Critical Contribution of Tyr15 in the HIV-1
Integrase (IN) in Facilitating IN Assembly
and Nonenzymatic Function through the
IN Precursor Form with Reverse
Transcriptase
Tatsuro Takahata,
aEri Takeda,
bMinoru Tobiume,
cKenzo Tokunaga,
cMasaru Yokoyama,
dYu-Lun Huang,
aAtsuhiko Hasegawa,
aTatsuo Shioda,
bHironori Sato,
dMari Kannagi,
aTakao Masuda
aDepartment of Immunotherapeutics, Graduate School of Medical and Dental Sciences, Tokyo Medical and
Dental University, Tokyo, Japana; Department of Viral Infections, Research Institute for Microbial Diseases,
Osaka University, Suita, Osaka, Japanb; Department of Pathology, National Institute of Infectious Diseases,
Tokyo, Japanc; Laboratory of Viral Genomics, Pathogen Genomics Center, National Institute of Infectious
Diseases, Tokyo, Japand
ABSTRACT
Nonenzymatic roles for HIV-1 integrase (IN) at steps prior to the
enzy-matic integration step have been reported. To obtain structural and functional
in-sights into the nonenzymatic roles of IN, we performed genetic analyses of HIV-1 IN,
focusing on a highly conserved Tyr15 in the N-terminal domain (NTD), which has
previously been shown to regulate an equilibrium state between two NTD dimer
conformations. Replacement of Tyr15 with alanine, histidine, or tryptophan
pre-vented HIV-1 infection and caused severe impairment of reverse transcription
with-out apparent defects in reverse transcriptase (RT) or in capsid disassembly kinetics
after entry into cells. Cross-link analyses of recombinant IN proteins demonstrated
that lethal mutations of Tyr15 severely impaired IN structure for assembly. Notably,
replacement of Tyr15 with phenylalanine was tolerated for all IN functions,
demon-strating that a benzene ring of the aromatic side chain is a key moiety for IN
assem-bly and functions. Additional mutagenic analyses based on previously proposed
te-tramer models for IN assembly suggested a key role of Tyr15 in facilitating the
hydrophobic interaction among IN subunits, together with other proximal residues
within the subunit interface. A rescue experiment with a mutated HIV-1 with RT and
IN deleted (ΔRT ΔIN) and IN and RT supplied in
trans
revealed that the
nonenzy-matic IN function might be exerted through the IN precursor conjugated with RT
(RT-IN). Importantly, the lethal mutations of Tyr15 significantly reduced the RT-IN
function and assembly. Taken together, Tyr15 seems to play a key role in facilitating
the proper assembly of IN and RT on viral RNA through the RT-IN precursor form.
IMPORTANCE
Inhibitors of the IN enzymatic strand transfer function (INSTI) have
been applied in combination antiretroviral therapies to treat HIV-1-infected patients.
Recently, allosteric IN inhibitors (ALLINIs) that interact with HIV-1 IN residues, the
lo-cations of which are distinct from the catalytic sites targeted by INSTI, have been
discovered. Importantly, ALLINIs affect the nonenzymatic role(s) of HIV-1 IN,
provid-ing a rationale for the development of next-generation IN inhibitors with a
mecha-nism that is distinct from that of INSTI. Here, we demonstrate that Tyr15 in the HIV-1
IN NTD plays a critical role during IN assembly by facilitating the hydrophobic
inter-action of the NTD with the other domains of IN. Importantly, we found that the
functional assembly of IN through its fusion form with RT is critical for IN to exert its
nonenzymatic function. Our results provide a novel mechanistic insight into the
non-enzymatic function of HIV-1 IN and its prevention.
Received6 October 2016Accepted7
October 2016
Accepted manuscript posted online19
October 2016
CitationTakahata T, Takeda E, Tobiume M,
Tokunaga K, Yokoyama M, Huang Y-L, Hasegawa A, Shioda T, Sato H, Kannagi M, Masuda T. 2017. Critical contribution of Tyr15 in the HIV-1 integrase (IN) in facilitating IN assembly and nonenzymatic function through the IN precursor form with reverse
transcriptase. J Virol 91:e02003-16. https:// doi.org/10.1128/JVI.02003-16.
EditorKaren L. Beemon, Johns Hopkins
University
Copyright© 2016 American Society for
Microbiology. All Rights Reserved. Address correspondence to Takao Masuda, [email protected].
OF VIRAL GENE EXPRESSION
crossm
on November 7, 2019 by guest
http://jvi.asm.org/
KEYWORDS
HIV-1, integrase, reverse transcriptase, viral ribonucleoprotein complex
I
ntegrase (IN) is a retroviral enzyme, the main catalytic function of which is to
integrate double-stranded viral DNA (vDNA) copies into host chromosomes (1). The
inhibitors that interact with the IN catalytic site and inhibit the IN enzymatic function
of vDNA strand transfer (INSTI) have been developed and approved for use in treating
HIV-1-infected patients (2). Although the potential anti-HIV-1 efficacy of INSTIs has been
demonstrated, INSTI-resistant mutants have emerged in patients treated with these
drugs (3), reinforcing the importance of developing novel inhibitors that act on IN or
other viral constituents through different mechanisms.
In addition to its main enzymatic function, accumulating evidence has suggested
that HIV-1 IN might have additional nonenzymatic roles at steps prior to viral
integra-tion (4–6). Previous studies from our group and others have demonstrated that amino
acid substitutions of the conserved residues within IN result in a lethal effect on HIV-1
that is accompanied with a severe reduction in vDNA synthesis (7–9). Further genetic
analyses of HIV-1 IN have revealed possible roles for IN in uncoating (10), reverse
transcription (11–16), and nuclear import of the viral genome (17–19).
To distinguish them from the mutations that specifically affect the IN catalytic
activity, which are known as class I IN mutations, the mutations that affect other steps
during HIV-1 infection are called class II IN mutations (20). Because the class II IN
mutations are lethal to HIV-1 and have a more profound inhibitory impact than the
class I mutations (7), the nonenzymatic functions of IN might serve as an attractive
target for the development of novel IN inhibitors (4–6). Compounds that interact with
IN and inhibit HIV-1 replication at steps other than integration, known as allosteric IN
inhibitors (ALLINIs), have been reported (21, 22). Subsequent studies have
demon-strated that ALLINIs induce an abnormal virus particle maturation accompanied with an
eccentric condensation of the virus contents and abortive vDNA synthesis (23–25) and
nuclear import of HIV-1 (24). More recently, cryo-electron tomography analysis
ele-gantly demonstrated that ALLINIs impair the proper packaging of the viral
ribonucle-oprotein (vRNP) complex outside the capsid core and the subsequent virus particle
maturation (26). These studies suggest a critical role for IN during vRNP assembly, which
might be essential for initiating early replication events, including reverse transcription,
providing a rationale for the development of a next-generation IN inhibitor that targets
the nonenzymatic role(s) of HIV-1 IN with a mechanism distinct from that of INSTI.
HIV-1 IN consists of three functional domains: the N-terminal domain (NTD), which
has a highly conserved HHCC motif; the catalytic core domain (CCD), which has a DDE
catalytic motif; and the C-terminal domain (CTD), which possesses a nonspecific DNA
binding activity (27). Structure analyses have revealed that each HIV-1 IN domain
(28–30) and the two domains composing the NTD-CCD (31) or the CCD-CTD (32)
assemble into either a dimer or a tetramer form. Recently, the entire structures of other
retroviral INs in a complex with their cognate viral DNA ends, referred to as intasomes,
have been successfully resolved (33–35). These structure analyses of intasomes
re-vealed the unprecedented assembly of INs into a tetramer form for the prototype
foamy virus (33) and octamer forms for the Rous sarcoma virus (34) and mouse
mammary tumor virus (35), providing new insights into the enzymatic role for IN during
retroviral DNA integration and the inhibitory results of INSTI (36).
In contrast to its enzymatic function, the structural basis for the nonenzymatic role
of IN remains largely unknown. Lead ALLINI compounds have been identified by
designing small compounds to inhibit the interaction between IN and the cellular
factor, lens epithelium-derived growth factor (LEDGF)/p75 (37). The ALLINIs interact
with residues within the CCD of HIV-1 IN, which is located at a site distinct from the DDE
catalytic sites (5). Similar to the impact of the ALLINIs, amino acid substitutions
introduced at conserved residues in the NTD (7, 38, 39) and CTD (40) of HIV-1 IN disrupt
the nonenzymatic functions of IN, providing rationales for using the NTD and CTD of
HIV-1 IN as alternative targets to develop a novel class of ALLINI.
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
A nuclear magnetic resonance (NMR) analysis of an isolated fragment consisting of
HIV-1 IN residues 1 to 55 showed that the NTD dimer exists in two conformational
states, E and D forms (30). The two dimer forms differ in the coordination of the zinc
ion by two histidines and two cysteines in the HHCC motif. Our previous NMR study
demonstrated that the NTD fragment with a replacement of the highly conserved
tyrosine at position of 15 (Tyr15) to an alanine (Y15A) folds correctly but takes only the
E form (41). The side chain containing Tyr15 in the D form is exposed to the solvent
more than that in the E form (30), and in the crystal structure of the NTD-CCD, Tyr15 is
located at the dimer-dimer interface to assemble the IN subunits into a tetramer form
(31). A subsequent study revealed that the Y15A mutation abolished IN enzymatic
activities (42). These structural and functional aspects suggest a critical role for Tyr15 in
forming the high-ordered assembly of HIV-1 IN required not only for its enzymatic
functions but also for its nonenzymatic roles. These results promoted us to further
analyze Tyr15 to delineate its contribution to the nonenzymatic roles of HIV-1 IN in the
context of a virus replication cycle.
Here, we performed genetic analyses of HIV-1 IN, focusing on the Tyr15 conserved
in the NTD, by introducing amino acid substitutions with several different residues. Our
results demonstrate that a benzene ring in the side chain containing Tyr15 is a critical
moiety for the nonenzymatic role of HIV-1 IN, providing a structural role in IN assembly
through its hydrophobic interactions with residues in other domains. Additionally, we
found that this critical role for Tyr15 might be exerted through a precursor form in
which IN is conjugated with reverse transcriptase. These findings provide a novel
insight into the nonenzymatic function of HIV-1 IN.
RESULTS
Nonenzymatic role of Tyr15 at the NTD of HIV-1 IN.
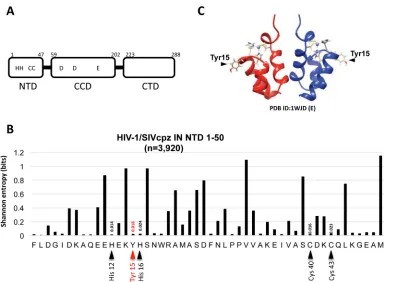
The Tyr15 residue is located
within the NTD of IN (Fig. 1A). Using a total of 3,920 HIV-1/SIVcpz (simian
immunode-ficiency virus of chimpanzees) sequences from the HIV Sequence Database (http://
www.hiv.lanl.gov/content/sequence/HIV/mainpage.html), Shannon entropy scores (43,
44) representing variations at individual amino acid positions from 1 to 50 of the NTD
were calculated. The Shannon entropy score for the Tyr15 residue is 0.018, which is as
low as the scores (0.014 to 0.024) for the HHCC residues (Fig. 1B). This analysis
demonstrated that the Tyr15 is highly conserved among HIV-1 subtypes and related
SIVcpz strains. Similarly, the residue corresponding to the Tyr15 in HIV-2/SIVsmm (SIV
of sooty mangabeys) strains was also highly conserved (Shannon entropy score of 0.14).
In the NTD dimer, the Tyr15 side chain is exposed to the solvent (30) (Fig. 1C), and it
was previously shown to be critical for maintaining an equilibrium state between two
NTD dimer conformations (41). To investigate an NTD functional role through Tyr15, we
generated HIV-1 IN mutant clones by introducing several amino acid substitutions at
Tyr15, and we evaluated the impacts of these substitutions in the context of virus
replication. An HIV-1 mutant lacking IN expression was previously shown to produce
viral particles with abnormal morphology and reduced viral DNA (vDNA) synthesis after
infection (8, 26). We generated a similar HIV-1 mutant lacking IN expression (ΔIN) by
introducing two
ochre
stop codons at the IN initiation site in the
pol
gene. Additionally,
to serve as a class I IN mutation control, the D116G mutant, in which one of the DDE
catalytic sites was mutated to specifically abolish IN enzymatic activity (7), was also
examined in parallel.
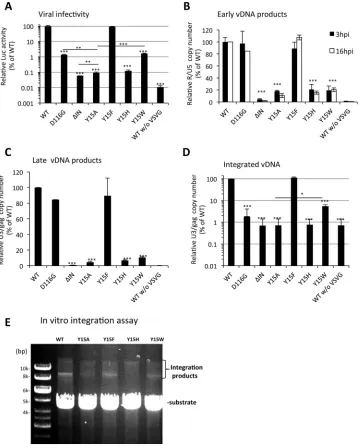
The infectivity of each HIV-1 mutant was determined in single-round infection assays
by monitoring the levels of the luciferase marker gene expression after infection (7).
Replacement of Tyr15 with alanine (Y15A) severely reduced virus infectivity to less than
0.1% of the parental HIV-1 clone (Fig. 2A). Importantly, as observed for the ΔIN
mutation, the Y15A mutation reduced both the early and late vDNA products to 2 to
10% of the parental HIV-1 pseudotype virus (wild-type [WT]) levels (Fig. 2B and C),
demonstrating its major impairment at or prior to reverse transcription. Meanwhile, the
D116G mutant generated both early and late vDNA products at comparable levels to
the WT (Fig. 2B and C) but had a significantly lower level of the integrated form, at less
on November 7, 2019 by guest
http://jvi.asm.org/
than 1% of the WT level (Fig. 2D). The substitutions of Tyr15 with histidine (Y15H) or
tryptophan (Y15W) also impaired HIV-1 infectivity (Fig. 2A) and was accompanied by a
severe reduction in vDNA synthesis (Fig. 2B and C). Notably, the HIV-1 mutant with the
Try15-to-phenylalanine substitution (Y15F) retained virus infectivity (Fig. 2A) with
effi-cient vDNA synthesis and integration (Fig. 2B to E). Interestingly, the Y15W mutant
showed significantly higher infectivity than the Y15A mutant (P
⬍
0.001). In addition,
the Y15W mutant showed significantly higher integration activity than other lethal
mutants (Fig. 2D), suggesting that the Y15W mutant might retain the IN enzymatic
function. To support this notion, an
in vitro
integration assay showed that the Y15A and
Y15H mutations severely abrogated integration activity, while the Y15F and Y15W
mutations retained the enzymatic activity (Fig. 2E). Taken together with the previous
in
vitro
study findings, which clearly demonstrated that the Y15A mutation abolished IN
enzymatic activities
in vitro
(42), our results demonstrate that a benzene ring in the
aromatic side chain, such as Tyr15 and Phe15, is a prerequisite moiety for both the
nonenzymatic and enzymatic functions of HIV-1 IN.
Impact of the Tyr15 mutations on viral particle constituents and morphology.
To delineate the underlying mechanism for the disruption of the nonenzymatic IN
role(s) by Tyr15 mutations, we examined the viral particle constituents produced by
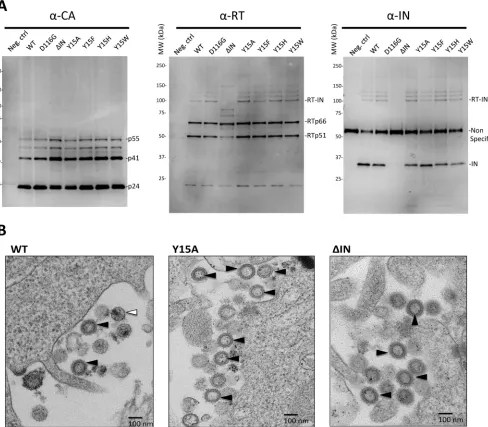
each mutant clone. The results of Western blot analyses of viral particle fractions
demonstrated that all of the Tyr15 mutants, as well as the ΔIN mutant, produced viruses
containing the fully processed capsid protein (p24) with traces of its precursor products,
p41 and p51, all of which were indistinguishable from those produced by the WT and
D116G clones (Fig. 3A, left). None of the Tyr15 mutations affected the level of reverse
FIG 1Tyr15 in the HIV-1 IN NTD. (A) Schematic diagram of HIV-1 IN domains (NTD, CCD, and CTD) are shown with the highly conserved HHCC motif and the catalytic DD35E motif. (B) Shannon entropy scores (43, 44) representing variations at individual amino acid positions were calculated using a total of 3,920 HIV-1/SIVcpz sequences (n ⫽3,916 and 4 sequences for HIV-1 and SIVcpz strains, respectively). The majority consensus amino acid sequences corresponding to the NTD (1 to 50) from HIV-1 are shown. The distribution of entropy scores in each residue spanning the IN NTD region (1 to 50) is shown along with amino acid positions. The Tyr15 is highlighted with a red arrowhead. The His12, His16, Cys40, and Cys43 comprising the HHCC motif are highlighted with a black arrowhead. An entropy score of 0 indicates absolute conservation, whereas a score of 4.4 indicates complete randomness. (C) Dimer structure for the HIV-1 IN NTD fragment [PDB ID 1WJD (E)] (30), which was created using UCSF Chimera. The location of the Tyr15 residue is indicated.
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.41.434.67.349.2]transcriptase (RT) subunits p66 and p51 (Fig. 3A, middle). However, the ΔIN mutant
clone produced a smaller amount of the p51 subunit than the WT clone, suggesting
that the presence of IN in the Pol precursor might be critical for its proper processing
to generate a mature RT heterodimer (p66/p51). The anti-RT antibody also detected a
slower-migrating band with an apparent molecular mass of
⬃
98 kDa (p98), the size of
which is compatible with the expected sum of RT (p66) and IN (p32). Furthermore, p98
FIG 2Effect of HIV-1 IN mutations on viral infection. (A to D) NL43lucΔenv/VSVG, an HIV-1 pseudotype virus possessing WT or D116G, ΔIN, Y15A, Y15F, Y15H, or Y15W IN mutation was prepared. HEK293T cells were infected with each HIV-1 pseudotype corresponding to 100 ng of p24. The cells were harvested and subjected to luciferase assays at 16 h postinfection (A), to qPCR to detect early reverse transcription products at 3 h and 16 h postinfection (B), or to late reverse transcription products at 16 h postinfection (C). To estimate integrated viral DNA, Alu-qPCR (17) was performed at 16 h postinfection (D). The level for each mutant is shown relative to the average value of the parental WT virus, which was set as 100%. All graphs represent the means⫾standard deviations (nⱖ3). WT without VSV-G virus was used for a negative control. The asterisk on the bar indicates thePvalue of each mutant compared with WT based on a two-tailed Student’sttest. ThePvalues between representative mutants are also shown with lines.*,P⬍0.05;**,P⬍0.01;***,P⬍0.001. (E) Anin vitrointegration assay was performed as described previously (61). The mini-HIV DNA substrate (ScaI digested pEGFP-U3U5) was incubated with each recombinant IN (WT, Y15A, Y15F, Y15H, or Y15W) as described in Materials and Methods. Products were resolved on a 0.8% agarose gel and visualized by ethidium bromide staining. The positions of the DNA substrate and its integration products are indicated.on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.44.404.72.520.2](Fig. 3A, RT-IN) was detected by an anti-IN antibody (Fig. 3A, right) and completely
absent in the ΔIN mutant particle, suggesting that p98 might correspond to an
uncleaved precursor protein of RT and IN (RT-IN). Notably, RT-IN was detected in all of
the Tyr15 mutants at the same level as in both WT and D116G. Taken together, none
of the Tyr15 mutations affected the proper processing of the Gag and Pol precursor
proteins within virus particles, at least from a quantitative aspect. We detected similar
levels of vRNA in all of the mutant virus particle fractions containing equivalent
amounts of capsid protein, which were comparable with the level in the WT particle
fraction (data not shown). Thus, none of the IN mutations significantly affected the
vRNA incorporation into virus particles. The transmission electron microscopy (TEM)
analysis revealed that the Y15A and ΔIN mutants predominantly produced immature
virions characterized by doughnut-shaped ring-form particles (Fig. 3B). The mature
particle characterized by a spherical core was hardly detected in either of the IN
FIG 3Impact of IN mutations on viral particle components and morphology. (A) The virus particle fractions from WT and D116G, ΔIN, Y15A, Y15F, Y15H, and Y15W pseudotypes were subjected to Western blot analyses. Gag-CA proteins were detected with a mouse monoclonal anti-p24 antibody (ab9071; Abcam). For the detection of RT and IN proteins, rabbit polyclonal anti-RT (ab63911; Abcam) and rabbit polyclonal anti-IN antibody (AB-INT100; XpressBio) were used, respectively. (B) HIV-1 pseudotype virus possessing WT or Y15A or ΔIN mutations was prepared as described for Fig. 2. At 40 h after transfection of HEK293T cells, the cells were harvested and prepared for TEM analysis. A representative picture for each sample is shown. Bar, 100 nm. An immature particle characterized by doughnut-shaped particles is indicated with the black arrowhead. A mature particle characterized by a spherical core is indicated with the white arrowhead.
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.50.538.74.501.2]mutants. These results suggested that lethal mutation on Tyr15, similar to the ΔIN
mutation, might affect proper virus particle generation. However, due to the low
frequency of the mature particle in our pseudotype preparation of the IN intact control
(Fig. 3B, WT), the impact of IN mutations on matured particle formation can hardly be
evaluated statistically.
Impact of the Tyr15 mutations on the functional aspects of viral constituents.
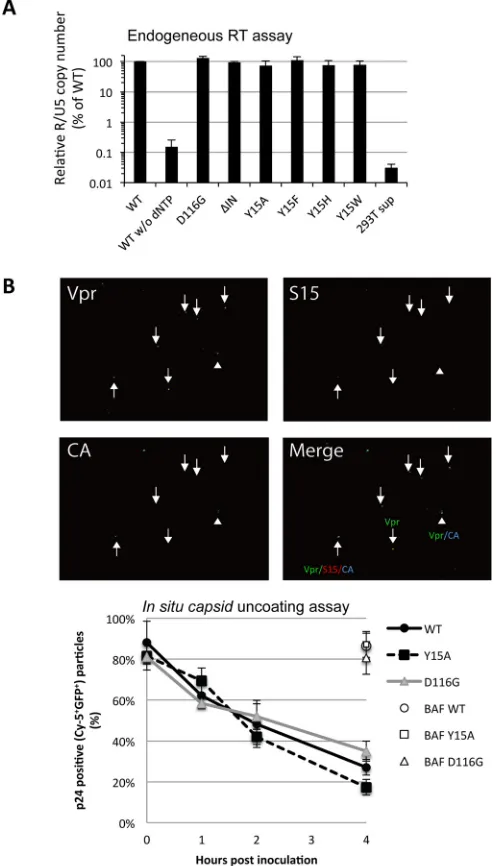
Our assessment of viral constituents failed to detect any apparent defect caused by the
Tyr15 mutations. In addition, all of the IN mutants possessed endogenous RT activities
(7, 45) in the virion-disrupted fractions, comparable with those in the WT (Fig. 4A). Thus,
all of the Tyr15 mutants produced virus particles that possessed the essential
constit-uents, enzymatically active RT and genomic vRNA, to initiate reverse transcription.
Together with our other results, the experiment described above shows that the
impact of Tyr15 on vDNA synthesis is only detected through a cell-based infection
assay. Therefore, we reasoned that the step(s) prior to reverse transcription, such as the
uncoating step (10) through the proper disassembly of the capsid core complex, might
be affected by Tyr15 mutations. We next addressed the fate of the virus capsid protein
(CA) complex shortly after entry into cells by performing
in situ
capsid uncoating assays
(46, 47). Briefly, HeLa cells were spinoculated with fluorescently labeled WT and mutant
viruses containing Vpr fused with green fluorescent protein (GFP-Vpr) and
S15-dTomato for 2 h. At various times after spinoculation, the total number of complexes
that entered and fused into the cytoplasm (Vpr-GFP-positive spots lacking
S15-dTomato) was counted (representative images are shown in Fig. 4B, upper images). The
number of the complexes that contained CA (coated) was compared with the number
of complexes lacking CA staining (uncoated). The data are graphed at each time point
as the percentage of the coated particles in the total fused GFP
⫹particles. Both the
Y15A and D116G mutants showed similar CA disassembly kinetics compared with those
of the WT control (Fig. 4B, lower graph). In our assay, a slight acceleration of capsid
disassembly was noticed for the Y15A mutant at 4 h postinoculation, but this
acceler-ation did not reach statistical significance. Although a possible contribution of IN for
proper uncoating (10) cannot be ruled out, we failed to detect a significant impact of
these IN mutations on the kinetics of HIV-1 CA disassembly, at least under our assay
conditions. The possible contribution of IN to efficient vDNA synthesis
in vivo, therefore,
might be independent from the proper disassembly of the CA complex shortly after
viral entry into cells.
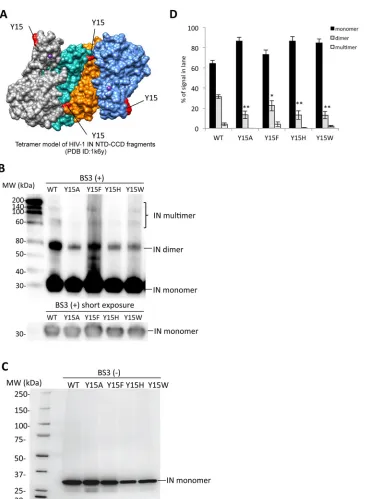
Biochemical properties of the Tyr15 mutant IN.
The assembly of IN subunits into
functional multimers plays a key role for the enzymatic function of IN (6). A crystal
structure analysis of the NTD-CCD of HIV-1 IN led to a proposed model for an IN
tetramer through the association of two IN dimers (31). Based on the tetramer model,
Tyr15 is located in the dimer-dimer interface that is formed through the interaction of
the NTD in one subunit with the CCD in the other subunit (Fig. 5A and 6A). The
intersubunit NTD-CCD interactions are maintained in the final energy-minimized
struc-ture model on the basis of the recent prototype foamy virus (PFV) IN X-ray crystal
structure analyses (36). A previous report clearly demonstrated that the Y15A mutation
abolished the IN enzymatic activities
in vitro
accompanied by severe reductions of IN
dimer and multimer formations (42). We next examined the effect of the Tyr15
mutations on the IN multimer formation. Recombinant proteins of full-length HIV-1 IN
(rIN) containing the Tyr15 mutations were prepared and subjected to cross-linking
analyses. The IN dimer and other high-order forms likely corresponding to IN trimer and
tetramer forms were detected for rIN with the wild-type control (Fig. 5B and C). The
Y15F mutation slightly reduced but retained the multimer formation. In contrast, rINs
carrying the lethal mutations at Tyr15 (Y15A, Y15H, and Y15W) produced much lower
levels of the IN dimer form (Fig. 5B and D). For other high-ordered forms of IN,
reduction was also evident in the Y15A and Y15H mutants (Fig. 5D). These results
suggest that a benzene ring of the aromatic side chain, like Tyr15 or Phe15, is a key
moiety for IN assembly through the functional dimer formation to exert the
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 4Functional aspects of virus particle components. (A) The virus particle fractions of HIV-1 pseu-dotypes (WT, D116G, ΔIN, Y15A, Y15F, Y15H, and Y15W) containing 10 ng of p24 were subjected to endogenous RT assays. The amount of cDNA product was determined by performing qPCR with R1-25/AA55 primer pairs. Values are shown as percentages relative to the average value for the WT. All graphs present the mean results⫾standard deviations (nⱖ3). Reactions using WT virus fraction without dNTP and mock-transfected HEK 293T culture supernatant (293T sup) were used as controls. (B)In situ
uncoating assays were performed as described previously (47). Briefly, HeLa cells were spinoculated with fluorescently labeled WT and mutant viruses containing GFP-Vpr or S15-dTomato for 2 h. At various times after the spinoculation, the total number of complexes that entered and fused into the cytoplasm (Vpr-GFP⫹, S15-dTomato⫺), the number of CA-coated complexes (Vpr-GFP⫹, CA-Cy-5⫹), and the number of CA-uncoated complexes that lost CA staining (Vpr-GFP⫹, CA-Cy-5⫺) were counted. Representative
images of GFP-Vpr (green), S15-Tomato (red), and HIV-1 CA (blue) and a merged image of these three colors taken at the 1-h time point are shown (upper). An upward arrow shows red, green, and blue triple fluorescent colocalization. An arrowhead shows green and blue double fluorescent colocalization. A downward arrow shows green fluorescence only. The data were graphed at each time point as the percentage of cytoplasmic coated particles (Vpr-GFP⫹, CA-Cy-5⫹) among the total number of the fused particles (Vpr-GFP⫹, S15-dTomato⫺). For BafA-treated samples, the percentage of Vpr-GFP⫹and
CA-Cy-5⫹complex among the total number of Vpr-GFP⫹particles at the 4-h time point is shown. Data at each time point represent the mean result⫾the standard deviation (n⫽4).
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.82.330.74.507.2]matic and enzymatic functions. The
in vitro
integration assay showed that the Y15W
mutant retained the enzymatic activity (Fig. 2E), suggesting that the aromatic side chain
of Tyr15 might have a role in maintaining the IN high-ordered structure required for the
enzymatic function.
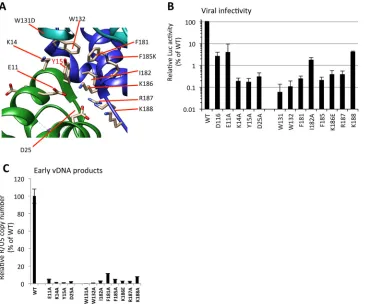
Based on the tetramer models proposed for HIV-1 IN (31, 36, 42), we generated
additional IN mutants by introducing alanine substitutions at the residues located
FIG 5Biochemical properties of the Tyr15 mutant IN. (A) The tetramer structure for the HIV-1 IN NTD-CCD fragment PDB ID (1k6y) (31) was created by using the UCSF Chimera program. The locations of the Tyr15 residues are indicated in red. (B) Twenty nanograms of each recombinant IN protein was cross-linked by BS3, and the resulting products were analyzed by Western blotting with mouse monoclonal anti-IN antibody (ab66645; Abcam). The gel fraction corresponding to the IN monomer with shorter exposure is shown below. (C) A representative blot for each rIN without BS3 is shown as a control. (D) Signal intensities of IN monomer, dimer, and multimer were quantified by using ImageJ software. The relative intensity (as a percentage) of each signal is shown; total signal intensity of each lane was set as 100%. Bars represent the mean results⫾standard deviations. The asterisk indicates thePvalue for each mutant versus WT by two-tailed Student’sttest (*,P⬍0.05;**,P⬍0.01).on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.43.434.64.563.2]around the Tyr15 within the NTD-CTD interface among the IN subunits (Fig. 6A). The
importance of these residues for virus infectivity has been previously reported for Lys14
(48) in the NTD and Trp131 (49, 50), Trp132 (50), Phe185 (51, 52), and Lys186 (11, 53)
in the CCD. Here, in addition to these previously evaluated residues, Glu11, Asp25,
Phe181, Ile182, Arg187, and Lys188 were analyzed. Severe reductions in virus
infectiv-ities (Fig. 6B) and vDNA levels (Fig. 6C) were observed for all of these IN mutants, to
various degrees. Among these IN mutations, W131A and W132A had the most
prom-inent impact on vDNA synthesis after infection. Within the NTD-CTD interface, the
hydrophobic interactions through Trp131/Trp132 might contribute to the assembly of
functional IN subunits. As reported in the previous model of the HIV-1 intasome (36, 42),
the intersubunit NTD-CCD interactions might involve salt bridges of Glu11-Lys186 and
Asp25-Lys188 and hydrogen bonds between the side chains of CCD residues Gln164
and Arg187 with the backbones of NTD residues Lys14 and Tyr15. In addition to these
salt bridges and hydrogen bonds, our results support the importance of Tyr15, along
with the proximal residues within the NTD and CCD, in the nonenzymatic role of HIV-1
IN. Importantly, the intersubunit NTD-CCD interaction proposed for the enzymatic role
of HIV-1 IN (42) may overlap, at least in part, with the structure required for its
nonenzymatic function.
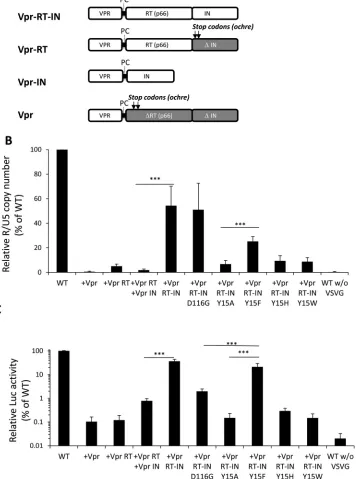
Rescue of HIV-1 IN functions by supplying RT and IN in
trans
.
Retrovirus IN is
originally expressed as a Gag-Pol fusion precursor protein, after which it is processed
into a mature form by viral protease within virus particles (54). We noticed that, in
addition to the fully processed form, a certain level of IN exists in the RT-IN conjugated
precursor form in the virus particle (Fig. 3A). We next addressed the possible
contri-FIG 6Analysis of IN mutants around Tyr15. (A) Alanine substitutions were introduced at critical residues located in the dimer-dimer interface of NTD-CCD subunits around Tyr15. The locations of residues Glu11, Lys14, and Asp25 in the NTD and Trp131, Trp132, Phe181, Ile182, Lys186, Lys187, and Lys188 in the CCD are indicated. (B and C) HEK293T cells were infected with HIV-1 pseudotypes (100 ng of p24). At 16 h postinfection, cells were lysed and subjected to luciferase assays (B), and the total DNA fraction was subjected to qPCR analysis with the R1-25/AA55 primer pair (C). The average WT value was set to 100%. The graph represents the mean results⫾standard deviation (nⱖ2).Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.43.414.71.377.2]bution of each IN form to the nonenzymatic role of IN by conducting a
trans-complementation analysis using RT and IN-defective HIV-1 through the
vpr-mediated
protein delivery system (51, 55). For this experiment, an HIV-1 mutant lacking RT-IN
expression (ΔRT ΔIN) was generated by the combined transfection of three different
expression vectors, each of which supplied the Vpr, Vpr-RT, Vpr-IN, or Vpr-RT-IN
proteins (Fig. 7A). To estimate the rescue efficacy of the HIV-1 ΔRT ΔIN mutant, the
levels of vDNA synthesis (Fig. 7B) and viral gene (luc) expression (Fig. 7C) were
determined after infection. The levels of vDNA and viral gene expression following the
expression of the Vpr-RT-IN were over 10-fold higher than that following the expression
of Vpr-RT and Vpr-IN separately (P
⬍
0.001). Moreover, the vDNA synthesis and viral
gene expression of the ΔRT ΔIN mutant were hardly recovered by the Vpr-RT-IN
carrying a Y15A, Y15H, or Y15W mutation. The rescue of vDNA by the Y15F mutant was
less efficient than with D116G or the WT control. Since the Y15F mutation was tolerable
when evaluated in the HIV-1 infection assay (Fig. 2), in which IN was originally
expressed in a
gag-pol
form, this reduction might be due to an unknown artificial effect
of the Y15F mutation when IN was expressed in fusion form with vpr-RT. Nonetheless,
statistical analysis showed that Vpr-RT-IN-Y15F possesses a significant rescue activity
compared with Vpr-RT-IN-Y15A in both cDNA synthesis and viral gene expression (P
⬍
0.001). In addition, viral gene expression following the expression of Vpr-RT-IN-Y15F
was significantly higher than that following the expression of Vpr-RT-IN-D116G (Fig. 7C)
(P
⬍
0.001). Thus, the Vpr-RT-IN carrying the tolerant mutation of Y15F efficiently
recovered the vDNA synthesis and viral gene expression of the ΔRT ΔIN mutant. Taken
together, these results demonstrate that the nonenzymatic roles of HIV-1 IN might be
exerted through the RT-IN fusion form.
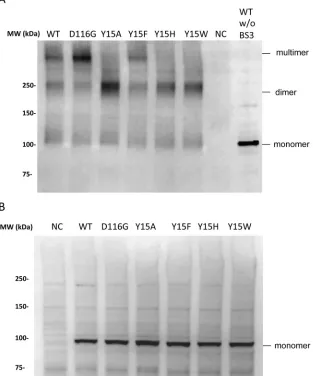
Impact of Tyr15 mutations on the assembly of RT-IN.
Finally, we examined the
impact of the Tyr15 lethal mutations on IN assembly in the RT-IN fusion form. To
address this point, the Vpr-RT-IN fusion protein was expressed in HEK293T cells, and the
cellular fraction was subjected to a cross-link analysis in the presence of synthetic HIV
RNA (56). After cross-linking, we detected slow-migrating bands corresponding to
dimers and higher-order assemblages of Vpr-RT-IN (Fig. 8). The assembly of RT-IN was
reproduced for the Vpr-RT-IN carrying the D116G or Y15F mutation. In contrast, the
Vpr-RT-IN carrying the lethal mutations of Tyr15 (Y15A, Y15H, and Y15W) resulted
predominantly in dimers, with minimal levels of higher-ordered assembly forms. These
results suggest that Tyr15 might be involved in forming the higher-ordered
assem-blages of RT-IN and that RT might be responsible for the dimer formation of RT-IN.
Although the specific contribution of vRNA to form the higher-ordered assembly of
Vpr-RT-IN remains to be determined, the Tyr15 in the HIV-1 IN NTD might play a critical
structural role during the assembly of RT and IN with vRNA through the RT-IN fusion
form.
DISCUSSION
Here, we have provided novel insights into the structural and functional roles of the
Tyr15 in the HIV-1 IN NTD for exertion of the nonenzymatic IN function through the
precursor RT-IN form. Importantly, a benzene ring in the side chain of Tyr15 was shown
to be a key moiety for the correct assembly of IN and RT-IN that might be prerequisite
for the proper course of vRNP complex formation within virus particles and the
subsequent vDNA synthesis after entry into cells.
Structural insight into the nonenzymatic function of IN.
Several mutations at
conserved amino acids within the NTD of HIV-1 IN are lethal to HIV-1 infectivity and
result in virus particle abnormalities and defects in vDNA synthesis (7, 38, 39). In
previous NMR analyses of these lethal mutations, we found that the NTD fragment with
a Y15A mutation folds correctly but only forms a single conformation (41). Here, in the
context of the HIV-1 infection cycle, we showed that the Y15A mutation abrogated virus
infectivity with severe impairment of vDNA synthesis. Thus, a subtle conformational
effect on the NTD by the Y15A mutation had a great impact on HIV-1 infection at the
steps before reverse transcription. Importantly, the side chain containing Tyr15 is
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 7trans-Complementation of HIV-1 IN functions through the Vpr-mediated protein delivery system. (A) Schematic diagrams of Vpr-RT-IN, Vpr-RT, Vpr-IN, and Vpr expressed by pNLTR-Vpr-RT-IN-RRE and its derivative expression vectors are shown. The protease cleavage motif (PC) derived from the HIV-1 protease and RT junction are indicated, and the locations of the two stops codons (ochre) that were introduced into the pNLTR-Vpr-RT-IN-RRE vector to express Vpr-RT or Vpr protein are shown with arrows. The deleted regions for RT (ΔRT) and IN (ΔIN) are shown with gray boxes. (B and C) HIV-1 pseudotype virus lacking RT and IN expression (ΔRT ΔIN) was generated by cotransfection with pNLTR-Vpr-RRE or its derivative expression vectors, each of which supplied the Vpr, Vpr-RT, Vpr-IN, or Vpr-RT-IN protein. HEK293T cells were infected with HIV-1 pseudotype (100 ng of p24). At 16 h postinfection, the cells were harvested and subjected to quantitative PCR or luciferase (luc) assays as described for Fig. 2. The values for the vDNA (B) and luc activity levels (C) of HIV-1 ΔRT ΔIN pseudotypes generated with each of the vpr expression vectors alone or in combination are shown as percentages relative to the level of WT HIV-1 pseudotypes (set as 100%). The graph represents the mean results⫾the standard deviation (nⱖ3). The three asterisks indicate thePvalue for the difference between representative expression constructs based on a two-tailed Student’sttest (P⬍0.001).
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.47.403.83.562.2]exposed to the solvent in an NTD dimer (30) (Fig. 1C). Together with previous
circum-stantial evidence, our current results suggest that the NTD plays a structural role in the
nonenzymatic function of HIV-1 IN that might be exerted through the intersubunit
interaction of the Tyr15 with other domains of IN. Indeed, the results of our
in vitro
cross-linking experiments of full-length rIN proteins demonstrated that the lethal
mutations Y15A, Y15H, and Y15W significantly reduced IN dimer formation, while the
Y15F mutation had less impact. These results suggest that the hydrophobic nature with
a benzene ring in the side chain at the Tyr15 position is necessary for functional IN
multimer conformations to exert the nonenzymatic function of HIV-1 IN.
Based on the tetramer models proposed for the NTD-CCD of HIV-1 IN (31, 36, 42), the
Tyr15 is located at the dimer-dimer interface formed by two subunits of the dimer. In
addition to Tyr15, our genetic analyses revealed that other residues located at the
dimer-dimer interface are also critical for the nonenzymatic function of HIV-1 IN (Fig. 6).
Judging from their hydrophobic nature and expected positions, Trp131, Trp132, and
Phe181 might be critical for forming a hydrophobic interaction with Tyr15, although
FIG 8Cross-link analysis of the RT-IN fusion protein. HEK293T cells were transfected with the pNLTR-Vpr-RT-IN-RRE plasmid to express Vpr-RT-IN fusion proteins carrying WT or the D116G, Y15A, Y15F, Y15H, or Y15W mutation of IN, together with pcDNA-Tat-HA and pRSV-rev. At 48 h posttransfection, the cell lysates were subjected to a cross-link analysis in the presence of 8.3 nM synthetic HIV-1 RNA (56). The resulting products were analyzed by Western blotting with mouse polyclonal anti-RT antibody (ab63911; Abcam). (A) The locations of Vpr-RT-IN in monomer, dimer, and multimer forms are indicated at the right. NC, negative control, consisting of mock-transfected HEK293T cell lysate. (B) The control blot for each product from the reaction in the absence of sRNA and BS3 is shown.on November 7, 2019 by guest
http://jvi.asm.org/
[image:13.585.51.364.79.455.2]the correct positions of the Trp131 and Phe185 in the tetramer model were not clear,
because these were replaced with Lys and Asp to increase the solubility the NTD-CCD
fragment for its crystallization. Meanwhile, previous genetic analyses of HIV-I IN have
shown that there are several critical residues within the CTD (40), mutations of which
result in the class II phenotype. Based on these combined findings, we believe that the
CTD, which is missing in the NTD-CCD model, might contribute to stabilizing the IN
dimer and subsequent higher-ordered assembly.
Implication for vRNP assembly.
vDNAs play a key role in intasome assembly,
which is required for HIV-1 IN to exert its enzymatic function (33–35). In line with this
finding, vRNA might play a critical role in vRNP complex assembly. A recent report
demonstrated that IN is an essential component for forming functional HIV-1 vRNP
complexes (26). In agreement with this idea, our genetic analysis of Tyr15 suggests that
HIV-1 IN plays a critical role in the proper assembly of the vRNP complex, which might
be a prerequisite for reverse transcription
in vivo. The physiological interaction of HIV-1
IN with RT through the IN CTD has been reported (15), and a subsequent study
demonstrated that IN has a direct impact on RT processivity
in vitro
(13). Recently, a
genetic study of HIV-1 IN found that altering the RT-binding surface on the IN CTD
disrupted both reverse transcription and viral replication (16), demonstrating the
biological significance of the HIV-1 RT and IN interaction
in vivo. The possible role of
HIV-1 IN NTD, through interaction with the CTD, therefore might also contribute the
nonenzymatic function of HIV-1 IN. Although the exact structure required for the IN
nonenzymatic function is largely unknown, our
trans-complement experiments
re-vealed that IN might exert a structural role through its precursor form with RT.
Importantly, the uncleaved form of RT-IN was detected in all of the Tyr15 mutants at the
same levels as in the WT, indicating that the Y15A mutation might hinder the
subse-quent steps toward the appropriate vRNP formation through the RT-IN. Within virus
particles, fully processed IN exists as a major form. Previous studies have shown a
significant rescue of particle morphology defects of ΔIN HIV-1 (26), vDNA synthesis and
infectivity of ΔRT ΔIN HIV-1 (51) by supplying IN protein separately from RT. We failed
to clearly show that Vpr-RT-IN fusion rescued the particle maturation in our TEM
analysis (data not shown), probably due to the low frequency of the mature particle in
our pseudotype preparation even in HIV-1 with the RT-IN intact control (Fig. 3B). It is,
therefore, difficult to conclude that RT-IN rescues reverse transcription via fixing particle
morphology under our assay conditions. Nonetheless, our results clearly demonstrated
that the level of vDNA synthesis (Fig. 7C) and infectivity (Fig. 7B) of ΔRT ΔIN HIV-1
following the expression of the Vpr-RT-IN was over 10-fold higher than that following
the expression of Vpr-RT and Vpr-IN separately. During the submission process of our
present report, Kessl et al. reported evidence suggesting the biological role of HIV-1 IN
binding to the viral RNA genome during virion morphogenesis (57). Therefore, it would
be interesting to evaluate the IN binding ability to viral RNA in the context of the RT-IN
precursor form. Meanwhile, we noticed that coexpression of Vpr-IN had a negative
effect on the vDNA synthesis of HIV-1 (data not shown). We reasoned that excess
amounts of the fully processed form of IN might hinder appropriate vRNP complex
formation. As demonstrated in previous reports (55), we also noticed that the rescue
efficacy by the Vpr-RT-IN was significantly higher for the ΔRT ΔIN HIV-1 mutant than
that for the ΔIN HIV-1 lacking only IN expression (data not shown). These data support
the idea that the nonenzymatic function of IN might be exerted through the RT-IN
precursor form.
A previous study showed that certain IN mutations (ΔIN and C130S) accelerated
uncoating (10), in which the CA level within cells was measured at a single time point
of 6 h postinfection. In our assay, we monitored the CA disassembly kinetics at earlier
time points (1 h to 4 h postinfection). A slight acceleration of capsid disassembly was
noticed for the Y15A mutant at 4 h postinfection, although we failed to show its impact
with statistical significance. Our results do not deny the notion that certain IN
muta-tions, including Y15A, affect stability of the capsid. Importantly, neither of these studies
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
directly measured a loss of CA from around the vRNP complex. The impact of IN
mutations on bona fide uncoating, therefore, remains to be determined.
Implication for a novel inhibitor targeting IN functions.
Tyr15 is also involved in
the enzymatic role of HIV-1 IN (42). Although it was difficult to evaluate their impact on
the IN enzymatic roles within cells due to the low levels of vDNA generated by the
Tyr15 mutants, the Y15W mutant had a higher level of
in vivo
integration activity than
the other lethal mutants of Tyr15 (Fig. 2D). We directly addressed the impact of these
mutations on the IN enzymatic function by performing
in vitro
integration assays. The
results of these assays demonstrated that the Y15A and Y15H mutations severely
abrogated integration activity, while the Y15F and Y15W mutations retained this
activity (Fig. 2E). Thus, Tyr15 might have a structural role in both the enzymatic and
nonenzymatic functions of HIV-1 IN, most probably through a benzene ring of the
aromatic side chain. Given the proximity of Tyr15 to His16, one of the
metal-coordinating HHCC residues, it is possible that the defects caused by the substitutions
at Tyr15 might involve effects of these mutations on the helix-turn-helix structure of the
IN NTD. In addition, the NTD-CCD interface formed by IN subunits through Tyr15 and
the surrounding residues could serve as possible targets for development of novel
inhibitors that disrupt the nonenzymatic, as well as the enzymatic, functions of HIV-1 IN.
MATERIALS AND METHODS
Plasmid DNAs.The pNL4-3lucΔenv plasmid, in which theenvgene has been deleted and thenef
gene replaced with a firefly luciferase gene, was used to generate HIV-1 pseudotype virus for single-round infection assays (7). We introduced IN mutations into the fragment spanning from the AgeI site (3487) to the SalI site (5786) of pNL4-3lucΔenv through PCR-based site-directed mutagenesis. The mutant fragments (AgeI to SalI) were replaced with the corresponding region of pNL4-3lucΔenv. To generate pNL4-3lucΔenv that lacks IN expression (pNL4-3lucΔINΔenv), twoochrestop codons were introduced 6 nucleotides downstream of the IN initiation site in thepolregion. To generate pNL4-3lucΔenv that lacks RT and IN expression (pNL4-3lucΔRTΔINΔenv), twoochrestop codons were introduced 6 nucleotides downstream of the RT initiation site in thepolregion of thevpr-defective version of pNL4-3lucΔenv (11). To prepare the bacterial expression vector for His-tagged HIV-1 IN (pET47b-NL43IN), DNA fragments containing the entire IN region were amplified with the primer pair 5=-GGA CCC GGG TTT TTA GAT GGA ATA GAT AAG-3=and 5=-AGC CTC GAG TTA ATC CTC ATC CTG TCT ACT-3=. The resulting PCR fragments were digested with XmaI and XhoI and ligated into a pET47b(⫹) vector (Novagen) using its XmaI and XhoI sites. pNL-Nh is a mutant of the NL4-3 proviral clone (58) in which an NheI restriction enzyme cleavage site was blunted and religated, introducing frameshift mutations in the env gene (59). S15-dTomato is a plasmid expressing the 15 N-terminal membrane-targeting amino acids of p60c-Src fused to red fluorescent protein dTomato (60). GFP-Vpr is a plasmid expressing GFP fused to the Vpr coding region of HIV-1 (46). The pEGFP-U3U5 plasmid carrying HIV-1 U3 and U5 ends was generated for the substrate of thein vitrointegration assay, as previously described (61). The DNA fragment of HIV-1 U3 and U5 ends derived from pNL4-3 was cloned into the EcoRI and KpnI site of pEGFP-1 (Clontech). HIV-1 long terminal repeat (LTR)-driven expression constructs for Vpr fusion forms of RT, IN, and RT-IN (pNLTR-Vpr-RRE and its derivatives) were generated as described previously (51, 55) with some modifi-cations. The Rev responsible element (RRE) containing XhoI or MluI sites at the 5=or 3=end was generated with primer pair 5=-TGC TCG AGC GGA GCG GCC GCA GGA GC-3=and 5=-GTA CGC GTC GCG GAA GCT TGT GTA AT-3=, using pMDLg/pRRE as a template (62). The Vpr fragment containing a BssII or SpeI site at the 5=or 3=end was generated with the primer pair 5=-TTG CGC GCA GCC GCC GCC ATG GAA CAA GCC CCA GAA GAC-3=and 5=-GCA CTA GTC CAG GAT CTA CTG GCT CCA TTT CTT GC-3=, using pNL4-3lucΔenv as a template. The RT-IN fragment containing SpeI or XhoI site at 5=or 3=end was generated with the primer pair of 5=-GAA CTA GTC TGT TGA CTC AGA TTG GCT GCA CT-3=and 5=-AGC CTC GAG TTA ATC CTC ATC CTG TCT ACT-3=using pNL4-3lucΔenv as a template. These three fragments of RRE, Vpr, and RT-IN were ligated in tandem and inserted into the BssHII to MluI fragment of pNL4-3lucΔenv, which contains the entire 5=-LTR and 3=-LTR regions (pNLTR-Vpr-RT-IN-RRE). Similarly, we generated a Vpr-IN fusion protein expression construct (pNLTR-Vpr-IN-RRE) using the primer pairs 5=-TCA CTA GTG GAA TCA GGA AAG TAC TAT TT-3=and 5=-AGC CTC GAG TTA ATC CTC ATC CTG TCT ACT-3=. To generate a Vpr-RT expression vector (pNLTR-Vpr-RT-RRE), the SpeI/XhoI fragment was prepared using the pNL4-3lucΔINΔenv vector, in which the stop codon was introduced at the IN initiation site. Thetat
expression vector of pcDNA-Tat-HA (63) was a kind gift from M. Peterlin. The lentivirus vector of pCSII-CMV-MCS (64),revexpression vector of pRSV-rev, and the vesicular stomatitis virus G protein (VSV-G) expression vector of pMD.G (65) were kind gifts from H. Miyoshi.
Analysis of amino acid variations.An alignment of 3,920 sequences of the full-length open reading frames of HIV-1/SIVcpz IN were obtained from a public database (HIV Sequence Database [http:// www.hiv.lanl.gov/content/sequence/HIV/mainpage.html]). Amino acid variations at each position of the IN NTD (1 to 50) were calculated as previously described (43, 44) on the basis of Shannon’s equation (66):
H共i兲⫽ ⫺兺
xi
p共xi兲log2p共xi兲(xi⫽G, A, I, V, . . .), whereH(i),p(xi), andiindicate the amino acid entropy score
of a given position, the probability of occurrence of a given amino acid at the position, and the number
on November 7, 2019 by guest
http://jvi.asm.org/
of positions, respectively. AnH(i) score of 0 indicates absolute conservation, whereas a score of 4.4 indicates complete randomness.
Preparation and infection of viruses.HEK293T (ATCC CRL-11268) and HeLa (ATCC CCL-2) cells were grown in Dulbecco’s modified Eagle medium containing 10% fetal calf serum, 100 units of penicillin G, and 0.1 mg/ml streptomycin. To prepare HIV-1 pseudotype (HIV-1/VSV-G) viruses, HEK293T cells (1.0⫻ 106) were transfected with 6g of pNL4-3lucΔenv plasmid together with 2g of pMD.G, which is a VSV-G expression vector, by using linear polyethylenimine (polyethylenimine max; Polysciences, Inc., Warrington, PA, USA). At 48 h posttransfection, the culture supernatants were filtered through a 0.45-m filter (Merck, Darmstadt, Germany) and treated with 100 U/ml DNase I (Worthington, Lakewood, NJ, USA) in the presence of 10 mM MgCl2for 1 h at 37°C to remove plasmid DNA contamination. The amount of HIV-1 in each virus preparation was determined by using an enzyme immunoassay (HIV-1 p24 enzyme-linked immunosorbent assay [ELISA] kit; Ryukyu Immunology Corp., Okinawa, Japan) according to the manufacturer’s instructions. Viruses produced in culture supernatants were concentrated by ultracen-trifugation at 4°C for 2 h (Beckman TLA110 rotor at 55,000 rpm, 126,000⫻g) through a TNE buffer–20% sucrose cushion (10 mM Tris-HCl [pH 7.5], 20% sucrose, 100 mM NaCl). The resulting pellets were suspended in phosphate-buffered saline (PBS) and stored at ⫺80°C. For infection experiments, the DNase-treated virus fraction containing 100 ng of p24 was inoculated into HEK293T cells. To monitor virus infectivity, the cells were harvested at 16 h postinfection and subjected to a luciferase assay (Promega).
Quantitative analysis of HIV-1 DNA after infection.To monitor vDNA synthesis after infection, HEK293T cells were harvested at 3 h or 16 h postinfection. After washing the cells with PBS, they were disrupted in urea lysis buffer (3.5 M urea, 1% sodium dodecyl sulfate [SDS], 175 mM NaCl, and 0.5 mM EDTA [pH 8.0]) and subjected to phenol-chloroform extraction and ethanol precipitation. The resulting DNA pellet was resolved in 100l of TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA [pH 8.0]). An aliquot of each DNA sample (100 ng) was subjected to quantitative PCR (qPCR) analysis using the real-time LightCycler detection system (Roche, Mannheim, Germany). To monitor the early virus cDNA products (vDNA), we used the primer pair R1-25/AA55, which is specific for the HIV-1 R-U5 region. To monitor the late cDNA products, U3/M661 specific for the U3-gagwas used, as described previously (56, 67). To monitor the integrated form HIV-1 DNA, sample DNA was subjected to Alu-PCR as described previously (17). Briefly, total DNA extract (100 ng) from virus-infected cells was amplified with the primer pair Alu primer (5=-TCC CAG CTA CTC GGG AGG CTG AGG-3=) and MLan1185rev (5=-GTA ATT TTG GCT GAC CTG GCT-3=) for 18 cycles using GoTaq DNA polymerase (Promega). The PCR products were then diluted 10-fold with TE buffer. The diluted samples were subsequently subjected to real-time qPCR analysis with the R1-25/AA55 primer set.
Endogenous RT assay.We performed endogenous RT (ERT) assays using a previously described method (7, 45) with some modifications. Reaction mixtures contained ERT buffer (50 mM Tris-HCl [pH 8.0], 5 mM MgCl2, 60 mM KCl, 0.5 mM EGTA, 0.01% Triton X-100, 0.5 mM each deoxynucleoside triphosphate [dNTP], 10 mM dithiothreitol [DTT]), and 10 ng of viral p24 in a final volume of 20l. The reaction mixture was incubated at 37°C for 16 h. For a negative control, a reaction mixture without dNTPs was performed in parallel. The reactions were stopped by adding 1l of 100 mM EDTA and boiling at 98°C for 5 min. The amount of the cDNA product was assessed by real-time qPCR with the primer pair R1-25/AA55.
Western blot analysis.Concentrated virions were solubilized in SDS sample buffer (60 mM Tris-HCl [pH 6.8], 2% SDS, 5% 2-mercaptoethanol [2-ME], 10% glycerol, 0.02% bromophenol blue) and then boiled at 98°C for 5 min. Next, samples were subjected to SDS-PAGE using a 5 to 20% gradient polyacrylamide gel, followed by transfer to a polyvinylidene difluoride membrane (0.2-m pore size). Target proteins on the membrane were reacted with specific primary antibodies, mouse monoclonal anti-p24 antibody (ab9071; Abcam), rabbit polyclonal anti-RT (ab63911; Abcam), or rabbit polyclonal anti-IN antibody (AB-INT100; XpressBio). After a subsequent reaction with horseradish peroxidase (HRP)-conjugated secondary antibodies, images of the reactions were obtained by using a chemiluminescence imaging system (Chemilum ImageQuant Las 400 mini; GE Healthcare, Japan).
In situcapsid uncoating assay.Thein situcapsid uncoating assay (46) was conducted as previously described (47). The labeled virus was generated by cotransfecting 9 g of pNL-Nh mutant proviral plasmid, 4g of S15-dTomato, 4g of VSV-G-expressing plasmid, and 1g of GFP-Vpr into 5⫻106of 293T cells by using polyethylenimine (molecular weight, 25,000; Polysciences). At 2 days posttransfec-tion, culture medium was collected and filtered through a 0.45-m filter. Viral titers were determined with the RETROtek antigen ELISA kit (ZeptoMetrix, Buffalo, NY, USA) according to the manufacturer’s instructions. HeLa cells in 24-well plates were spinoculated with the labeled virus for 2 h at 16°C in the presence or absence of 40 nM bafilomycin A (BafA; Sigma). Virus-containing supernatant then was removed and replaced with 37°C medium in the presence or absence of BafA, and the cells were fixed with 3.7% formaldehyde (Polysciences) in 0.1 M PIPES [piperazine-N,N=-bis(2-ethanesulfonic acid); pH 6.8] at each indicated time point postinfection. The fixed HeLa cells were permeabilized with blocking solution (PBS, 10% normal donkey serum [Jackson ImmunoResearch Laboratories], 0.01% Triton X-100, 0.01% NaN3) for 5 min at room temperature, stained with anti-p24 monoclonal antibody (MAb) AG3.0 (NIH AIDS Research and Reference Reagent Program) in blocking solution without Triton X-100 for 1 h at room temperature for primary staining and secondarily stained with Cy-5-labeled donkey anti-mouse antibodies (Jackson ImmunoResearch Laboratories) for 30 min at room temperature. Images were collected and deconvoluted with an Eclipse TE2000-E inverted microscope and NIS-Elements AR soft-ware (Nikon). Following deconvolution, images were blinded for identity to remove bias during counting.
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
The number of GFP-positive cytoplasmic virion particles was assessed at each time point, and each particle was individually inspected for punctate dTomato fluorescent signal and p24 Cy-5 signal.
Preparation of recombinant HIV-1 IN.TheEscherichia coliBL21 derivative strain SoluBL21 (Gen-lantis, San Diego, CA, USA) was transformed with pET47b–NL4-3IN vector. His-tagged IN was expressed in 400 ml of LB culture with 0.3 mM isopropyl--D-thiogalactopyranoside for 4 h at 25°C. For protein purification, each bacterial pellet was treated with 10 ml of BugBuster protein extraction reagent containg benzonase nuclease (Novagen) for 20 min at room temperature. The suspension was sonicated in 80 ml of sonication buffer {50 mM HEPES-NaOH [pH 8.5], 1.2 M NaCl, 10 mM MgCl2, 10M ZnCl2, 10 mM CHAPS, 10% glycerol; CHAPS is 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} in the presence of 1⫻protease inhibiter cocktail (Roche), 14.4 mM 2-ME, and 20 mM imidazole, and then centrifuged at 12,000⫻gfor 30 min at 4°C. The supernatant was incubated with Ni-nitrilotriacetic acid–agarose (Qiagen) at 4°C for 2 h with gentle mixing. After extensively washing the resin with wash buffer (20 mM HEPES-NaOH [pH 8.0], 500 mM NaCl, 5 mM CHAPS, 10% glycerol, 10 mM imidazole, 5 mM DTT), His-tagged IN was eluted with elution buffer (20 mM HEPES-NaOH [pH 8.0], 500 mM NaCl, 350 mM imidazole, 5 mM CHAPS, 10% glycerol, 5 mM DTT). The His tag was removed by treatment with Turbo3C protease (Accelagen) at 4°C for 20 h, and then the sample was subjected to ion exchange column purification (HiTrap SP HP column; GE Healthcare). Eluted proteins were concentrated with Amicon Ultra (molecular weight cutoff [MWCO], 10,000; Millipore) and dialyzed against IN dialysis buffer (20 mM HEPES [pH 7.5], 20% glycerol, 1 M NaCl, 5 mM CHAPS, 5 mM DTT) using a nitrocellulose membrane dialysis cup (MWCO, 12,000; BioTech) for 24 h at 4°C. The resulting product was stored at⫺80°C, and the protein concentration was determined using the Bradford assay (Bio-Rad).
In vitrointegration assay with the miniviral DNA substrate.Anin vitrointegration assay (61) was performed with the reaction mixtures (20 mM HEPES-NaOH [pH 7.5], 15% polyethylene glycol 8000, 500 ng of ScaI-digested pEGFP-U3U5, and 150 ng rIN) incubated at 37°C for 16 h. Each reaction was quenched by addition of SDS to 0.5%. After incubation at 72°C for 10 min, samples were analyzed by 0.8% agarose gel electrophoresis.
Recombinant IN cross-linking.Recombinant IN (20 ng) was suspended in 18l of cross-linking buffer (20 mM HEPES-NaOH [pH 7.0], 150 mM NaCl, 1 mM MgCl2, 1M ZnCl2). The cross-linking reaction was initiated by adding 2l of 2 mM bis-sulfosuccinimidyl suberate (BS3), and the samples were incubated at 25°C for 20 min. The reaction was stopped by the addition of 5⫻SDS sample buffer (250 mM Tris-HCl [pH 6.8], 30% glycerol, 10% sodium dodecyl sulfate, 5% 2-ME, 0.02% bromophenol blue). Samples were analyzed by Western blotting using Nu-PAGE 4 to 12% gradient bis-Tris gels with morpholineethanesulfonic acid (MES)-SDS running buffer (Invitrogen). IN was detected with a mouse monoclonal anti-IN antibody (ab66645; Abcam). After a subsequent reaction with HRP-conjugated secondary antibodies, images of the reactions were obtained by using a chemiluminescence imaging system (Chemilum ImageQuant Las 400 mini; GE Healthcare, Japan). Signal intensity was measured with ImageJ software.
Incorporation of Vpr-fusion protein into virus. HEK293T cells were transfected with pNL4-3lucΔRTΔINΔenv and pMD.G together with 3g of Vpr-fusion protein expression plasmids (pNLTR-Vpr-RRE and its derivatives). At 48 h posttransfection, the culture supernatants were harvested and treated with DNase I as described above.
Expression of Vpr-RT-IN and cross-linking.HEK293T cells were transfected with the pNLTR-Vpr-RT-IN-RRE expression plasmid together with pcDNA-Tat-HA and pRSV-rev. At 48 h posttransfection, the cells were harvested, and the cell lysate was prepared for cross-linking analyses as described previously (68) with some modifications (14). Cells were lysed with cell lysis buffer for cross-linking (20 mM MES-NaOH [pH 7.0], 10% [wt/vol] sucrose, 14.4 mM 2-ME, 1 mM MgCl2, 400 mM NaCl, and 0.5% Nonidet P-40). Synthetic HIV-1 RNA was prepared by using the T7 in vitro transcription system (RiboMAX large-scale RNA production systems; Promega) using the pCSII-CMV-MCS vector as described previously (56). Cell lysates were then incubated with 8.3 nM HIV-1 RNA at 4°C for 20 min, followed by reaction with 2 mM BS3 at room temperature for 20 min or at 4°C for 6 h. The cross-linking reaction was terminated by the addition of 5⫻SDS sample buffer.
TEM.HEK293T cells were transfected with pNL4-3lucΔenv (WT-IN, Y15A, ΔIN, or ΔRT ΔIN) and pMD.G with or without Vpr-fusion protein expression plasmids (pNLTR-Vpr-RRE and its derivatives) as described above. At 40 h after transfection, the cells were harvested with a cell scraper and washed twice with ice-cold PBS. Transfected cells were then prefixed with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4, for 2 h at room temperature, postfixed in 1% osmium tetroxide, and embedded in Epon 812 (TAAB Laboratories). Ultrathin sections were stained with uranyl acetate and lead citrate and then observed under a transmission electron microscope (HT7700; Hitachi) at 80 kV.
ACKNOWLEDGMENTS
We thank I. S. Y. Chen for providing pNL4-3lucΔenv, H. Miyoshi for providing
pCSII-CMV-MCS, pRSV-rev, and pMD, and T. Okamoto and M. Peterlin for providing
pcDNA-Tat-HA. We also thank the Research Institute for Microbial Diseases (BIKEN),
Osaka University, and the National Institute of Infectious Diseases for use of their
experimental facilities and for technical assistance.
We declare that we have no competing interests.
This work was supported by grants for research on HIV/AIDS from the Japan Agency
on November 7, 2019 by guest
http://jvi.asm.org/
for Medical Research and Development (AMED) and by the Japan Society for the
Promotion of Science (JSPS; KAKENHI 15K15139).
T.M. conceived the study. T.T., E.T., M.T., K.T., M.Y., Y.H., A.H., T.S., H.S., M.K., and T.M.
performed the experiments and/or participated in the experimental design. T.T. and
T.M. wrote the manuscript. E.T., M.Y., H.S., A.H., T.S., and M.K. edited the manuscript.
REFERENCES
1. Brown PO. 1997. Integration, p 131–203. In Coffin JM, Hughes SH, Varmus HE (ed), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
2. Hazuda DJ. 2012. HIV integrase as a target for antiretroviral therapy. Curr O p i n H I V A I D S 7 : 3 8 3 – 3 8 9 . h t t p s : / / d o i . o r g / 1 0 . 1 0 9 7 / C O H .0b013e3283567309.
3. Grobler JA, Hazuda DJ. 2014. Resistance to HIV integrase strand transfer inhibitors: in vitro findings and clinical consequences. Curr Opin Virol 8:98 –103. https://doi.org/10.1016/j.coviro.2014.07.006.
4. Masuda T. 2011. Non-enzymatic functions of retroviral integrase: the next target for novel anti-HIV drug development. Front Microbiol 2:210. https://doi.org/10.3389/fmicb.2011.00210.
5. Engelman A, Kessl JJ, Kvaratskhelia M. 2013. Allosteric inhibition of HIV-1 integrase activity. Curr Opin Chem Biol 17:339 –345. https://doi.org/ 10.1016/j.cbpa.2013.04.010.
6. Feng L, Larue RC, Slaughter A, Kessl JJ, Kvaratskhelia M. 2015. HIV-1 integrase multimerization as a therapeutic target. Curr Top Microbiol Immunol 389:93–119. https://doi.org/10.1007/82_2015_439.
7. Masuda T, Planelles V, Krogstad P, Chen IS. 1995. Genetic analysis of human immunodeficiency virus type 1 integrase and the U3 att site: unusual phenotype of mutants in the zinc finger-like domain. J Virol 69:6687– 6696.
8. Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R. 1995. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J Virol 69:2729 –2736.
9. Leavitt AD, Robles G, Alesandro N, Varmus HE. 1996. Human immuno-deficiency virus type 1 integrase mutants retain in vitro integrase activity yet fail to integrate viral DNA efficiently during infection. J Virol 70: 721–728.
10. Briones MS, Dobard CW, Chow SA. 2010. Role of human immunodefi-ciency virus type 1 integrase in uncoating of the viral core. J Virol 84:5181–5190. https://doi.org/10.1128/JVI.02382-09.
11. Tsurutani N, Kubo M, Maeda Y, Ohashi T, Yamamoto N, Kannagi M, Masuda T. 2000. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells. J Virol 74:4795– 4806. https://doi.org/10.1128/JVI.74.10.4795-4806.2000. 12. Zhu K, Dobard C, Chow SA. 2004. Requirement for integrase during reverse transcription of human immunodeficiency virus type 1 and the effect of cysteine mutations of integrase on its interactions with reverse transcriptase. J Virol 78:5045–5055. https://doi.org/10.1128/JVI .78.10.5045-5055.2004.
13. Dobard CW, Briones MS, Chow SA. 2007. Molecular mechanisms by which human immunodeficiency virus type 1 integrase stimulates the early steps of reverse transcription. J Virol 81:10037–10046. https:// doi.org/10.1128/JVI.00519-07.
14. Nishitsuji H, Hayashi T, Takahashi T, Miyano M, Kannagi M, Masuda T. 2009. Augmentation of reverse transcription by integrase through an interaction with host factor, SIP1/Gemin2 is critical for HIV-1 infection. PLoS One 4:e7825. https://doi.org/10.1371/journal.pone.0007825. 15. Wilkinson TA, Januszyk K, Phillips ML, Tekeste SS, Zhang M, Miller JT, Le
Grice SF, Clubb RT, Chow SA. 2009. Identifying and characterizing a functional HIV-1 reverse transcriptase-binding site on integrase. J Biol Chem 284:7931–7939. https://doi.org/10.1074/jbc.M806241200. 16. Tekeste SS, Wilkinson TA, Weiner EM, Xu X, Miller JT, Le Grice SF, Clubb
RT, Chow SA. 2015. Interaction between reverse transcriptase and inte-grase is required for reverse transcription during HIV-1 replication. J Virol 89:12058 –12069. https://doi.org/10.1128/JVI.01471-15.
17. Ikeda T, Nishitsuji H, Zhou X, Nara N, Ohashi T, Kannagi M, Masuda T. 2004. Evaluation of the functional involvement of human immunodefi-ciency virus type 1 integrase in nuclear import of viral cDNA during acute infection. J Virol 78:11563–11573. https://doi.org/10.1128/JVI .78.21.11563-11573.2004.
18. Ao Z, Huang G, Yao H, Xu Z, Labine M, Cochrane AW, Yao X. 2007. Interaction of human immunodeficiency virus type 1 integrase with cellular nuclear import receptor importin 7 and its impact on viral replication. J Biol Chem 282:13456 –13467. https://doi.org/10.1074/jbc .M610546200.
19. Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, Debyser Z. 2008. Transportin-SR2 imports HIV into the nucleus. Curr Biol 18:1192–1202. https://doi.org/ 10.1016/j.cub.2008.07.079.
20. Engelman A. 1999. In vivo analysis of retroviral integrase structure and function. Adv Virus Res 52:411– 426. https://doi.org/10.1016/S0065 -3527(08)60309-7.
21. Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, Van der Veken NJ, Van Remoortel B, Strelkov SV, De Maeyer M, Chaltin P, Debyser Z. 2010. Rational design of small-molecule inhib-itors of the LEDGF/p75-integrase interaction and HIV replication. Nat Chem Biol 6:442– 448. https://doi.org/10.1038/nchembio.370. 22. Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S,
Smets W, Chaltin P, Westby M, Debyser Z, Pickford C. 2012. Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization. Antimicrob Agents Chemother 56:4365– 4374. https://doi.org/10.1128/AAC.00717-12. 23. Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W,
Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A. 2013. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc Natl Acad Sci U S A 110: 8690 – 8695. https://doi.org/10.1073/pnas.1300703110.
24. Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Van Remoortel B, Hofkens J, De Rijck J, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z. 2013. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 10:57. https://doi.org/10.1186/1742-4690-10-57. 25. Balakrishnan M, Yant SR, Tsai L, O’Sullivan C, Bam RA, Tsai A,
Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T. 2013. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse tran-scription block in target cells. PLoS One 8:e74163. https://doi.org/ 10.1371/journal.pone.0074163.
26. Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, Steven AC. 2015. Distribution and redistribution of HIV-1 nucleo-capsid protein in immature, mature, and integrase-inhibited virions: a role for integrase in maturation. J Virol 89:9765–9780. https://doi.org/ 10.1128/JVI.01522-15.
27. Li X, Krishnan L, Cherepanov P, Engelman A. 2011. Structural biology of retroviral DNA integration. Virology 411:194 –205. https://doi.org/ 10.1016/j.virol.2010.12.008.
28. Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR. 1994. Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266:1981–1986. https:// doi.org/10.1126/science.7801124.
29. Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hard K. 1995. The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat Struct Biol 2:807– 810. https://doi.org/10.1038/nsb0995-807. 30. Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM. 1997.
Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat Struct Biol 4:567–577. https://doi.org/10.1038/nsb0797-567.
31. Wang JY, Ling H, Yang W, Craigie R. 2001. Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein. EMBO J 20:7333–7343. https://doi.org/10.1093/emboj/ 20.24.7333.
32. Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM. 2000. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci U S A 97:8233– 8238. https://doi.org/10.1073/pnas.150220297.
Takahata et al. Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
33. Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464: 232–236. https://doi.org/10.1038/nature08784.
34. Yin Z, Shi K, Banerjee S, Pandey KK, Bera S, Grandgenett DP, Aihara H. 2016. Crystal structure of the Rous sarcoma virus intasome. Nature 530:362–366. https://doi.org/10.1038/nature16950.
35. Ballandras-Colas A, Br