Combined PI3K and CDK2 inhibition induces
cell death and enhances
in vivo
antitumour
activity in colorectal cancer
Gary Beale
1, Emma J Haagensen
1, Huw D Thomas
1, Lan-Zhen Wang
1, Charlotte H Revill
2, Sara L Payne
2,
Bernard T Golding
2, Ian R Hardcastle
2, David R Newell
1, Roger J Griffin
2,{and Celine Cano*
,21Newcastle Cancer Centre, Northern Institute for Cancer Research, Paul O’Gorman Building, Medical School, Newcastle
University, Framlington Place, Newcastle-upon-Tyne NE2 4HH, UK and2Newcastle Cancer Centre, Northern Institute for Cancer Research, School of Chemistry, Newcastle University, Bedson Building, Newcastle NE1 7RU, UK
Background:The phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) pathway is commonly deregulated in human cancer, hence many PI3K and mTOR inhibitors have been developed and have now reached clinical trials. Similarly, CDKs have been investigated as cancer drug targets.
Methods:We have synthesised and characterised a series of 6-aminopyrimidines identified from a kinase screen that inhibit PI3K and/or mTOR and/or CDK2. Kinase inhibition, tumour cell growth, cell cycle distribution, cytotoxicity and signalling experiments were undertaken in HCT116 and HT29 colorectal cancer cell lines, andin vivoHT29 efficacy studies.
Results:2,6-Diaminopyrimidines with anO4-cyclohexylmethyl substituent and a C-5-nitroso or cyano group (1,2,5) induced cell cycle phase alterations and were growth inhibitory (GI50o20mM). Compound1, but not2or5, potently inhibits CDK2 (IC50¼0.1
nM) as well as PI3K, and was cytotoxic at growth inhibitory concentrations. Consistent with kinase inhibition data, compound1
reduced phospho-Rb and phospho-rS6 at GI50concentrations. Combination of NU6102 (CDK2 inhibitor) and pictilisib (GDC-0941;
pan-PI3K inhibitor) resulted in synergistic growth inhibition, and enhanced cytotoxicity in HT29 cells in vitroand HT29 tumour growth inhibitionin vivo.
Conclusions:These studies identified a novel series of mixed CDK2/PI3K inhibitors and demonstrate that dual targeting of CDK2 and PI3K can result in enhanced antitumour activity.
Insights into the molecular pathology of cancer have resulted in the development of targeted approaches for cancer therapy. The phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/ mTOR) pathway is one of the most commonly deregulated pathways in human cancer in which signals are transduced via a cascade of lipid and serine/threonine kinases resulting in diverse effects on proliferation, apoptosis, protein translation, DNA repair, survival and transformation (for review see (Mayer and Arteaga, 2015)). Activation of receptor tyrosine kinases (RTKs) stimulates the PI3K pathway via adapter proteins, and PI3K phosphorylates phosphoinositide-3,4-biphosphate
(PIP2) to generate PIP3 that activates AKT(PKB). AKT has many functions including roles in cell migration, apoptosis and glucose uptake, and the activation of mTOR via downregulation of the TSC1/ Rheb complex. mTOR in a complex with Raptor (to form mTORC1) signals via p70S6 kinase and 4EBP1 to regulate the translation of proteins and cell growth. mTOR also complexes with Rictor (to form mTORC2) to activate AKT. Negative feedback occurs at many nodes in the PI3K/mTOR cascade, most notably by PTEN and by S6 kinase-mediated regulation of RTK signalling, for example, by downregulation of IRS-1 (Harringtonet al, 2005).
*Correspondence: Dr C Cano; E-mail: celine.cano@ncl.ac.uk
{
Deceased.
Revised 31 May 2016; accepted 14 July 2016; published online 16 August 2016
&2016 Cancer Research UK. All rights reserved 0007 – 0920/16
Keywords:CDK2; PI3K; combination; colorectal; cancer; synergy
Activating mutations in the PIK3CA (p110a) isoform of PI3 kinase are observed in a wide range of tumours including breast, colon, glioblastoma and ovarian cancers (Cancer Genome Atlas Research Network, 2008; Stemke-Hale et al, 2008; Mayer and Arteaga, 2015), and inactivation of the tumour suppressor protein PTEN has been documented in, among others, glioblastoma, breast cancer, prostate cancer and melanoma (Cairnset al, 1997; Garcia et al, 1999; Celebi et al, 2000; Cancer Genome Atlas Research Network, 2008). More rarely AKT mutations or amplifications have been detected in colorectal, head and neck, pancreatic and breast tumours, and large a proportion of cancers have activation of mTORC1 as demonstrated by increased phosphorylation of proteins such as S6 kinase, 4EBP1, ribosomal S6 protein and mTOR itself (reviewed in (Menon and Manning, 2008)).
Considerable effort has been focussed on the development of PI3K/mTOR pathway inhibitors, many of which have now reached clinical trials (Yap et al, 2015). Rapamycin, discovered as an antifungal agent from Streptomyces hygroscopicus, was the first mTOR inhibitor characterised and a number of the trial candidates are rapamycin analogues (rapalogues). Rapamycin binds to FK-binding protein 12 and therefore only inhibits the mTORC1 complex and not mTORC2. Rapamycin has been approved for the treatment of advanced renal cell carcinoma and, following promising Phase III trials, the rapalogues temsirolimus and everolimus have been approved for renal cell carcinoma and soft tissue/bone sarcomas, respectively (Chiarini et al, 2015). In addition, compounds are in clinical trials that block both mTORC1 and mTORC2 by competitive inhibition at the ATP-binding site (Chiariniet al, 2015).
ATP competitive inhibitors of PI3 kinase have also entered clinical trials (Chiariniet al, 2015). These compounds mainly target the p110aisoform of PI3K, but some have been developed that have selectivity for other isoforms, for example, idelalisib inhibits p110d but notaorb(Lannuttiet al, 2011). Compounds such as dactolisib target PI3K and mTOR at similar potency, and others such as pictilisib are pan-PI3K inhibitors (Brachmannet al, 2009). A review of published early clinical trial data indicates that PI3K inhibitors are well tolerated and as single agents they can induce disease stabilisation, and in some cases regression (Brachmannet al, 2009; Engelman, 2009). Preclinical data suggest that combinations of PI3K inhibitors with other targeted therapies, for example, MEK inhibitors (Engelman et al, 2008; Haagensen et al, 2012; Haagensen et al, 2013), can result in significant synergism and clinical trials to explore such combinations are ongoing.
Cyclin-dependent kinases (CDKs) regulate cell cycle progression and transcription via sequential interaction with specific cyclins. In some cancers defects in the expression of CDKs and their regulatory proteins, for example, loss of CDK inhibitors or upregulation of cyclin expression, leads to aberrant regulation of the cell cycle (Satyanarayana and Kaldis, 2009). CDK2 has an important role in the G1 to S-phase transition and also has a role in S-phase progression. In particular, phosphorylation of Retino-blastoma protein (Rb) by cyclin D/CDK4 and 6 complexes, followed by the cyclin E/CDK2 complex, inactivates Rb releasing E2F and thereby promoting S-phase entry and progression (Harbouret al, 1999; Aleemet al, 2004). Numerous small molecule inhibitors of CDKs have been evaluated in clinical trials (Shapiro, 2006; Asgharet al, 2015), some of which are pan-CDK inhibitors such as flavopiridol, whereas other compounds are more selective. Preliminary clinical data with CDK inhibitors suggests they are generally well tolerated but rarely induce marked anti-proliferative toxicity or tumour regressions when given as single agents (Shapiro, 2006; Asghar et al, 2015). Thus, with the exception of flavopiridol for the treatment of chronic lymphocytic leukaemia (Linet al, 2009), where the transcriptional kinase CDK9 is thought to be the primary target, and palbociclib for the management of ER-positive HER2-negative breast cancer, where CDK4/6 is the
target (Rocca et al, 2014), it has yet to be determined whether CDK-specific or pan-CDK inhibitors are preferable (Shapiro, 2006), or indeed if this compound class generally possesses widespread antitumour activity.
In a counter-screen of compounds designed to inhibit CDK2 we identified a number of molecules that were also mTOR and/or PI3 kinase inhibitors. These compounds provided an opportunity to evaluate the impact of simultaneous modulation of cell cycle and survival/growth signalling, via dual CDK and PI3K/mTOR inhibition, which revealed that potent CDK2 inhibition combined with PI3K inhibition resulted in cell cycle and growth inhibition, and cytotoxicity. In vitroresults with a dual CDK/PI3K inhibitor were recapitulated using a combination of a selective CDK2 inhibitor (NU6102) and a pan-PI3K inhibitor (pictilisib), and extended toin vivostudies that showed enhanced tumour growth inhibition with the drug combination.
MATERIALS AND METHODS
Materials. Dactolisib and pictilisib were purchased from Stratech Scientific (Suffolk, UK) and rapamycin from Merck (Nottingham, UK). Anti phospho-4EBP1 (Cat# 2855S), 4EBP1 (Cat# 9644), AKT (Cat# 4060S), AKT (Cat# 4691S), phospho-ribosomal S6 (Cat# 4858S) and phospho-ribosomal S6 (Cat# 2217S) antibodies were purchased from NEB (Hitchin, UK). Anti phospho-Rb (Cat# 44–582 G) was supplied by Invitrogen (Paisley, UK) and anti-Rb (Cat# 554136) by BD Biosciences (Oxford, UK). All other reagents were analytical grade where available and supplied by Sigma (Gillingham, UK).
Synthesis of compounds. The following compounds were synthe-sised as previously described:1 (Sayleet al, 2003),2(Arris et al, 2000), 3and4(Mesguicheet al, 2003), 5and6(Marchettiet al, 2010). 7 and 8 were synthesised as described in Supplementary Material. NU6102 (4-[[6-(cyclohexylmethoxy)-9H-purin-2-yl]a-mino]-benzenesulfonamide) was synthesised as previously described (Davieset al, 2002).
Enzyme assays. PI3Ks (Condliffe et al, 2005) and CDK2 (Arris et al, 2000) assays were performed as previously described. The inhibition of mTOR kinase activity at 10mM ATP was measured by TR-FRET according to the manufacturer’s instructions (http://tools.invitrogen.com/Content/SFS/lanthascreen/ PV4753%20FRAP1%20Assay%20Validation.pdf).
Cell culture. HCT116 and HT29 cells were obtained from ATCC (Teddington, UK), and grown in DMEM supplemented with 10% (v/v) fetal bovine serum, 100 units ml1 penicillin, 0.1 mg ml1 streptomycin and 2 mML-glutamine.
Cell growth inhibition assays. Cells were trypsinised and plated in 96-well plates at a seeding density of 1000 cells per well. Twenty-four hours later cells were incubated for 72 h with 0.001–100mMof each inhibitor dissolved in DMSO with a final DMSO concentra-tion of 1% (v/v). Cells were fixed with 50% (w/v) TCA and stained with 0.4% (w/v) sulforhodamine B (SRB) for 30 min before washing with 1% (v/v) acetic acid. SRB was solubilised with 10 mM Tris pH 10.5, the absorbance read at 570 nM, and cell growth expressed as % of the growth of cells exposed to 1% (v/v) DMSO. The concentration of compound that resulted in 50% growth inhibition (GI50) was calculated by interpolation from the growth inhibition/concentration curves using GraphPad Prism software (La Jolla, USA). Combination growth inhibition studies of NU6102 and pictilisib were performed as previously described (Haagensen et al, 2012).
concentration from the SRB assay for the pyrimidine compounds
1–8was added to the cells for 8 or 72 h. Cells were washed twice with PBS before the addition of standard growth medium. Following incubation to allow colony (450 cells) formation, cells were fixed with Carnoy’s solution and stained with 0.4% (w/v) crystal violet solution for 1 h. Colonies were washed with 1% (v/v) acetic acid and counted using an Oxford Optronix Colcount (Oxford, UK). Single agent and combination cytotoxicity studies with NU6102 and pictilisib at 1 or 10mMwere performed using the same method with cells being exposed to drugs for 72 h before plating out for colony formation. Statistical analyses were performed using ANOVA.
Flow cytometry. Cells were seeded in six-well plates at a density of 4105per well and allowed to adhere overnight. Incubation of the cells with the GI50concentration of each compound for up to 24 h, was followed by washing with PBS and cell removal with trypsin. Trypsinised cells were centrifuged, washed with PBS and fixed with 75% ethanol/25% PBS (v/v) overnight. Pellets were rehydrated with PBS and stained using 1 mg ml-1 RNase and 400 mg ml1 propidium iodide for 4 h, and analysed using a Becton Dickinson FACSscan and Cellquest software (BD biosciences) with data evaluation using WinMdi software (http://facs.scripps.edu/ software.html) and ANOVA statistical analysis.
Western blotting. Cells were seeded in six-well plates at a density of 4105 per well and allowed to adhere overnight. Cells were incubated with 0.5, 5 or 50mM1or5for 4, 8 or 24 h before being washed and extracted with Phosphosafe buffer (Merck). Protein concentrations were determined by bicinchoninic acid assay (Thermo Scientific, Cramlington, UK) and 30mg of total protein for each sample was loaded into the wells of a 4–20% tris-glycine Novex gel (Invitrogen). Proteins were resolved at 120 V forB2 h before being transferred to Hybond-C nitrocellulose (GE Health-care, Chalfont St Giles, UK). Membranes were probed with the appropriate dilution of antibody overnight and, after washing, the appropriate secondary antibody used at a dilution of 1 : 1000, followed by analysis using Supersignal West Dura enhanced chemiluminescence substrate (Thermo Scientific).
In vivostudies. All of thein vivoexperiments were reviewed and approved by the institutional animal welfare committee, and performed according to the United Kingdom Coordinating Committee on Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in Experimental Neoplasia (second edition) and national law. CD-1 nude mice (Charles River, Ramsgate, Kent, UK) were implanted with 1107 HT29 cells, in a mixture of Matrigel/RPMI cell culture media (1 : 1 v/v), subcutaneously on the right flank. Tumours were allowed to develop until they were palpable (B0.50.5 cm, 10–12 days after implantation) and randomised into four treatment groups (six animals per group).
Mice were treated with vehicle (control), NU6301 120 mg kg1i.p. (a water soluble prodrug of NU6102 (Thomas et al, 2011); 120 mg /kg1NU6301 being equivalent to 100 mg kg1NU6102) twice daily, 100 mg kg1 pictilisib p.o. once daily, or the combination of twice daily NU6301 (120 mg kg1i.p.) and once daily pictilisib (100 mg kg1p.o.) for 10 days. Tumour volume was calculated from two-dimensional calliper measurements using the equationa2b/2, whereais the smallest measurement andbthe largest. Data are presented as median relative tumour volumes (RTV), where the tumour volume on the initial day of treatment (day 0) is assigned an RTV value of 1 in accordance with the formula: RTV¼tumour volume on day of observation/tumour volume on day 0. Statistical analyses of the effects of drug treatment was undertaken using the Mann–Whitney test.
RESULTS
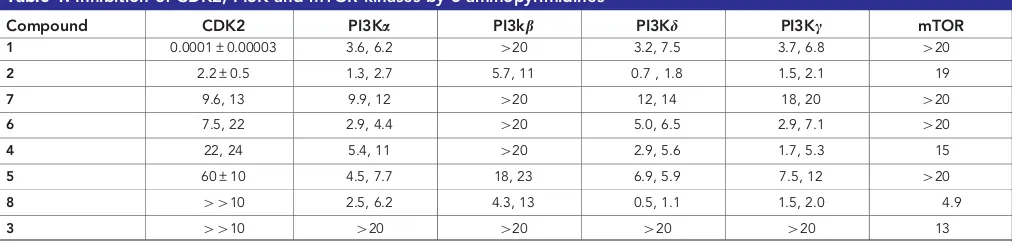
The structures of the compounds tested are shown in Supplementary Figure 1 and their activity as inhibitors of CDK2, the four PI3K isoforms and mTOR in purified kinase assays is shown in Table 1; ranked in order of potency as inhibitors of CDK2. Sulphonamide1is an extremely potent inhibitor of CDK2 (IC500.1 nM) and a low mMinhibitor of all of the PI3K isoforms with the exception of PI3Kb. However,1did not inhibit mTOR at the highest concentration tested (20mM). The parent compound in this series, 2, inhibited CDK2 and all of the PI3K isoforms at similar concentrations (1–10mM), but was somewhat less potent against mTOR. The cyano compound 7 has IC50 values in the range 10–20mMfor CDK2 and the PI3Ka,dandgisozymes, but like 1 is less active against PI3Kb and mTOR. The nitroso compound6is similar to7, although somewhat less active against CDK2 and more active against PI3Ks a,dandg. The aldehyde4
and cyano compound 5are similar to6, although4is distinct in having an IC50for mTOR of 15mM,vso5% inhibition for6and5 at 20mM. Also, 5 is less active than 6 and 4 against CDK2. Importantly, theO4-(4-chlorobenzyl) compound8did not inhibit CDK2 but is an inhibitor of all four PI3K isozymes and mTOR, whereas3is only active against mTOR (IC5013mM).
[image:3.595.45.551.605.726.2]The cell growth inhibitory activity of each compound was tested using the SRB assay in HCT116 and HT29 cells. GI50values were calculated from the mean of three separate assays for each compound, and are given in Table 2. Rapamycin, dactolisib (NVP-BEZ235), pictilisib (GDC-0941; pictrelisib) and NU6102 were used as positive controls for mTOR inhibition, dual PI3K and mTOR inhibition, pan-PI3K inhibition and CDK2 inhibition, respectively. Compounds1,2and5were equipotent in both cell lines, similar in potency to rapamycin, but markedly less potent than the dual PI3K/mTOR inhibitor dactolisib. Inhibitors 4, 7 and8 also had similar GI50values, but were 5–10-fold less potent at inhibiting cell
Table 1.Inhibition of CDK2, PI3K and mTOR kinases by 6-aminopyrimidines
Compound CDK2 PI3Ka PI3kb PI3Kd PI3Kc mTOR
1 0.0001±0.00003 3.6, 6.2 420 3.2, 7.5 3.7, 6.8 420
2 2.2±0.5 1.3, 2.7 5.7, 11 0.7 , 1.8 1.5, 2.1 19
7 9.6, 13 9.9, 12 420 12, 14 18, 20 420
6 7.5, 22 2.9, 4.4 420 5.0, 6.5 2.9, 7.1 420
4 22, 24 5.4, 11 420 2.9, 5.6 1.7, 5.3 15
5 60±10 4.5, 7.7 18, 23 6.9, 5.9 7.5, 12 420
8 4410 2.5, 6.2 4.3, 13 0.5, 1.1 1.5, 2.0 4.9
3 4410 420 420 420 420 13
growth than1,2and5. Growth inhibition induced by3and6did not reach 50% at 100mM, inducing a maximum of 35% reduction in growth at 100mMin HCT116 cells.
The effects of the compounds on cell cycle phase distribution at GI50 concentrations, or at 100mM for 6 and3, in asynchronous cultures were evaluated by flow cytometry in HCT116 cells (Figure 1A). An increase in the G1 population was observed, of varying magnitude, with 2, 4and 5producing greater G1 phase accumulation than rapamycin or dactolisib (NVP-BEZ235). No effect on cell cycle phase distribution was observed with 100mM3. Compound 1was unique in inducing the formation of a sub-G1
fraction, indicative of apoptosis (Figure 1). Further analysis of the effect of 1 on the cell cycle indicated that the sub-G1 fraction became apparent from 3 h onwards in HCT116 cells (Figure 1B). The increase in the sub-G1 fraction was significant after 3 h (P¼0.013) and remained significant up to 8 h (Po0.002). The G1 fraction was reduced after 4 h exposure to 1, but was only significant at the 10% level (Po0.09), whereas the S-phase fraction was significantly reduced at 4 h (Po0.016). The increase in the G2/ M phase fraction was significant at both 4 and 6 h (Po0.011). In HT29 cells, 1 induced a time-dependent increase in the G2/M fraction, with a sub-G1 fraction becoming apparent from 6 h onwards (Figure 1C), which was concentration-dependent at 24 h (Supplementary Figure 2). Although a sub-G1 fraction was apparent at 6 and 8 h, this was not significant (P¼0.183 at 8 h); whereas the G1 fraction was significantly reduced after 4 h (Po0.002). The S-phase fraction in HT29 cells was significantly reduced after 6 h (Po0.04) treatment with 1, and the G2/M fraction was significantly increased after 4 h (Po0.002).
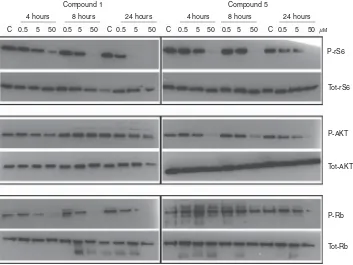
To determine whether the sub-G1 fraction observed following treatment with1reflected cytotoxicity, clonogenic survival assays were performed in HCT116 and HT29 cells following an 8 h exposure. Compound1reduced the clonogenic survival of cells in a concentration-dependent manner in both HCT116 and HT29 cells (Figure 2A and B), with 50% inhibition of colony formation at 5mM. Consistent with the lack of a sub-G1 peak in flow cytometry experiments, the other pyrimidine inhibitors were not cytotoxic in either cell line even after 72 h exposure (HCT116 cells; Figure 2C). To explore the contribution of CDK2 inhibition to the cytotoxicity of 1, the effects of 1 and 5 on target protein phosphorylation were examined in HCT116 (Figure 3) and HT29 (Supplementary Figure 3) cells following 4, 8 and 24 h exposure. Compound 5 was selected as being equipotent to 1
[image:4.595.46.292.217.381.2]against PI3Ks and mTOR, with similar growth inhibitory potency in both cell lines but 600 000-fold less active against CDK2 Table 2.Colorectal tumour cell growth inhibition by
6-aminopyrimidines
Compound GI50HCT116 (lM) GI50HT29 (lM)
1 4.3±2.6 12±3
2 6.2±1.8 12±4
7 73±28 4100mM(30%)
6 4100mM(35%) 4100mM(1%)
4 64±18 44±5
5 6.7±1.8 14±6
8 52±11 59±20
3 4100mM(30%) 4100mM(17%)
Dactolisib (NVP-BEZ235) 0.016±0.008 0.014±0.001
Rapamycin 2.3±1.5 4.2±1.4
NU6102 5.7±0.4 13.3±3.4
Pictilisib (GDC-0941) 1.7±0.3 0.25±0.13
Values are the mean GI50±s.d., or the highest concentration tested and the % growth inhibition observed at that concentration.
150
Sub-G1 G1 S phase G2/M
Sub-G1 G1 S phase G2/M
Sub-G1 G1 S phase G2/M 100
% of cells gated
% of cells gated % of cells gated
50
0
150
B C
A
150 125 100 75 50 25 0 175
100
50
0
Control1 2 3 Control
Hours Hours
4 6 8 1 2 3 4 6 8
1
Rapam ycin
NVP-BEZ235 Control
2 7 6 4 5 8 3
Figure 1. Cell cycle phase analysis following exposure to 6-aminopyrimidine inhibitors measured by flow cytometry.(A) HCT116 cells were
treated with GI50concentrations of inhibitors (or 100mM, Table 2) for 24 h then fixed, stained with propidium iodide and analysed by flow
cytometry. Cell cycle phase distribution was analysed by WinMDI software, and the mean cell cycle fraction of cells gated into each phase of the cell cycle shown. (B) HCT116 cells were treated with the GI50concentration of1for up to 8 h and fixed at time points indicated before being
stained with propidium iodide and analysed by flow cytometry. (C) HT29 cells were treated with the GI50concentration of1for up to 8 h and fixed
[image:4.595.109.487.426.670.2](Tables 1 and 2). Compound1induced concentration-dependent inhibition of Rb phosphorylation in HCT116 cells, with inhibition at a GI50concentration of 5mMbeing observed at all time points, and at 24 h in HT29 cells. In contrast, the effect of 5 on Rb
phosphorylation was less pronounced and only clearly observed after 24 h exposure to 50mM. Compounds 1 and 5 had similar effects on rS6 phosphorylation in both cell lines, and only 5
consistently reduced AKT phosphorylation at 50mM.
120 125
175
150
125
100
75
50
25
0
Con 2 6 4 5 3 7 8
100
75
50
25
0 110
100 90 80 70 60
% sur
viv
a
l
% sur
viv
al
% sur
viv
al
50 40 30 20 10 0
0.0 2.5 5.0 7.5 10.0
Compound 1 (M) Compound 1 (M)
12.5 0.0 2.5 5.0 7.5 10.0 12.5 B
[image:5.595.117.478.50.304.2]C A
Figure 2. Effects of 6-aminopyrimidine inhibitors on the clonogenic survival of HCT116 and HT29 colorectal cancer cells.(A) HCT116 cells were
treated with increasing concentrations of1for 8 h. The medium was replaced with normal growth medium and the cells allowed to form colonies for 1 week. Colonies were stained with 0.4% (v/v) crystal violet and counted. (B) HT29 cells were treated as described in (A). (C) HCT116 cells were treated with the GI50concentration (or 100mM, Table 2) of the 6-aminopyrimidine inhibitors for 72 h before being re-plated for analysis of colony
formation as described in (A). All experiments were performed in triplicate and the data are the mean±s.d.
Compound 1
4 hours
C 0.5 5 50 5 C 0.5 5 50 C 0.5 5 50 0.5 5 50 C 0.5 5 50
P-rS6
Tot-rS6
P-AKT
Tot-AKT
Tot-Rb P-Rb M 50
0.5
8 hours 24 hours 4 hours 8 hours 24 hours Compound 5
Figure 3. Effects of compounds 1 and 5 on the phosphorylation of rS6, AKT and Rb in HCT116 cells.HCT116 cells were treated with1or5for 4,
[image:5.595.122.475.380.645.2]The results obtained with compound1in comparison with the other compounds suggested that dual CDK2 and PI3K inhibition resulted in cytotoxicity, and to confirm this suggestion studies were performed with the potent CDK2 inhibitor NU6102 (4-[[6-(cyclohexylmethoxy)-9H-purin-2-yl]amino]-benzenesulfonamide) (Davies et al, 2002) in combination with the pan-PI3K inhibitor pictilisib. As shown in Figure 4A, the combination of NU6102 and pictilisib was synergistic in growth inhibition studies in both the HCT116 and HT29 cell lines. Furthermore, the combination of the two drugs at 10mM was cytotoxic to HT29 cells, which was significantly enhanced compared with either compound alone at this concentration (Figure 4B; Pp0.0001). However, in the HCT116 cell line, there was only a significant increase in cytotoxicity with the combination compared to pictilisib alone (Pp0.0001), as NU6102 was cytotoxic as a single agent (Figure 4B).
Anin vivotumour growth inhibition study was performed using the HT29 model treated for 10 days with the NU6102 prodrug NU6301 at 120 mg kg1 twice each day (equivalent to 100 mg kg1 NU6102), pictilisib (GDC-0941) at 100 mg kg1 per day or the combination of the two drugs at these doses, which
were based onin vivopharmacodynamic data from earlier studies (Thomaset al, 2011; Haagensenet al, 2013). Compound1was not suitable forin vivostudies as it contains a 5-nitroso group, which is notoriously metabolically labile and potentially reactive. As shown in Figure 4C, RTV in mice treated with the combination of NU6301 and pictilisib (GDC-0941) were significantly lower in comparison with either of the drugs alone (P¼0.008 and P¼0.018, respectively, Mann–Whitney test) at the end of treatment (day 10). There was no tumour growth in the combination group while on treatment, whereas single-agent treatment at the doses used only generated growth delays, which were not significant in the case of NU6301 alone (time to RTV4vs control –P¼0.13, Mann–Whitney test), and not highly significant in the case of pictilisib (GDC-0941; time to RTV4 vscontrol – P¼0.026, Mann–Whitney test). In contrast, growth delay was highly significant for the combination therapy (time to RTV4 vs control – P¼0.0043, Mann–Whitney test). Although a formal toxicology study was not performed, the combination treatment was well tolerated with no consistent impact of single-agent or combination drug treatment on body weights or clinical condition.
100 mg kg
–1 pictilisib po d × 10
120 mg kg
–1 NU6301 ip bid × 10 +
100 m g k
g–1 pictilisib po d × 10
120 m g k
g–1 NU6301 ip bid × 10 Control
P= 0.008
P= 0.08
P= 0.032
P= 0.008
P= 0.75 P= 0.018 6
4
2
0
Relativ
e tumour v
o
lume (Da
y
10)
C
2.6
HT29
80
80 100
60
60 40
% Sur
viv
a
l
Combination inde
x (CI)
% Sur
viv
a
l
40
20 20
0 0
Treatment Treatment
10 M pictilisib
10 M pictilisib
10 M NU6102
10 M NU6102
10 µ M pictilisib /10
M NU6102
10 M pictilisib /10
M NU6102 HCT116
2.4 2.2 2.0 Antagonism
Moderate antagonism
Nearly additive Moderate synergism Synergism Strong synergism Very strong synergism
1.8 1.6 1.4
1.4 1.2
1.2 1.0
1.0 0.8
0.8 0.6
0.6
Fraction affected by dose 0.4
0.4 0.2
0.2 0.0
0.0
[image:6.595.82.519.46.421.2]A B HCT116 HT29
Figure 4. The impact of combined CDK2 and PI3K inhibition on colorectal tumour cell growth and survivalin vitro, and HT29 tumour growth
in vivo.(A) HCT116 or HT29 cells were exposed to combinations of NU6102 and pictilisib at fixed ratios (0.25, 0.5, 1, 2 and 4) of their respective GI50concentrations (NU6102: HCT116 5.7mM, HT29 13mM; pictilisib: HCT116 1.7mM, HT29 0.25mM) and cell growth inhibition evaluated by
median effect analysis. (B) Cytotoxicity of 1mMor 10mMNU6102 and pictilisib alone and in combination in HCT116 and HT29 colorectal tumour cell lines.
(C) Efficacy of the NU6102 prodrug NU6301 and pictilisib, alone and in combination, in HT29 human colorectal tumour xenograft bearing mice. Mice were treated with vehicle (control), NU6301 120 mg kg1i.p. (equivalent to 100 mg kg1NU6102) twice daily, 100 mg kg1pictilisib p.o. once daily, or
DISCUSSION
Inhibitors of mTOR, PI3K and CDK2 are currently in various stages of clinical development. Extensive preclinical studies indicate that combinations of targeted agents may demonstrate greater antitumour activity and selectivity than single agents, the combination of PI3K and MEK inhibitors being an excellent example (Engelmanet al, 2008; Haagensenet al, 2012; Haagensen et al, 2013). Counter-screening of compounds synthesised to target CDK2 identified a number of pyrimidines that were able to inhibit PI3K and/or mTOR, and we evaluated inhibitors with a range of potencies against CDK2, PI3K and mTOR for their effects on cell growth, cell cycle phase distribution and cytotoxicity.
In the two colorectal cancer cell lines studied, HCT116 and HT29, three of the compounds had GI50values of o20mM:1, 2 and 5. The wide range of potencies of these compounds as inhibitors of CDK2 (0.1, 2200 and 60 000 nM, respectively) and the lack of activity against mTOR of two of the compounds (o5% inhibition at 20mM: 1 and 5), yet similar potency for PI3K inhibition, is not inconsistent with growth inhibition owing to activity against the latter targets. The reduced growth inhibitory properties on4,6,7and8, despite similar potency against PI3Ks to
1,2and5, may be due to reduced cellular uptake resulting from the major structural changes at the 4-position (replacement of the cyclohexylmethyl group in 6,7,8) or 5-position (replacement of the nitroso group with formyl in4). In this latter regard, removal of the 5-nitroso group (3) resulted in complete loss of activity against CDK2 and PI3Ks; however, interestingly, modest mTOR inhibition was retained. Nevertheless, despite the mTOR inhibitory activity of3, the compound did not inhibit cell growth. The reason for the lack of CDK2 inhibition by 3 can be explained by the absence of an intra-molecular hydrogen bond as in1(i.e., between the 5-nitroso and the 6-amino group), which is critical for maintaining the correct orientation of the 6-amino group for hydrogen bonding to Glu-81 of CDK2 (Arriset al, 2000). A similar effect may explain the lack of activity of3against PI3Ks, although there are no structural data to support this contention.
When asynchronous cell cultures were treated with GI50 concentrations of the growth inhibitory compounds the predomi-nant effect was an accumulation of cells in the G1 phase of the cell cycle. Both rapamycin and dactolisib (NVP-BEZ235) also caused a G1 arrest at their GI50 concentrations, an effect that has been previously reported with PI3K and/or mTOR inhibitors (Mitaet al, 2003; Serraet al, 2008). In contrast to the other growth inhibitory compounds,1initially either had no effect on the fraction of cells in the G1 phase (HCT116) or reduced the size of the G1 fraction (HT29), before rapidly (p6 h) inducing a sub-G1 population in both cell lines indicative of apoptosis, although only significantly in the HCT116 cells. The impact of the apoptosis induced by1was revealed by clonogenic cytotoxicity studies where the compound was shown to be cytotoxic to both HCT116 and HT29 cells following exposure for 8 h at growth inhibitory concentrations. In marked contrast, exposure of HCT116 cells for 72 h to all of the other pyrimidines failed to induce significant cytotoxicity. Together, these cellular data suggest that the potent CDK2 inhibitory activity of 1contributes to the cytotoxic activity of the compound.
To evaluate whether the relative potencies of compounds against purified kinases reflected their activity in a cellular context, western blotting experiments were performed with1and the non-CDK2 inhibitory control compound, 5. As expected, 1 inhibited the phosphorylation of the CDK2 substrate Rb at earlier time points and at lower concentrations than 5, and these data are consistent with the hypothesis that the apoptosis and cytotoxicity induced by1is related to CDK2 inhibition. Interestingly, despite good inhibition of rS6 phosphorylation, compound 1 did not
inhibit the phosphorylation of AKT, which is likely to reflect phosphorylation at this site by other pathways, for example, IKK has recently been implicated in promoting AKT phosphorylation at Ser473 (Danet al, 2016).
To confirm that the cytotoxicity observed with1was due to dual CDK2 and PI3K inhibition, combination studies were performed with the potent and selective CDK2 inhibitor NU6102 in combination with the pan-PI3K inhibitor pictilisib. In growth inhibition studies, a synergistic as opposed to an additive interaction was observed, particularly in the HCT116 cell line, and in cytotoxicity studies combination of the two inhibitors was markedly more cytotoxic in HT29 cells than either compound alone. Importantly, the superior activity of the combination of the CDK2 and the PI3K inhibitor was recapitulated in an in vivo antitumour study without any marked increase in host toxicity, as evaluated by body weight changes and clinical observations. The specificity of NU6102 as a CDK2 inhibitor has been well-established (Davies et al, 2002) and confirmed subsequently in a study using CDK2 WT and KO MEFs (Thomaset al, 2011). At the concentrations and doses of NU6102 used in the current study (10mMin vitro and 100 mg kg1 in vivo), these previous studies have demonstrated CDK2 specificity (Davies et al, 2002; Thomas et al, 2011). Pictilisib is a very widely used and well-characterised PI3K inhibitor, and was used at doses and concentrations in line with previous published studies (Haagensenet al, 2012; Haagensen et al, 2013). Consequently, these tool compounds can be used to show that dual CDK2 and PI3K inhibition may represent an approach to improving antitumour selectivity with these two classes of targeted agents.
Mechanistically, the potential of dual CDK2 and PI3K pathway inhibition has been highlighted by a number of studies. Initially, Maddika et al, 2008 demonstrated that nuclear translocation of AKT results in phosphorylation of CDK2/cyclin A, leading to cytoplasmic redistribution of CDK2/cyclin A, which was required for S to G2-M cell cycle phase progression. Subsequently, a landmark paper (Liuet al, 2014) reported that phosphorylation of AKT by CDK2/cyclinA2 at S477/T479 was a critical priming event that precedes AKT activation by phosphorylation at the canonical S473 locus, and that by this mechanism CDK2/cyclinA1 activity is a prerequisite for the oncogenic activity of AKT and hence more generally the PI3K pathway. These recent mechanistic insights into the interplay between CDK2 and PI3K signalling provide a rationale for the improved activity of dual CDK2 and PI3K inhibition described in the current paper. Furthermore, the effects of the dual pyrimidine-based inhibitor 1 and combined treatment with the selective CDK2 antagonist NU6102 and the pan-PI3K inhibitor pictilisib described here are consistent with the results using alternative small molecule CDK and PI3K inhibitors, and siRNA (Cheng et al, 2012). In this latter study, increased apoptosis in vitro and enhanced tumour growth delay in vivo were observed when both CDK2 and PI3K, specifically PI3Ka, were modulated by chemical and/or molecular genetic means.
In summary, we have synthesised and characterised a series of 6-aminopyrimidines identified from a kinase screen that are inhibitors of PI3K and/or mTOR and/or CDK2. In HCT116 and HT29 cell lines, PI3K inhibition is associated with cell growth inhibition, regardless of the activity against CDK2. Likewise, the level of mTOR inhibition does not influence cell growth inhibitory potency. Importantly, the mixed CDK2 and PI3K inhibitor 1
ACKNOWLEDGEMENTS
PI3K and mTOR assays were performed at UCB Celltech and we thank UCB Pharmaceuticals scientists Andrew Gill and Tom Crabbe for the development of these assays. GB, HDT, L-Z W, BTG, IRH, DRN, RJG and CC were funded by a CR UK Drug Discovery Programme (C240/A7409), CHR was a PhD Student funded by CR UK (C8120/A8000), SLP was funded by a CR UK Medicinal Chemistry Training Programme (C129/A6963) and EJH was an MRC CASE Award PhD Student supported by UCB Celltech.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
Aleem E, Berthet C, Kaldis P (2004) Cdk2 as a master of S phase entry: fact or fake?Cell Cycle3(1): 35–37.
Arris CE, Boyle FT, Calvert AH, Curtin NJ, Endicott JA, Garman EF, Gibson AE, Golding BT, Grant S, Griffin RJ, Jewsbury P, Johnson LN, Lawrie AM, Newell DR, Noble ME, Sausville EA, Schultz R, Yu W (2000) Identification of novel purine and pyrimidine cyclin-dependent kinase inhibitors with distinct molecular interactions and tumor cell growth inhibition profiles. J Med Chem43(15): 2797–2804.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES (2015) The history and future of targeting cyclin-dependent kinases in cancer therapy.Nat Rev Drug Discov14(2): 130–146.
Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C (2009) PI3K and mTOR inhibitors: a new generation of targeted anticancer agents.Curr Opin Cell Biol21(2): 194–198.
Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D (1997) Frequent inactivation of PTEN/ MMAC1 in primary prostate cancer.Cancer Res57(22): 4997–5000. Cancer Genome Atlas Research Network (2008) Comprehensive genomic
characterization defines human glioblastoma genes and core pathways. Nature455(7216): 1061–1068.
Celebi JT, Shendrik I, Silvers DN, Peacocke M (2000) Identification of PTEN mutations in metastatic melanoma specimens.J Med Genet37(9): 653–657.
Cheng CK, Gustafson WC, Charron E, Houseman BT, Zunder E, Goga A, Gray NS, Pollok B, Oakes SA, James CD, Shokat KM, Weiss WA, Fan QW (2012) Dual blockade of lipid and cyclin-dependent kinases induces synthetic lethality in malignant glioma.Proc Natl Acad Sci USA109(31): 12722–12727.
Chiarini F, Evangelisti C, McCubrey JA, Martelli AM (2015) Current treatment strategies for inhibiting mTOR in cancer.Trends Pharmacol Sci 36(2): 124–135.
Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, Vanhaesebroeck B, Turner M, Webb L, Wymann MP, Hirsch E, Ruckle T, Camps M, Rommel C, Jackson SP, Chilvers ER, Stephens LR, Hawkins PT (2005) Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood106(4): 1432–1440.
Dan HC, Antonia RJ, Baldwin AS (2016) PI3K/Akt promotes feed-forward mTORC2 activation through IKKalpha.Oncotarget7(16): 21064–21075.
Davies TG, Bentley J, Arris CE, Boyle FT, Curtin NJ, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Hardcastle IR, Jewsbury P, Johnson LN, Mesguiche V, Newell DR, Noble ME, Tucker JA, Wang L, Whitfield HJ (2002) Structure-based design of a potent purine-based cyclin-dependent kinase inhibitor.Nat Struct Biol9(10): 745–749.
Engelman JA (2009) Targeting PI3K signalling in cancer: opportunities, challenges and limitations.Nat Rev Cancer9(8): 550–562.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, Garcia-Echeverria C, Weissleder R,
Mahmood U, Cantley LC, Wong K-K (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers.Nat Med14(12): 1351–1356.
Garcia JM, Silva JM, Dominguez G, Gonzalez R, Navarro A, Carretero L, Provencio M, Espana P, Bonilla F (1999) Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype.Breast Cancer Res Treat57(3): 237–243.
Haagensen EJ, Kyle S, Beale GS, Maxwell RJ, Newell DR (2012) The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition.Br J Cancer106(8): 1386–1394.
Haagensen EJ, Thomas HD, Wilson I, Harnor SJ, Payne SL, Rennison T, Smith KM, Maxwell RJ, Newell DR (2013) The enhanced in vivo activity of the combination of a MEK and a PI3K inhibitor correlates with [18F]-FLT PET in human colorectal cancer xenograft tumour-bearing mice. PLoS One8(12): e81763.
Harbour JW, Luo RX, Dei Santi A, Postigo AA, Dean DC (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1.Cell98(6): 859–869.
Harrington LS, Findlay GM, Lamb RF (2005) Restraining PI3K: mTOR signalling goes back to the membrane.Trends Biochem Sci30(1): 35–42. Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ,
Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA (2011) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability.Blood117(2): 591–594.
Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, Andritsos LA, Blum KA, Flynn JM, Jones JA, Hu W, Moran ME, Mitchell SM, Smith LL, Wagner AJ, Raymond CA, Schaaf LJ, Phelps MA, Villalona-Calero MA, Grever MR, Byrd JC (2009) Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease.J Clin Oncol27(35): 6012–6018. Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P,
Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, Yuan M, Wang Z, Gygi SP, Lee TH, Lu KP, Toker A, Pandolfi PP, Asara JM, Kirschner MW, Sicinski P, Cantley L, Wei W (2014) Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus.Nature508(7497): 541–545.
Maddika S, Ande SR, Wiechec E, Hansen LL, Wesselborg S, Los M (2008) Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis.J Cell Sci121(Pt 7): 979–988.
Marchetti F, Cano C, Curtin NJ, Golding BT, Griffin RJ, Haggerty K, Newell DR, Parsons RJ, Payne SL, Wang LZ, Hardcastle IR (2010) Synthesis and biological evaluation of 5-substituted O4-alkylpyrimidines as CDK2 inhibitors.Org Biomol Chem8(10): 2397–2407.
Mayer IA, Arteaga CL (2015) The PI3K/AKT pathway as a target for cancer treatment.Annu Rev Med67: 11–28.
Menon S, Manning BD (2008) Common corruption of the mTOR signaling network in human tumors.Oncogene27(Suppl 2): S43–S51.
Mesguiche V, Parsons RJ, Arris CE, Bentley J, Boyle FT, Curtin NJ, Davies TG, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Jewsbury P, Johnson LN, Newell DR, Noble ME, Wang LZ, Hardcastle IR (2003) 4-Alkoxy-2,6-diaminopyrimidine derivatives: inhibitors of cyclin dependent kinases 1 and 2.Bioorg Med Chem Lett13(2): 217–222.
Mita MM, Mita A, Rowinsky EK (2003) The molecular target of rapamycin (mTOR) as a therapeutic target against cancer.Cancer Biol Ther 2(4 Suppl 1): S169–S177.
Rocca A, Farolfi A, Bravaccini S, Schirone A, Amadori D (2014) Palbociclib (PD 0332991): targeting the cell cycle machinery in breast cancer.Expert Opin Pharmacother15(3): 407–420.
Satyanarayana A, Kaldis P (2009) Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms.Oncogene 28(33): 2925–2939.
Sayle KL, Bentley J, Boyle FT, Calvert AH, Cheng Y, Curtin NJ, Endicott JA, Golding BT, Hardcastle IR, Jewsbury P, Mesguiche V, Newell DR, Noble ME, Parsons RJ, Pratt DJ, Wang LZ, Griffin RJ (2003) Structure-based design of 2-arylamino-4-cyclohexylmethyl-5-nitroso-6-aminopyrimidine inhibitors of cyclin-dependent kinases 1 and 2.Bioorg Med Chem Lett 13(18): 3079–3082.
inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations.Cancer Res68(19): 8022–8030. Shapiro GI (2006) Cyclin-dependent kinase pathways as targets for cancer
treatment.J Clin Oncol24(11): 1770–1783.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT (2008) An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer.Cancer Res68(15): 6084–6091.
Thomas HD, Wang LZ, Roche C, Bentley J, Cheng Y, Hardcastle IR, Golding BT, Griffin RJ, Curtin NJ, Newell DR (2011) Preclinical in vitro and
in vivo evaluation of the potent and specific cyclin-dependent kinase 2 inhibitor NU6102 and a water soluble prodrug NU6301.Eur J Cancer 47(13): 2052–2059.
Yap TA, Bjerke L, Clarke PA, Workman P (2015) Drugging PI3K in cancer: refining targets and therapeutic strategies.Curr Opin Pharmacol 23: 98–107.
This work is published under the standard license to publish agree-ment. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.