(e.g.,12%
polyethyleneglycol
or30%
ammoniumsulfate).
TheCaCl-precipitated
virus wassus-pended
in the high-pH buffer in thepresence of25 mM EDTA, and the soluble
virus-containing
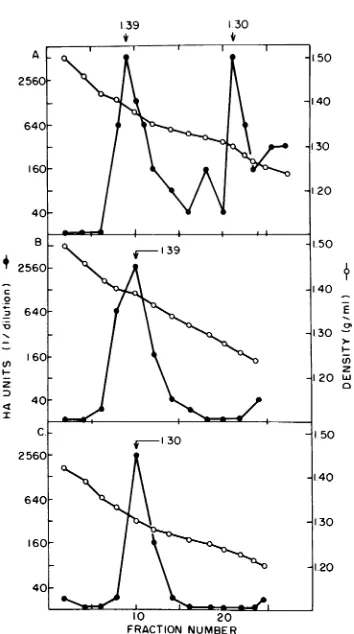
material was then subjected to discontinuous CsCldensity centrifugation (Fig. 2). Asdetected by HA, viral antigen appeared in two major, well-resolved peaksatbuoyantdensities of 1.39 and 1.30
g/ml (Fig.
2A). These twopeaks
wereroutinely
seenin different fetal preparations andusually
contained
equal
amounts ofhemaggluti-nating activity. To further enrich for these two viral antigen-containing species, we subjected
the
pooled
fractions of eachdensity
peak
to asecond cycle of discontinuous CsCl
centrifuga-tion.The
1.39-g/ml
densityparticles
were run ona
density
gradient
identical to the firstgradient
(Fig. 2B), and the lighter
1.30-g/ml
particles werecentrifugedin aslightlylessdensegradient (Fig.2C).
Ineach case, asingle
HApeak of viral antigen wasobserved,banding
atthe same den-sity as found in the first densitygradient
run.The
HA-positive
fractions of both the 1.39- andthe
1.30-g/ml particles
were individually pooledand
dialyzed
against
TE buffer.VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
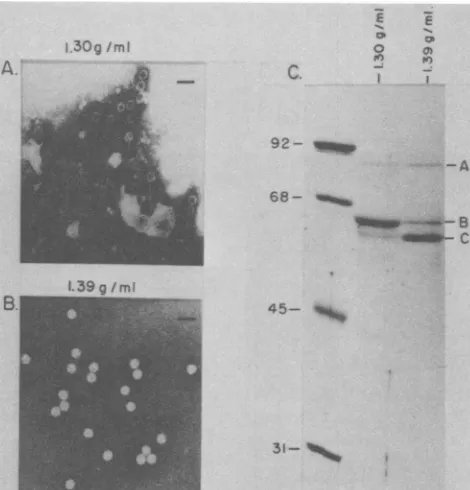
The
purity
of these two viralantigen-contain-ing fractions was assessed by negative-contrast electron microscopy (Fig. 3A) of samples and
Coomassie brilliant
bluestaining
inSDS-con-taining 10% polyacrylamide gels electropho-resed(Fig. 3C). Particles banding at a density of 1.39 g/ml had a diameter of approximately 20 nm. They excluded the phosphotungstic acid stain (Fig. 3A) and therefore appeared as charac-teristic "full" (DNA-containing) parvovirus viri-ons. Particles of similar size were found in the 1.30-g/ml density band together with varying amounts
of
debris (Fig. 3B). However, these particles appeared to largely take up the nega-tive stain; therefore, they most likely represent "empty" (DNA-lacking) virion capsids. These results appear to be similar to those obtainedfrom
other, more thoroughly studied parvovirus systems (35). Wehave not attempted to identify viral particles that may be present atintermedi-ate
densities
or that may represent variousim-mature or partial genome-containing virions.
The polypeptide
composition
of thematerial
present in the 1.39- and
1.30-g/ml
density bands
was analyzed by
SDS-polyacrylamide
gelelec-trophoresis (Fig.
3C). Bothpreparations
ap-peared
tocontain three
predominant
protein
bands with
molecular
weights
of83,000, 64,000,
and 60,000.
The threeproteins
of
the "full"particles appeared
tobe
highly enriched,
where-as
minor, presumably
contaminating protein
species
were present in the"empty"
particle
preparations.
The presenceof
threepolypep-tides
of
theobserved molecular
weights
inhighly
purified virus
preparations
is verycharacteristic
of
parvoviruses (5, 10, 11, 26-28, 33),
and wetherefore conclude
that these threepolypeptides
are
the structural
proteins
of
PPV. Thestructur-al
proteins of parvoviruses
havepreviously
been
termed A, B,
and C in
descending order of
molecular
weight
(32),although VP1, VP2,
andVP3
have also been
used
(19, 24).
The
relative
distribution of the three
PPVstructural
proteins should be noted.
Inthe
1.39-g/ml
particles,
the60-kilodalton
(kd) protein (C
orVP3) appears to be most
abundant,
whereas in the1.30-g/ml particles,
the64-kdprotein
(Bor VP2) is present in the greatest amounts(Fig.
3C).
Although this difference
in the B and Cproteins
betweenfull and emptyvirion
particles
exists,
the amount of the 83-kdprotein
(A orVP1) appearsto
remain
constant(approximately
10%
of thetotal).
This observation has been made with severalindependent
PPV prepara-tions and has also been observed in otherparvo-virus
systems(5, 24).Structural relatedness of PPV
polypeptides.
Previous studies of
parvovirus structural
pro-teins have indicated that the
A, B,
and Cpoly-peptides
are veryclosely
related asdetermined
4
(0
z
I
E
-p
(0) z 0a
FRACTION NUMBER
FIG. 2. CsCl density centrifugation of PPV. PPV
was precipitated with CaC12 from virus-containing homogenates ofinfected swinefetuses, suspended in
TEbuffer, and prepared for discontinuous CsCl densi-ty centrifugation as described in the text. Gradient fractions were assayed for PPV-specific hemaggluti-nating activity (HA) and density (A). The two peak regions ofHAactivitywereindividually pooled. The1.39-g/mldensity peak wassubjected to discontin-uousCsCl centrifugation asecond time in agradient identical tothat used the first time (see the text) and assayedsimilarly (B).The1.30-g/mldensity peakwas
centrifuged in a second gradient of slightly lower density than the first. The pooled fractions from the first CsClgradient wereadjusted to afinalCsCl
density of1.25g/ml and layeredonto anequal volume
ofCsCl solutionhavingadensity of1.30g/ml. Centrif-ugation and subsequent gradient fraction assay (C)
wereperformedasdescribed inthe text.
by
variouspeptide
mapping procedures (12, 19,
22,
34). In these systems,it
appears that thepeptides
of the Cprotein
are contained withinthe
peptides
of the Bprotein,
whichareinturn asubset of those of the A
polypeptide.
To deter-mine whetherasimilarsituation existed
with thePPV
structural
proteins,
weperformed
one-dimensional
partial proteolysis mapping
studiesonthe three PPV
polypeptides.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.496.267.444.68.385.2]PPV: PURIFICATION AND ANTIGENIC PROPERTIES
i,30g/ml
A.
B
E '
0 oL
C. i I
92- ,
-A 68- -_
_- C
[image:6.496.127.362.76.321.2]45- _
FIG. 3. Analysis of CsCldensity gradientfractionscontainingPPV-specifichemagglutination activity. The 1.39-and1.30-g/mlpeaks of HAactivity(Fig.2BandC,respectively)wereindividually pooledandextensively dialyzedagainst TE buffer. Portions of thedialyzedmaterialswereanalyzed bynegative
staining
inthe electron microscope(see thetext).(A)1.30-g/ml
densitymaterial. (B)1.39-g/mldensity
material.Bar,40nm.Another portionof thedialyzedmaterials wassubjectedtoelectrophoresisinanSDS-l0o
polyacrylamide gel.Thegel wasthenstained with Coomassie brilliant blue and destained(C).Molecularweightstandardswereincluded inanadjacenttrack in thegel(numbersrepresentkilodaltons).
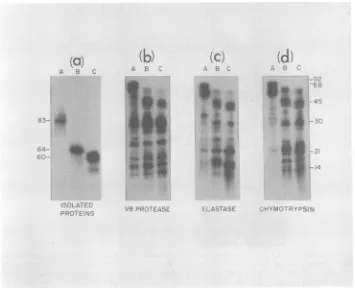
The
proteins
of
highly purified
PPV
(1.39-g/ml
particles)
wereradiolabeled in vitro with 1251 and
separated
by SDS-polyacrylamide
gel
electro-phoresis. To
ensurethe
purity of each
polypep-tide, the three proteins
wereindividually
sub-jected
to asecond cycle of polyacrylamide gel
electrophoresis before
anyanalyses
wereper-formed. A
portion
of each
of the
twice-gel-purified proteins
waselectrophoresed
on athird
polyacrylamide
gel (Fig.
4a).
Using
the
partial
proteolytic
mapping
proce-dure
originally
described
by
Cleveland
etal.
(4),
we
compared the
partial digests of the
1251_
labeled PPV A, B, and C
proteins by using three
different
proteases:S.
aureusV8
protease,elas-tase, and
chymotrypsin
(Fig. 4). The V8prote-ase
patterns
werenearly identical for
thethree
polypeptides.
Theelastase and
chymotrypsin
patterns of
eachof the
threeproteins
werealsovery
similar, with
manypeptides from the threeproteins
migrating identically.
Itappeared,how-ever, that
several of the
peptide bands derived
from protein
Amigrated
moreslowly than the
similar
peptides in the B protein digest.
Similar-ly,
certain peptides in the B
protein pattern
migrated
moreslowly than the
corresponding
C
protein
peptides (Fig. 4).
This suggeststhat
these
fragments
areterminal
portions of the
respective
proteins, which
areotherwise
verysimilar in
primary
structure.Use of individual PPV structural
proteins
to generatePPV-specific
antisera.The data
report-ed
above
suggestclose structural similarities
among the
three virion
polypeptides A, B, and C
of PPV. To further compare
these
polypeptide,
we
felt it would be useful
toprepareantisera
toeach individual SDS-denatured
protein. To
thisend, highly purified
PPVpreparations
weredis-rupted and
subjected
topreparative
SDS-poly-acrylamide gel
electrophoresis
to separate thethree viral
proteins.
After
localization
of the
protein bands,
each was excised and then wassubjected
to asecond
cycle of gel
electrophore-sis
as wasdone
for
theabove-described
125I1
labeled
proteins. The
protein
bandsof
thesec-ond
polyacrylamide
gel
werelocalized
andextracted from the
gel pieces
asdescribed
above.
Figure
5shows
aCoomassie
blue-stained
polyacrylamide gel
of
aliquots
of each of the
gel-purified
proteins
and
asample
of the
starting
virus preparation. After these
procedures,
weroutinely
wereunable
todetect
contamination
ofVOL. 45,1983 847
on November 10, 2019 by guest
http://jvi.asm.org/
848 MOLITOR, JOO, AND COLLETT
(a)
r. C:
.; A
0. *
i.-(b)
II
.
0
43
*
i
1**
(c)
S
.
*9.
O..
*.0
9.
I;
_(d)
sg
-U
F~rA. '¼-;;..; F
FIG. 4. Partialproteolysis mapping of
125I-labeled
PPVstructural proteins. Purified virions (1.39 g/ml) were disrupted and radiolabeled with 1251 as described in the text. The three individual polypeptides, A, B, and C, were gel purified twice (see the text), and a portion of each was re-electrophoresed in a third SDS-10% polyacrylamidegel to assess theirpurity (panel a). Portions of each polypeptide were then subjected to partial proteolytic hydrolysis during electrophoresis in SDS-15%polyacrylamide gels as described previously (4), using (b) V8 protease, 0.1 ,ug per track; (c) elastase, 1 ,g per track; and (d) chymotrypsin, 5 ,ug per track. After electrophoresis, gels were dried and exposed to X-ray film. The numbers in the margins represent approximate molecular weights (in kilodaltons).one protein
with
theothers,
even when1251
labeled
proteins
weresimilarly
purified
(Fig. 4).
However,
wedo
feel that
contamination below
the
sensitivity of
our testscould have existed.
Each
of these
gel-purified proteins
wasused
toimmunize
rabbits.
Inaddition,
intact PPV
wasused as an
immunogen.
At3 and 7weeks
post-immunization,
therabbits
receiving
thegel-purified
proteins
wereboosted with additional
protein.
Atvarious times after
theprimary
im-munization, small samples of
serawereobtained
from each
rabbit and assayed for PPV-specific
antibodies
by
anHAI
assay. The immunere-sponses of each rabbit are shown in
Fig.
6. Allof
the
immunogens elicited
aPPV-specific
re-sponse;
however, it
appeared that intact virus
was
able
to generatethe strongest and
mostrapid
response. It must benoted
here, however,that
only
onerabbit
was usedfor
eachimmuni-zation.
Wefeel
thatthis
precludes
anyconclu-sions
concerning
theantigenic
strength of
thevarious immunogen
preparations.
A summaryof
the
various antisera used in this
study
is
provid-ed
in Table
1.Serological
testscomparing
variousPPV-specif-ic antisera. A
variety of standard
serological
tests was
performed, using
conventional
anti-sera toPPV and
antisera
generated by
theSDS-denatured,
gel-purified
viralproteins.
There-sults of
thesetests aresummarized
inTable
2.Although
theantisera
generated against
thegel-purified proteins
exhibited lower HAI titers
thanantisera
produced
from intact virus
(Fig.
6;
Table
2), these antisera did
reactwith viral
antigens
ininfected
cells in an IFA assayand did
contain
precipitating
antibodies
asdetermined
inan
AGID
assay.Ability of antisera to
recognize
denatured and native PPVantigens.
Wewent ontocompare thereactivities of
thevarious
PPVantisera
toboth
denatured
viralproteins
andnative,
intactvirus
particles.
Becauseantisera
weregenerated
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.496.81.436.76.364.2]PPV: PURIFICATION AND ANTIGENIC PROPERTIES 849
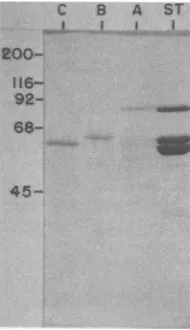
C B A ST
L..L. .-..II. i
200- 116-
92-
68-IN~
_
45-FIG. 5. Purity of PPV polypeptides A, B, and C used forrabbit immunizations. Purified virions (1.39g/ ml) were disrupted by boiling in SDS-sample buffer and electrophoresed in preparative SDS-101% poly-acrylamide gels. After localization (see the text), the threepolypeptides A, B, and C were excised and then wereindividually subjected to a second cycle of elec-trophoresis. A portion of the three protein prepara-tions obtained after gel elution anddialysis (see the text) was thenelectrophoresedin anothergel together with a sample of the starting viruspreparation(ST). Thegel was stainedwith Coomassie brilliant blue and destained.
2 4 6 8 10 12 14
POST-IMMUNIZATION TIME (WEEKS)
FIG. 6. Immuneresponse of rabbits injected with
intact virions and isolated PPV structural proteins. Individual rabbits were immunized with 50 ,ug of
intact, CsCl-purified virus (1.39-g/ml particles), gel-purified polypeptide /A, gel-purified polypeptide B,
andgel-purifiedpolypeptideCasdescribed in thetext. Rabbits receiving gel-purified polypeptides were
boostedat3and 7weeksafter the initialinjection,and
the rabbit receiving intact virus was boosted at 3
weeksonly.Atvarious times aftertheprimary
immu-nization, blood samples werecollected, and the sera were assayed for PPV-specific antibodies by
hemag-glutination inhibition (see the text).
TABLE 2. Serological tests comparing the immune responseof rabbits and swine immunized with either
intact PPV or gel-purified structuralproteins
Antiserum Serologicaltest
resulta
HAI IFA AGID
Normal pig <4 -
-Adultpiga PPV 8,096 + +
Fetalpig a PPV 8,096 + +
Normalrabbit serum <4
Rabbit aintact PPV 8,096 + +
Rabbit a A polypeptide 1,024 + +
Rabbit a Bpolypeptide 32 + +
RabbitaCpolypeptide 2,048 + +
a The serological assays, HAI (hemagglutination inhibition), IFA (indirect fluorescent-antibody), and AGID(agargelimmunodiffusion),aredescribed inthe text. -, Nodetectable reactivity; +,clearly positive reactivity. As the IFA and AGID are difficult to evaluate quantitatively, no attempt is made here to distinguish antisera reactivities.
against the
SDS-gel-purified proteins,
wemight
expect
these
sera tocontainantibodies directed
more toward linear determinants on the
pro-teins, whereas antisera produced by
immuniza-tion
withintact
virions
might contain antibodies
to
higher-order
structuraldeterminants. PPV
a-(f) 0- Go u
z cr : c
I2 4 5
FIG. 7.
Reactivity
of variousantiseratodenaturedPPV
polypeptides.
Punified PPV wasdisrupted by
boiling
inSDS-sample
buffer, and the viralpolypep-tideswereresolvedon anSDS-10%
polyacrylamide gel.
The
proteins
werethenelectrophoretically
transferredtonitrocellulosepaper(2),
strips
werecut, andindivid-ual
strips
were incubated with various antisera (seeTable1), followedbyreaction with
"N-I-abeled
protein A(seethetext).The washedstrips
werethenexposedto
X-ray
film. The exposure time for allstrips
shownwas thesame(10 min). Upon
longer
exposure(5to6h), a
qualitatively
similarreactivity
of R aPPV withthe PPV
proteins
wasrevealed; however,noreactivity
wasobserved with NRS.VOL. 45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.496.95.190.77.242.2] [image:8.496.251.441.394.552.2] [image:8.496.44.232.399.558.2]particles
were disrupted by boiling in SDS and2-mercaptoethanol,
resolved by electrophoresis inan
SDS-polyacrylamide
gel, and transferred tonitrocellulose
paper.Identical
strips of
thepro-tein-containing nitrocellulose
paper were thenincubated with
thevarious antisera, followed by
incubation
with125I-labeled
protein A tolocal-ized bound immunoglobulin.
The results of this"Western
blot" analysis
are shownin Fig.
7.Normal
rabbit
serumshowed
noreactivity
tothe
PPV proteins (Fig. 7, track 1). The individual antisera generated against each of the viral pro-teinsshowed very strong reactivity with all three PPV structural proteins (Fig. 7, tracks 3 to 5).
Antisera obtained by immunization with intact
virus reacted
verypoorly
withthe denatured
antigens (Fig. 7, track
2). However,longer
auto-radiographic
exposures (10times) did
revealsome
reactivity
to the threeviral proteins.
Simi-larly, porcine
antisera obtained from
naturallyinfected
adultpigs
orinfected fetuses
reactedpoorly
with thedenatured
PPVpolypeptides
(data
notshown). Thus,
as expected,antisera
raised against
theSDS-gel-purified proteins
re-acted
verywell with
thedenatured polypeptides.
It is
of
interest here that all antisera
reacted to allthree viral structural proteins.
Eventhough
anti-serumwas
generated by
immunization with
eachindividual viral
protein, each antiserum,
inaddi-tion
torecognizing
theimmunizing antigen,
rec-ognized equally
wellthe
other twoviral
pro-teins. This result is consistent with
theclose
structural
similarities
among the threeproteins
(Fig.
4).However,
analternate
interpretation
ofthese
resultsis
that ourimmunogen preparations
of
the
gel-purified proteins
weresufficiently
con-taminated with
theother
polypeptides
that animmune
response wasalso
mountedagainst
these contaminant
proteins.
To
determine whether the various antisera
were
able
torecognize
native, intact virus
parti-cles,
wetested their
ability
to causetheimmuno-specific aggregation (lattice formation) of
PPVvirions
in animmunoelectron microscopy
assay(7). Antisera
weremixed with
purified virus,
andany aggregates formed were collected by
centrif-ugation.
Thepelleted
material was thenana-lyzed after negative staining in the electron
microscope. Immunospecific
latticeformation
occurred with antisera
generated against
nativevirus
(Fig. 8, panel 2) and with the antiseragenerated
against
theindividual gel-purified,
de-natured proteins
(Fig.
8, panels 3 to 5), but not withnormal, preimmune serum (Fig. 8, panel 1). Ability of antisera to neutralize virus infectiv-ity. The resultsdescribed
above indicate thatantisera produced
by
immunization
withSDS-gel-purified
PPVproteins
are able to recognizenative, intact virus particles
as well as lineardeterminants on the denatured viral
polypep-tides.
Wefinally
asked whether these antiseracontained antibodies
capable
ofneutralizing
vi-rus infectivity. Using a direct immunofluores-cenceassay,wefound that antiserumgenerated
by each of the
gel-purified
proteins did indeed containneutralizing
antibodies (Fig.9; Table
3).
It appears,however, that the titer of the neutral-izing antibodies in each of the antiersa
produced
against the individual proteins was lower than that found in antisera generated against the intact virus. The neutralizing titers of the sera paralleled the
HAI
titers described above(Table
2).
DISCUSSION
Swine
fetuses, either naturally
exposed to or experimentally inoculated with PPV, actively support virusreplication,
which ultimately re-sults in fetal death. As PPV isoften difficult
to grow tohigh titers in cells culturedin vitro,
we initially concentrated on characterizing themo-lecular
features of
PPV aspropagatedin infected
animals. Recently, a brief report on certain aspects of PPVreplication in culturedfetal
por-cine
kidney cells has appeared
(29). Theavail-ability
of both
an animal modeland
an in vitro cell culture system for viruspropagation
pro-vides an excellent opportunity for the detailed and controlled study of the molecular, immuno-logical, and pathogeniccharacterization
of PPV.Propagation of
PPVinswine fetuses
providesanabundant source
of
virus material.Employing
procedures used
for
thepurification of
previous-ly
studied parvoviruses
(33), we have been ableto obtain
highly enriched
virionpreparations.
These
preparations contained
twopredominant
forms
of
the 20-nmvirus
particle:
"full"(DNA-containing) particles with
adensity
inCsCl
of1.39
g-ml, and "empty"
(DNA-deficient)
parti-cles with the
lighter density
of1.30g/ml.
Threemajor
polypeptides, designated
A,B,
andC,
were
consistently found
in both of these virusspecies, having
molecularweights of 83,000,
64,000,
60,000,
respectively.
Otherparvoviruses
have been shown to consist of three structural proteins with molecular
weights
in thefollowing
ranges: A, 93,000 to 73,000; B, 80,000 to64,000;
C,
67,000 to56,000(32).
Wetherefore conclude that theseproteins are the structuralproteins
ofPPV.
Polypeptide
Aroutinely represented
ap-proximately 10%
of the total viralprotein
inboth the full and the empty viral
particles.
Polypeptide
C appeared most abundant in the"full"
particles,
whereas the Bprotein
was present in the greatest relative amounts in the"empty" particles (Fig.
3). All of these resultsare
consistent
withprevious observations
inother
studied
parvovirus
systems(35).
Earlierreports on work with rodent
on November 10, 2019 by guest
http://jvi.asm.org/
PPV: PURIFICATION AND ANTIGENIC PROPERTIES 851
4
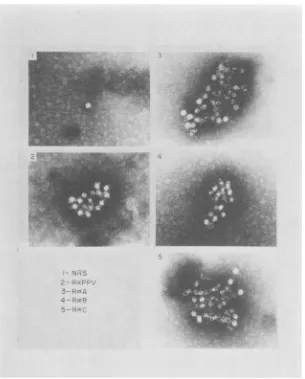
-FIG. 8. Immunoelectron microscopy of
purified
PPV reacted with various antisera. Various antisera (see Table 1)wereincubated withpurified, intact virusparticlesand tested for theirabilitytoform immunespecific
virus-antibody latticesasdescribed in thetext.
ruses(34), the defective adeno-associated virus-es(12, 19), and,morerecently,witha
densonu-cleosisvirus(22) have shownthat theA, B,and C viral structural proteins in their respective systems have extensive sequence homology withoneanother. Itnow appearstobeaccepted that the lower-molecular-weight polypeptides
aresubsets of thelarger proteins; however, the meansof their derivationand the mechanism of
putative RNAorprotein processingremain
ob-scure. Inour limited structural analyses of the
PPV proteins, we found that the three viral
proteins were also very closely related to each other inprimary sequence.
Avariety of antiseraweregeneratedto inves-tigate certain antigenic properties of PPV and its constituent proteins. Ourgreatest interest
cen-tered
onthe
antisera
that were raised againstthe
SDS-denatured, gel-purified
viral structuralproteins. Antibodies elicited by
theindividual
gel-purified polypeptides
werequalitatively
sim-ilar
toantibodies in
serafrom naturally infected
adult
pigs
orrabbits
experimentally immunized
with intact virus particles in several standard
serological
assays,including
HAI, an IFA test,and an
AGID
assay(Table
2).However,
theantisera
generatedagainst
thedenatured,
gel-purified
proteins
reacted more avidly with thedenatured
antigens
thandid
theconventional
antisera
(Fig. 7). Still,
theseantisera
were allable
torecognize
andimmunospecifically
aggre-gate
native
intact virion particles (Fig. 8).
Final-ly,
allantisera
directed against PPV,whether
produced by
naturalinfection, immunization
VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
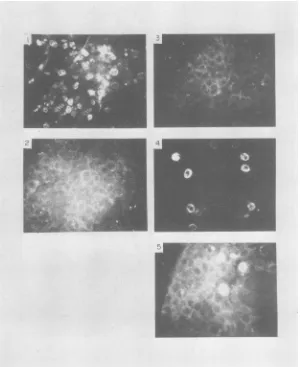
[image:10.496.98.403.72.451.2]852
FIG. 9. Virusinfectivityneutralizationbyvarious antisera detected by direct immunofluorescence. Various
sera
(diluted
1:10inPBS)weremixed andincubated with PPV and then were placed on cultures of ST cells, asdescribed in thetext.After4days, PPVcellularantigensweredetected with afluorescent-antibody conjugate andviewed under afluorescentmicroscope(see the text). The seraused were (1) NRS; (2) R a PPV; (3) R a A; (4) R a B; (5) R a C (see Table 1 forabbreviations).
with intact virus
particles,
orimmunization
withthe
individual, SDS-gel-purified
viral structural
proteins,
wereable
toneutralize virus
infectivity
(Fig. 9; Table 3). Antisera prepared
against
individual
parvovirus structural
proteins
in othersystems have
failed
toelicit
avirus-neutralizing
response
(6,
9). Several
explanations
may ac-countfor this
apparentdiscrepancy.
Wefeel
thatthe method of
antigen preparation
may be animportant factor.
We
find it
interesting
that each
of the antisera
generated
against
theindividually gel-purified
viral
proteins reacted with all
three structuralproteins and that each viral
polypeptide
wascapable of
eliciting
virus-neutralizing
antibody.
These results
areconsistent
with the
sequencehomology
among thethree
proteins
and suggestthat
thevirus-neutralizing
determinant(s)
is
present on
all
threeviral
structuralproteins.
However,
wefeel that due
tothe
possible
minor
contamination of each
immunogen preparation
with the
other structural
proteins, and
tothe
fact
that the
neutralizing
antibody
responsecould
have been
aresult of
suchcontamination,
anyconclusions
concerning
the
natureof
PPV-neu-tralizing
antigenic
determinants is
unwarrantedat
this time. The
generation
of monoclonal
anti-bodies
to theseproteins should
provide
moreconclusive
information
concerning
the number
and
distribution of
virus-neutralizing antigenic
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:11.496.109.409.83.450.2]PPV: PURIFICATION AND ANTIGENIC PROPERTIES TABLE 3. Viral infectivity neutralization of various
antisera
Culture fluid Fluorescent
antigen
Antiseruma (HA)b cells (%)C
1:2 1:10 1:2 1:10
Noserum 256 256 >80 >80
Novirus <4 <4 0 0
Normal pig serum 256 256 >80 >80
Fetal
pig
aPPV <4 <4 0 0Normal rabbit serum 256 256 >80 >80 Rabbit a intact PPV <4 <4 0 0
Rabbit a A <4 <4 0 0-5
RabbitaB <4 32 0 30-40
Rabbit aC <4 16 0 5-10
aFor standardization, rabbit a A, a B, and a C
antisera were normalized to the same HAI titer and thenwere diluted either 1:2 or 1:10 in PBS.
bCellculture fluids were tested at 3 days postinfec-tion for the presence of extracellular (culture fluid) virus by hemagglutination of guinea pig erythrocytes (seethe text).
cAcetone-fixed cells were stained at 3 days postin-fection with PPV-fluorescent antibody conjugate for thepresence of intracellular virus as described in the text.The percentageof fluorescent cells was estimated fromatleastfiveindependent fields of view.
determinants on the PPV structural polypep-tides.
ACKNOWLEDGMENTS
We thank Dennis Anderson, Shirley Halling, and Allen Lemanforassistance andnumerousenlighteningdiscussions, SusanWells forcontinuing support, andCindy Kosman for manuscriptpreparation.
Portionsofthis work weresupported bygrant MIN-26-074 from theMinnesotaAgricultureExperiment Station.
LITERATURECITED
1. Bier, M., R. M. Cuddeback, and A. Kopivillen. 1977. Preparative plasmaproteinfractionationby isotachophor-esis inSephadexcolumns. J.Chromatogr. 132:437-450. 2. Bittner, M., P. Kupferer, and C. F. Morris. 1980.
Electro-phoretic transfer of proteinsand nucleic acids from slab gelstodiazobenzyloxymethylcelluloseornitrocellulose sheets. Anal.Biochem. 102:459-471.
3. Bommeli, W., M. Kihn, F. Zindel, and H. Fey. 1980. Enzyme-linkedimmunosorbent assay andfluorescent an-tibody: techniques in thediagnosis of viral diseases using StaphylococcusproteinAinsteadofanti-a-globulin.Vet. Immunol. andImmunopathol. 1:179-193.
4. Cleveland, D. W., S. G. Fisher, M. W. Kirschner, and U. K.
Laemmnl.
1977.Peptide mapping bylimited proteol-ysisinsodiumdodecyl sulfate andanalysis by gel elec-trophroesis.J. Biol.Chem. 252:1102-1106.5. Clinton, G. M., and M. HayashL. 1975. The parvovirus MVM:particleswith altered structuralproteins. Virology 66:261-267.
6. Croft, G. F., M.D. Hoggan, and F. B.Johnson. 1974. Production and reactivity of immune sera specific for HADENviruspolypeptide antigens.J. Virol. 13:608-613. 7.England, J.J.,andD. E. Reed. 1980.Negativecontrast
electron microscopic techniques for diagnosis ofviruses of veterinary importance. Cornell Vet. 70:125-136. 8. Hunter, W. M. 1971. Radioimmunoassay, p. 17.1-17.36.
In D. W. Weis (ed.), Immunochemistry, vol.I. Blackwell Scientific, Oxford, England.
9. Johnson, F. B., N. R. Blacklow,and M. D. Hoggan. 1972. Immunological reactivity of antisera prepared against the sodium dodecyl sulfate-treated structural polypeptides of adenovirus-associated virus. J. Virol. 9:1017-1026. 10. Johnson, F. B., H. L. Ozer, and M. D. Hoggan. 1971.
Structural proteins of adenovirus-associated virus type 3. J.Virol. 8:860-863.
11. Johnson, F. B., and M. D. Hoggan. 1973. Structural proteins of Haden virus. Virology 51:129-137.
12. Johnson, F. B., T. A. Thomson, P. A. Taylor, and D. A. Vbzny. 1977. Molecular similarities among the adenovi-rus-associated virus polypeptides and evidence for a precursor protein. Virology 82:1-13.
13. Joo, H. S., C. R. Donaldson-Wood, and R. H. Johnson. 1976. Observations of the pathogenesis of porcine parvo-virusinfection. Arch. Virol. 51:123-129.
14. Joo, H. S., C. R. Donaldson-Wood, and R. H. Johnson. 1976. A standardizedhemagglutination-inhibition test for porcine parvovirus. Aust. Vet. J. 52:422-424.
15. Joo, H. S., and R. H. Johnson. 1977. Serological responses in pigs vaccinated with inactivated porcine parvovirus. Aust. Vet. J.53:550-552.
16. Joo, H. S., R. H. Johnson, and P. C. Watson. 1978. Serological procedures to determine time of infection of pigs with porcineparvovirus. Aust. Vet. J. 54:125-127. 17. Laemmli, U. K. 1970. Cleavage of structural proteins
during the assembly of the head ofbacteriophage T4. Nature (London)227:680-685.
18. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 192:265-275.
19. Lubeck, M. D., H. M. Lee, M. D. Hoggan, and F. B. Johnson. 1979. Adenovirus-associated virus structural protein sequencehomology. J. Gen. Virol. 45:209-216. 20. Mengeling, W. L., T. T. Brown, P. S. Paul, and D. E.
Gutenkunst. 1979.Efficacy of an inactivated virus vaccine for prevention of porcine parvovirus-induced reproduc-tivefailure. Am. J. Vet. Res.40:204-207.
21. Mengeling, W. L., and R. C.Cutlip. 1975. Pathogenesis of in utero infection:experimental infection of five-week old porcine fetuses with porcine parvovirus. Am. J. Vet. Res. 36:1173-1177.
22. Moore, N. F., and D. C.Kelly. 1980. Interrelationship of theproteins of two insect parvoviruses (densonucleosis virustypes 1 and 2). Intervirology 14:160-166. 23. Morimoto, T., Y.Fujisaki,Y. Ito, and Y. Tanaka. 1972.
Biological and physiochemical properties of porcine par-vovirus recoveredfrom stillborn pigs. Inst. Animal Health Quart. 12:137-144.
24. Paradiso, P. R. 1981. Infectious process of the parvovirus H-1:correlation of the protein content, particle density, andviral infectivity. J. Virol. 39:800-807.
25. Redman, D. R., E. H. Bohl, and L. C. Ferguson. 1974. Porcine parvovirus: natural and experimental infections of theporcine fetus and prevalence in mature swine. Infect. Immun. 10:718-723.
26. Rose, J. A., J. V. Maizel, Jr., J. K. Inman, and A. J. Shatkin. 1971. Structural proteins of adenovirus-associat-edviruses. J. Virol. 8:766-770.
27. Salo, R. J., and H. S. Mayor. 1977. Structural polypep-tides of parvoviruses. Virology 78:340-345.
28. Salzman,L. A., and W. L. White. 1970. Structural pro-teins of Kilham rat virus. Biochem. Biophys. Res. Com-mun.41:1551-1556.
29. Shahrabi, M. S., J. Lynch, H. J. Cho, and R. G. Marusk. 1982.Studies on the multiplication of a porcine parvovi-rus.Vet. Microbiol. 7:117-125.
30. Sterzl, J., J.Rejnik,and J.Travnicek.1966. Impermeabil-ity of pig placenta for antibodies. Folia Microbiol. (Prague) 11:7-10.
853 VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
31. Symington,J., M.Green, and K.Bragemann. 1981. Im-munoautoradiographic detection of proteins after electro-phoretic transfer from gels to diazo-paper: analysis of adenovirus encoded proteins. Proc. Natl. Acad. Sci. U.S.A. 78:177-181.
32. Tattersall,P.1978.Parvovirus proteinstructureandvirion maturation, p. 53-72.In D.C. Ward and P. Tattersall (ed.), Replication of mammalian parvoviruses. Cold Spring Harbor Laboratory, ColdSpring Harbor, N.Y.
33. Tattersall,P., P. J.Cawte,A.J. Shatkin,andD.C. Ward. 1976.Three structuralpolypeptidescoded forbyminute virus ofmice,aparvovirus, J. Virol. 20:273-289.
34.Tattersall, P., A. J. Shatkin, and D.C. Ward. 1977. Sequence homologybetween the structuralpolypeptides ofminute virus of mice. J. Mol. Biol.111:375-394. 35. Ward, D.C.,andP.Tattersall (ed.). 1978.Replication of
mammalian parvoviruses. Cold Spring Harbor Labora-tory,ColdSpring Harbor,N.Y.
J. VIROL.