0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
High-Level Hepatitis B Virus Replication in Transgenic Mice†
LUCA G. GUIDOTTI,
1,2BRENT MATZKE,
1HEINZ SCHALLER,
3AND
FRANCIS V. CHISARI
1*
Department of Molecular and Experimental Medicine, Scripps Research Institute, La Jolla, California 92037
1;
Istituto di Patologia Generale, Universita’ degli studi di Parma, 43100 Parma, Italy
2; and Zentrum fur
Molekulare Biologie, Universitat Heidelberg, Heidelberg 69120, Germany
3Received 23 May 1995/Accepted 30 June 1995
Hepatitis B virus (HBV) transgenic mice whose hepatocytes replicate the virus at levels comparable to that
in the infected livers of patients with chronic hepatitis have been produced, without any evidence of
cytopa-thology. High-level viral gene expression was obtained in the liver and kidney tissues in three independent
lineages. These animals were produced with a terminally redundant viral DNA construct (HBV 1.3) that starts
just upstream of HBV enhancer I, extends completely around the circular viral genome, and ends just
downstream of the unique polyadenylation site in HBV. In these animals, the viral mRNA is more abundant
in centrilobular hepatocytes than elsewhere in the hepatic lobule. High-level viral DNA replication occurs
inside viral nucleocapsid particles that preferentially form in the cytoplasm of these centrilobular hepatocytes,
suggesting that an expression threshold must be reached for nucleocapsid assembly and viral replication to
occur. Despite the restricted distribution of the viral replication machinery in centrilobular cytoplasmic
nucleocapsids, nucleocapsid particles are detectable in the vast majority of hepatocyte nuclei throughout the
hepatic lobule. The intranuclear nucleocapsid particles are empty, however, suggesting that viral nucleocapsid
particle assembly occurs independently in the nucleus and the cytoplasm of the hepatocyte and implying that
cytoplasmic nucleocapsid particles do not transport the viral genome across the nuclear membrane into the
nucleus during the viral life cycle. This model creates the opportunity to examine the influence of viral and host
factors on HBV pathogenesis and replication and to assess the antiviral potential of pharmacological agents
and physiological processes, including the immune response.
The hepatitis B virus (HBV) contains a small (3.2-kb),
cir-cular, double-stranded DNA genome and causes acute and
chronic hepatitis, cirrhosis, and hepatocellular carcinoma (10).
Because of the limited host range of HBV and the inability to
grow it in vitro, much of what is known about the HBV life
cycle has come from studies of other members of the
hepad-navirus family, especially the woodchuck hepatitis virus and the
duck hepatitis B virus, which infect their native species and, in
the case of the duck hepatitis B virus, can be grown in tissue
culture (34).
On the basis of circumstantial evidence, it is generally
ac-cepted that HBV is not directly cytopathic for the hepatocyte
and that the antiviral immune response is responsible for the
associated liver disease during HBV infection (5). This concept
has not been directly testable until recently, however, because
the immunobiology of woodchuck hepatitis virus and that of
duck hepatitis B virus are not well defined because of the
outbred nature of their natural hosts. We and others,
there-fore, set out several years ago to develop HBV transgenic mice
to permit these, and other, complex biological questions to be
addressed in a well-defined, inbred, small-animal system.
As a result of those studies, many transgenic lineages that
express the HBV envelope (2, 3, 6, 7, 11), core (13, 25), precore
(26) and X (20, 22) proteins under the control of HBV or
cellular liver-specific promoters have been described in recent
years. Much has been learned about the assembly, transport,
secretion, and other functional properties of these proteins and
about the induction, the characteristics, and the consequences
of the corresponding immune responses in those animals. Such
animals are not suitable, however, for studies of viral
replica-tion, since they express only a single gene product and cannot
replicate the virus.
To address this important question, other investigators
pro-duced transgenic mice using constructs structurally capable, as
linear molecules, of transcribing all of the viral gene products
simultaneously (1, 9). While these animals clearly
demon-strated that HBV can replicate in the murine hepatocyte, their
further experimental value has been limited because they
rep-licate the virus at very low levels (1, 9) and because in one
instance (9) the transgene was not transmitted through the
germ line.
Over the past several years, we have produced transgenic
mice with a series of terminally redundant,
greater-than-ge-nome length constructs with the hope of identifying a DNA
structure that would drive HBV expression and replication at
levels comparable to that in naturally infected liver. All
con-structs shared a common 3
9
terminus at nucleotide (nt) 1982,
just downstream of the unique polyadenylation site in the HBV
genome, and they were made to differ in overall length by
positioning their 5
9
termini either just upstream of the
nucleo-capsid promoter and enhancer II (1.1 genome eq), in the
middle of the X gene (1.2 genome eq), or just upstream of the
X promoter and enhancer I (1.3 genome eq).
We now report that very-high-level HBV replication has
been achieved in the liver and kidney tissues of three
indepen-dent lineages containing the 1.3 genome eq construct while
only low-level, kidney-preferential replication occurred with
the shorter constructs. This article describes the salient
char-acteristics of this model. The results illustrate that high-level
HBV replication is not cytopathic for the murine hepatocyte,
and they highlight several important aspects of the HBV life
cycle that are not widely appreciated. Perhaps most
impor-tantly, this model creates the opportunity to examine many
* Corresponding author. Mailing address: Department of Molecular and Experimental Medicine, Scripps Research Institute, 10666 N. Tor-rey Pines Rd., La Jolla, CA 92037. Phone: (619) 554-8228. Fax: (619) 554-6134.
† Scripps Research Institute manuscript 9401-MEM.
6158
on November 9, 2019 by guest
http://jvi.asm.org/
aspects of HBV immunobiology and pathogenesis that have
not been heretofore analyzable.
MATERIALS AND METHODS
Transgenic mice.The HBV transgenic mice used in these studies were pro-duced by microinjection of the HindIII-SacI fragment excised from plasmids pT-HBV1.1, pT-HBV1.2, and pT-HBV1.3 (Fig. 1) into either (C57BL/6 3
SJL)F2embryos or inbred B10.D2 embryos by conventional technology as pre-viously described (2). Plasmids pT-HBV1.1, pT-HBV1.2, and pT-HBV1.3 con-tain overlength HBV genomes, differing 59terminally by the size of the redun-dancy but all terminating at a unique SacI site introduced downstream of the polyadenylation signal (17). Each HBV genome was preceded by a unique HindIII cleavage site introduced at position 1640 in pT-HBV1.1, position 1393 in pT-HBV1.2, and position 1068 in pT-HBV1.3. Founder animals were screened by analysis of serum for hepatitis B surface antigen (HBsAg) and HBeAg using commercially available reagents (Abbott Laboratories, Abbott Park, Ill.) as pre-viously described (6), and animals that were positive for both antigens were bred for analysis. Lineages 1.1.42 (official designation, Tg[HBV 1.1 genome]Chi42), 1.1.223 (Tg[HBV 1.1 genome]Chi223), 1.1.370, (Tg[HBV 1.1 genome]Chi370), 1.1.390 (Tg[HBV 1.1 genome]Chi390), 1.1.399 (Tg[HBV 1.1 genome]Chi399), 1.2.23 (Tg[HBV 1.2 genome]Chi23), 1.3.2 (Tg[HBV 1.3 genome]Chi2), 1.3.32 (Tg[HBV 1.3 genome]Chi32), and 1.3.46 (Tg[HBV 1.3 genome]Chi46) were expanded by repetitive backcrossing against either the C57BL/6 or the B10.D2 parental strain according to the genetic background of the founder.
In selected experiments, liver tissue derived from transgenic-mouse lineages 1.2HBBS (1) and PC21 (9), generously provided by Kenichi Yamamura (Insti-tute for Medical Genetics, Kumamoto University Medical School, Kumamoto,
Japan) and by Christine Pourcel (Institut de Biologie, Nantes, France), respec-tively, were also studied. Lineage 1.2HBBS was produced by microinjection of a terminally redundant, greater-than-genome length fragment representing 1.19 copies of the HBV genome that extends between nt 1272 and 1891 (in which the unique XhoI site is designated nt 1) of HBV (adr4 subtype) (1). Lineage PC21 contains a HindIII-PstI fragment representing a complete dimer of the HBV genome (ayw subtype) in which the entire HBV genome was cloned in a head-to-tail arrangement at the EcoRI site (nt 1 in Galibert sequence terminology) of pBR322 (9).
Serum from transgenic lineage pFC80-219 (11) (official designation, Tg[HBs, HBV]Chi 219) was also used for comparative purposes in selected assays. This lineage expresses only the 2.1-kb HBV mRNA and secretes HBsAg at a high level in the serum (11).
In all experiments, mice were anesthetized with Metophane (Pitman-Moore, Mundelein, Ill.) prior to phlebotomy from the retro-orbital plexus and before they were sacrificed by cervical dislocation.
Human liver tissue.Autopsy-derived liver tissue from six HBV-infected pa-tients with chronic active hepatitis and cirrhosis were provided by John Brems (Scripps Clinic and Research Foundation), Michael Gerber (Tulane University Medical Center), and David Shafritz (Albert Einstein College of Medicine). All tissues were stored at2708C until use.
HBV DNA analysis. (i) DNA isolation and southern blot analysis.Southern blot analysis was performed on total genomic DNA by agarose gel electrophore-sis of 20mg of restricted genomic DNA as previously described (23). Before electrophoresis, all DNA samples were digested with RNase A (Boehringer Mannheim, Indianapolis, Ind.) at 10mg/ml for 1 h at 378C. Nylon filters were hybridized either with HBV-specific32P-radiolabeled DNA probes as previously described (12) or with33P-radiolabeled strand-specific RNA probes that were also used for in situ hybridization analysis (described below).
(ii) PCR analysis of the integrated transgene.Total liver genomic DNA (1mg) from lineages 1.3.32 and 1.3.46 was analyzed by PCR using HBV-specific primers (59-CTAAGCAGGCTTTCACTTTC [sense, nt 1077 to 1096] and 59-CAGA AAGGCCTTGTAAGTTG [antisense, nt 1120 to 1101]). Twenty-microliter samples of the products from direct PCR amplifications were analyzed by elec-trophoresis on a 1% agarose gel in the presence of 0.5mg of ethidium bromide per ml. DNA bands were visualized by UV fluorescence.
(iii) Subcellular localization of replicative forms of HBV DNA.Viral DNA contained in the nucleus was analyzed as follows. Nuclei from liver samples were purified exactly as described previously (15). Briefly, liver samples were homog-enized by 10 to 20 strokes in a Potter-Elvehjem tissue grinder in 5 ml of buffer A (60 mM KCl, 15 mM NaCl, 0.15 mM spermine, 0.5 mM spermidine, 14 mM 2-mercaptoethanol, 0.5 mM EGTA [ethylene glycol-bis(b-aminoethyl ether)-N,N,N9,N9-tetraacetic acid], 2 mM EDTA, 0.3 M sucrose, 15 mM Tris Cl [pH 7.5]), and centrifuged over a 5-ml cushion of buffer B (60 mM KCl, 15 mM NaCl, 0.15 mM spermine, 0.5 mM spermidine, 14 mM 2-mercaptoethanol, 0.5 mM EGTA, 2 mM EDTA, 0.85 M sucrose, 15 mM Tris Cl [pH 7.5]) at 100,0003g for 20 min at 48C. The pellet was resuspended in 3 ml of buffer C (60 mM KCl, 15 mM NaCl, 0.15 mM spermine, 0.5 mM spermidine, 14 mM 2-mercaptoetha-nol, 0.1 mM EGTA, 0.1 mM EDTA, 2 M sucrose, 15 mM Tris Cl [pH 7.5]) and centrifuged over an 8-ml cushion of buffer C in a Beckman SW40 rotor at 36,000 rpm for 1 h at 48C. Nuclei were counted and resuspended in 1 ml of Tris-EDTA buffer (10 mM Tris-acetate [pH 8], 10 mM EDTA), and Nonidet P-40 (Sigma, St. Louis, Mo.) (final concentration, 0.5%) was added for 30 min on ice. After magnesium acetate (final concentration, 5 mM) was added, the nuclei were digested with RNase A (Boehringer Mannheim) at 10mg/ml and with 20 U of DNase I (Boehringer Mannheim) per ml for 1 h at 378C. Subsequently, EDTA (final concentration, 10 mM), sodium dodecyl sulfate (SDS) (final concentration, 1%), NaCl (final concentration, 0.1 M), and pronase E (Sigma) (final concen-tration, 0.5 mg/ml) were added sequentially, and the samples were incubated for 3 to 4 h at 428C. The samples were then extracted with phenol-chloroform, ethanol precipitated and quantitated, and DNA extracted from 83106nuclei was loaded into the gel and analyzed by Southern blot (23).
DNA contained in the cytoplasm was analyzed essentially as described previ-ously (31). Briefly, 120 mg of liver tissue was homogenized by 10 to 20 strokes in a Potter-Elvehjem tissue grinder in 2 ml of ice-cold Tris-EDTA buffer. One milliliter was used for analysis of viral cytoplasmic DNA, and the other milliliter was used for analysis of covalently closed circular DNA (cccDNA) (see below). Nonidet P-40 (final concentration, 0.5%) was added to 1 ml of liver homoge-nates, and after a 30-min incubation on ice the samples were centrifuged to remove the nuclei and cellular debris. Magnesium acetate (final concentration, 5 mM) was then added, and the supernatants were incubated for 30 min at 378C with 20 U of DNase I per ml. Subsequently, EDTA (final concentration, 10 mM), SDS (final concentration, 1%), NaCl (final concentration, 0.1 M), and pronase E (final concentration, 0.5 mg/ml) were added sequentially, and the samples were incubated for 3 to 4 h at 428C. The samples were then extracted with phenol-chloroform, ethanol precipitated, quantitated, and digested with RNase A, and an amount of cytoplasmic extract derived from 43105
liver cells was analyzed by Southern blot.
[image:2.612.58.300.69.424.2](iv) Analysis of HBV cccDNA by Southern blot.Isolation of cccDNA was performed essentially as described elsewhere (4). Briefly, SDS was immediately added (final concentration, 2%) to 1 ml of the liver extract described above. Cellular DNA, proteins, and viral protein-bound DNA were precipitated with FIG. 1. A schematic representation of the HBV genome (ayw subtype) used
in this study is shown at the top. Schematic representations of the HBV-derived constructs that contain terminally redundant, greater-than-genome length (1.1-, 1.2-, 1.3-eq) copies of the complete HBV genome used for the generation of HBV transgenic mice are shown below. Enh. and En, enhancer; Poly A, poly-adenylation signal; X, C, PS, and S, X, core, pre-S, and S promoter, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
KCl (final concentration, 0.5 M). After centrifugation, the supernatant was extracted with phenol-chloroform, ethanol precipitated, quantitated, digested with RNase A, and analyzed by Southern blot.
(v) Structural analysis of replicative forms of HBV DNA.Viral DNA con-tained in cytoplasmic core particles was quantitated and digested with RNase A as described above and either heated at 1008C for 5 min, treated with Klenow polymerase (Boehringer Mannheim) for 1 h at 378C according to the manufac-turer’s instructions, or digested with selected restriction enzymes before South-ern blot analysis.
Analysis of HBV gene expression. (i) Northern blot analysis.Frozen tissues were mechanically pulverized and extracted by the acid-guanidium–phenol-chlo-roform method (8). Total RNA (20mg) was analyzed for HBV and glyceralde-hyde-3-phosphate dehydrogenase (GAPDH) expression by Northern (RNA) blot as previously described (12).
(ii) Primer extension analysis.Total liver RNA was extracted as described above. Fifty micrograms of total liver RNA was subjected to primer extension with 10 U of Moloney murine leukemia virus reverse transcriptase (Boehringer Mannheim) and 5 ng of gel-purified oligonucleotides that were32P labeled at the 59end with T4 polynucleotide kinase (New England Biolabs, Beverly, Mass.). The samples were electrophoresed on a denaturing 6% polyacrylamide gel and exposed to X-ray film (Kodak, Rochester, N.Y.). The primer for the HBV 2.4-kb mRNA (59CCTTGTTGGGATTGAAGTCCCAATCTGGATTTGCG, nt 1395 to 1361) is designed to give an extension product of 153 bases, while the primer for the HBV 0.7-kb mRNA (59TTGGCAGCACAGCCTAGCAGCCATGGAA ACGATGT, nt 2392 to 2368) should produce several extension products ranging from about 178 to 130 bases in keeping with the multiple start sites previously mapped for these transcripts (32).
(iii) In situ hybridization.Snap frozen liver sections (thickness, 5mm) were transferred to polylysine-coated slides and fixed in a 4% paraformaldehyde– phosphate-buffered saline (PBS) solution for 20 min at room temperature. The fixed sections were briefly washed in PBS at room temperature and dehydrated by sequential transfer through 30, 70, and 90% ethanol baths. After digestion with pronase E (Sigma) at 0.35 mg/ml in PBS for 10 min at room temperature, the slides were refixed in 4% paraformaldehyde–PBS as above and acetylated by 0.25% acetic anhydride (Sigma) in 0.1 M triethanolamine (Sigma). The sections were then washed in PBS and dehydrated by sequential transfer through 30, 70, 90, and 100% ethanol baths. Hybridization buffer (50% formamide, 12.5% dex-tran sulfate, 23SSC [13SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 13 Denhardt’s solution, 10 mM dithiothreitol [DTT], 1 mM EDTA, 250mg of yeast tRNA per ml containing 203106
cpm of alkaline digested probe per ml) was added to each section, and the samples were covered with Parafilm and incu-bated overnight at 508C in a humidified chamber. The slides were then washed for 3 h at 528C in buffer A (50% formamide, 23SSC, 13Denhardt’s solution, 10 mM DTT) and for 15 min at 378C in buffer B (1 M Tris-HCl [pH 8], 0.5 M EDTA, 0.5 M NaCl). The sections were digested with RNase A (20mg/ml) in buffer B for 30 min at 378C and then washed in buffer B for 30 min at 378C. Remaining free radioactivity was removed by washing in 23SSC for 30 min at room temperature and 0.13SSC for 30 min at room temperature. The sections were then dehydrated in graded ethanol as above, dipped in Kodak NTB-2 emulsion (diluted 1:1 with 0.6 M ammonium acetate), developed after 24 to 48 h of exposure, and counterstained with hematoxylin and eosin. The33P-labeled RNA probes used for in situ hybridization were generated by in vitro transcrip-tion (Transcriptranscrip-tion kit; Boehringer Mannheim) of 1mg of the DNA template. A 0.75-kb fragment which spans sequences on the noncoding and coding strands of HBV between residues 1243 and 1948 was subcloned from an EBO vector previously described (14) into the KpnI-SacI site of pBluescript SKII. The plas-mid was linearized with KpnI or SacI to yield the sense and antisense probes. A 0.66-kb fragment which spans sequences on the noncoding strand of HBV be-tween residues 2139 and 2800 was subcloned from an EBO vector previously described (14) into the XbaI-SalI site of pBluescript SKII. The plasmid was linearized with NotI or EcoRI to yield the sense and antisense probes. The 33P-labeled RNA probes were digested in an alkaline buffer (10 mM EDTA, 10 mM Tris-HCl [pH 7.4], 0.2% SDS, 12.5 mM DTT, 100 mM NaCl, 250 mM NaOH) for 45 min on ice and extracted with phenol-chloroform. Free radioac-tivity was removed by passing the probes through a Sephadex G-50 column (Pharmacia Biotech, Uppsala, Sweden), and the probes were ethanol precipi-tated and resuspended in diethyl pyrocarbonate-treated water.
(iv) Immunoblot analysis.Serum and organ homogenates were prepared as described elsewhere (24). Total soluble protein was determined by Coomassie blue G-250 binding (Bio-Rad). Precore, core envelope, and X proteins were analyzed by Western blot (immunoblot) as previously described (1, 13). Briefly, 100mg of total liver protein extract was separated by SDS-15% polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Amersham, Arlington Heights, Ill.). To detect core and precore antigens, rabbit anti-HBc/ eAg (Dako, Carpinteria, Calif.) primary antiserum was applied at a 1:500 dilu-tion, and then a125
I-labeled anti-rabbit whole antibody (Amersham) was used. To detect envelope antigens, goat anti-HBsAg (Dako) primary antiserum was applied at a 1:500 dilution, and then a biotinylated anti-goat whole antibody (Dako) and streptavidin-horseradish peroxidase conjugate (Amersham) was used. To detect the HBV X protein, mouse monoclonal anti-X antibodies (11/ S1/G5 and 11/4/80, generously provided by Claus H. Schro¨der, Angewandte Tumorvirologie, Heidelberg, Germany) were applied at a 1:100 dilution, and
then a125I-labeled anti-mouse whole antibody (Amersham) was used. Total liver protein extracts from nontransgenic littermates were used as negative controls. Recombinant HBeAg and recombinant HBcAg (Sorin, Saluggia, Italy), total liver protein extracts from lineage 50-4, which overexpresses all envelope pro-teins (6), and total protein extracts from JY cells infected with recombinant vaccinia virus encoding the HBV X protein were used as positive controls for HBe/cAg, HBsAg, and HBV X protein, respectively.
(v) Histological analysis.Tissue samples were fixed in 10% zinc-buffered formalin (Anatek, Battle Creek, Mich.), embedded in paraffin, sectioned (3mm), and stained with hematoxylin and eosin as described elsewhere (6).
(vi) Immunohistochemical analysis.The intracellular distribution of HBcAg was assessed by the labeled-avidin-biotin detection procedure exactly as de-scribed elsewhere (13). Briefly, paraffin-embedded sections in PBS, pH 7.4, were treated for 10 min at 378C with 3% hydrogen peroxide and washed with PBS. After the sections were blocked with normal goat serum for 30 min at room temperature, rabbit anti-HBc/eAg (Dako) primary antiserum was applied at a 1:100 dilution for 60 min at 378C (HBcAg). After a wash with PBS, a secondary antiserum consisting of biotin-conjugated goat anti-rabbit immunoglobulin G F(ab9)2(Sigma) was applied at a 1:100 dilution for 30 min at 378C. The antibody-coated slides were washed with PBS, treated with the streptavidin-horseradish peroxidase conjugate (Extravidin; Sigma) at a 1:600 dilution for 30 min at 378C, stained with 3-amino-9-ethyl carbazole (AEC; Shandon-Lipshaw, Pittsburgh, Pa.), and counterstained with Mayer’s hematoxylin before being mounted. The intracellular distribution of HBsAg was assessed by the indirect immunoperox-idase method, using 3-amino-9-ethyl carbazole as a coloring substrate, as previ-ously described (6).
(vii) Serological analysis.Serum HBsAg and HBeAg concentrations were determined as follows. Two pools of serum were prepared from 30 age- and sex-matched transgenic mice from lineages 1.3.32 and 1.3.46 and analyzed for HBsAg and HBeAg by using commercially available reagents (Abbott Labora-tories). HBsAg and HBeAg were quantitated by comparison with serial dilutions of known standards (Abbott Laboratories and Sorin).
The concentration of pre-S1 in serum was determined as follows. Serum was pooled as described above and analyzed for the presence of the HBV pre-S1 antigen by enzyme-linked immunosorbent assay. Briefly, plates (Falcon 3912; Becton Dickinson, Oxnard, Calif.) were coated with 100ml of a mouse mono-clonal anti-pre-S1 antibody (MA18/7, generously provided by Wolfram Gerlich, Universitat Giessen, Giessen, Germany) at 1mg/ml in PBS for 1 h at 378C. After being blocked for 15 min at room temperature with 1% bovine serum albumin (BSA) in PBS, the wells were washed twice with 1% BSA in 0.05% Tween 20–PBS, and 50ml of transgenic-mouse serum, HBV-infected human serum, or recombinant small or recombinant large envelope protein (Merck, West Point, Pa.) was added for 1 h at 378C. The wells were washed as above, and 100ml of 125
I-labeled anti-HBsAg antibody (provided in a commercially available kit [Ab-bott Laboratories]) was added for 1 h at 378C. The wells were then washed three times with 1% BSA in 0.05% Tween 20–PBS and counted forg-emission in a Packard gamma counter (Packard, Downers Grove, Ill.). HBV-infected human serum was also used for comparative purposes in selected assays.
(viii) Quantitation of the HBV DNA concentration in serum.The concentra-tion of HBV DNA in transgenic-mouse serum was determined by two different quantitative dot blot procedures in our laboratories. In the first procedure (per-formed in La Jolla, Calif.), 300ml of filtered transgenic-mouse serum pooled from lineages 1.3.32 and 1.3.46 was incubated for 1 h at 378C with 50ml of 50 mM magnesium acetate, and 200 U of DNase I. As a negative control, filtered transgenic-mouse serum pooled from a lineage (pFC80-219) that does not rep-licate the viral genome was also included but not digested with DNase. EDTA (final concentration, 10 mM), SDS (final concentration, 1%), NaCl (final con-centration, 0.1 M), and pronase E (final concon-centration, 0.5 mg/ml) were added sequentially, and the samples were incubated for 3 to 4 h at 428C. The samples were then extracted with phenol-chloroform, ethanol precipitated, resuspended in Tris-EDTA, and applied with vacuum to a nylon membrane. After denatur-ation with 0.5 N NaOH, the filters were neutralized and hybridized with a specific radiolabeled HBV DNA probe. The hybridization signal was compared with the signal of a serially diluted plasmid DNA (pFC80) that contains four complete HBV genomes (ayw subtype) (11) and quantitated by the Matrix 96 Beta Counter (Packard).
Independently, in Heidelberg, the concentration of HBV DNA in a separate pool of transgenic-mouse serum from lineage 1.3.32 was compared with that in acutely HBV-infected-chimpanzee serum (35). Briefly, 100-ml samples of trans-genic-mouse and HBV-infected-chimpanzee sera were cleared by centrifugation at 13,000 rpm for 1 min, and viral particles were pelleted by ultracentrifugation in a TLA 45 rotor at 44,000 rpm for 1 h. The pellet was dissolved in 100ml of 10 mM Tris HCl, (pH 7.6)–0.1 M NaCl, denatured with NaOH (final concentration, 0.1 M), and adsorbed by filtration to a nylon membrane. The filters were neu-tralized and hybridized with a specific32
P-labeled HBV DNA probe. The hy-bridization signal was compared with the signal of a serially diluted plasmid HBV DNA standard (ayw subtype).
(ix) Serum endogenous polymerase activity.The endogenous polymerase ac-tivity of transgenic-mouse serum was compared with that in serum from an acutely HBV-infected chimpanzee (35) with a known HBV DNA concentration (see above). One milliliter of pooled transgenic-mouse serum from lineage 1.3.32 and 100 ml of HBV-infected-chimpanzee serum were pelleted as described
on November 9, 2019 by guest
http://jvi.asm.org/
above, and DNA was32P radiolabeled by the endogenous polymerase reaction exactly as described elsewhere (27). The32P-radiolabeled endogenously elon-gated HBV DNA products were analyzed by electrophoresis in a 1.5% agarose gel containing 1% SDS before and after digestion with selected restriction enzymes and analyzed by 1.5% agarose gel containing 1% SDS.
(x) Electron microscopy.Intracellular nucleocapsid particles were analyzed exactly as described elsewhere (13). Briefly, thin liver sections were fixed over-night at 48C in 4% paraformaldehyde–0.1% glutaraldehyde in PBS. Sections were then postfixed in 1% OsO4in cacodylate buffer (pH 7.4) for 1 h at room temperature, dehydrated in gradient ethanol, and embedded in epoxy resin (TAAB 812; Emmer Green, Reading, England). Sections were cut on an LKB Ultratome III, mounted on copper grids, stained in uranyl acetate and lead citrate, and viewed with a Hitachi HU 12-A electron microscope.
Serum-derived virus particles were analyzed as follows. Five hundred micro-liters of transgenic-mouse serum was centrifuged (200,0003g) overnight at 148C through 2.4 ml of 10% sucrose into 0.8 ml of 60% sucrose. Fractions (100ml) were collected from the bottom, and the first fraction (positive for endogenous polymerase activity) was incubated for 45 min at room temperature on MA18/ 7-coated (100mg/ml) Parlodion-carbon-coated nickel grids that were subse-quently drained with a piece of filter paper and washed twice in PBS. The grids were then fixed in 1.5% glutaraldehyde for 2 to 5 min, washed twice in H2O, negatively stained with uranyl acetate, and viewed with a Hitachi HU 12-A electron microscope. As positive control, Dane particles (see Results) purified from infected human sera (generously provided by John Gerin, Georgetown University, Washington, D.C.) were subjected to the same procedure.
RESULTS
Founder animals.
A total of nine founder animals whose
sera were positive for both HBsAg and HBeAg were produced
with the 1.1 (five founders), 1.2 (one founder), and 1.3 (three
founders) HBV genome constructs (Fig. 1). By Northern and
Southern blot analysis of tissues from the 1.1 and 1.2 genome
animals, only low-level, kidney-preferential HBV expression
and replication were observed (not shown), and these animals
were not studied further. Because we observed high-level HBV
gene expression and viral replication in the liver in the three
lineages containing the 1.3 genome construct (designated
lin-eages 1.3.2, 1.3.32, and 1.3.46), these animals were studied in
greater detail. Founder 1.3.2 was infertile, so all subsequent
analysis was performed with the progeny of founders 1.3.32
and 1.3.46, produced by mating the founders to nontransgenic
C57BL/6 and B10.D2 mice, respectively.
Analysis of the integrated transgene. (i) Transgene
integra-tion site.
Total DNA was isolated from liver and kidney tissues
of lineages 1.3.32 and 1.3.46, digested with an enzyme
(Hin-dIII) that does not cut within the transgene, and analyzed by
Southern blot. As shown in Fig. 2A, we detected a single slowly
migrating band for lineages 1.3.32 and 1.3.46, indicating that
the transgenes are integrated at a single site. The estimated
size of the integrated transgene in lineage 1.3.32 was less than
8 kb (Fig. 2), suggesting that a single copy of the microinjected
1.3 HBV construct (4,096 bp) may be integrated in this lineage
(see below). Abundant lower-molecular-weight species
repre-senting replicating forms of HBV DNA were also detected in
these tissues and will be discussed below.
(ii) Transgene structure and copy number.
To determine
the structure and copy number of the integrated transgene in
lineages 1.3.32 and 1.3.46, we carried out a PCR-based strategy
using HBV-specific sense and antisense nonoverlapping
prim-ers located at the 5
9
end of the terminal redundancy in the
microinjected construct. These primers amplified in lineage
1.3.32 only a 3.2-kb product (not shown), indicating that a
single uninterrupted copy of the construct is integrated at a
single site in the mouse genome (see also above). When the
same primers were used to analyze lineage 1.3.46, in addition
to this 3.2-kb band we observed a
;
950-bp product, indicating
that two or more uninterrupted copies of the transgene are
integrated in a head-to-tail orientation in the mouse genome
(not shown). Since the bands corresponding to the integrated
transgene in lineages 1.3.32 and 1.3.46 show comparable signal
intensities by Southern blot (Fig. 2A), it is likely that lineage
1.3.46 contains only two or three copies of the integrated
trans-gene. These structures contain the regulatory and structural
elements required for expression of all of the viral gene
prod-ucts and for replication of the viral genome.
HBV replicative intermediates.
As can be seen in Fig. 2A,
Southern blot analysis of total DNA extracted from liver and
kidney tissues of lineages 1.3.32 and 1.3.46 shows a smear of
low-molecular-weight HBV DNA molecules, in which it is
pos-sible to distinguish three bands that correspond to the expected
sizes of the relaxed circular DNA, the double-stranded linear
DNA, and the single-stranded DNA. As mentioned above,
lineage 1.3.32 contains a single copy of the integrated
trans-gene per cell (and two or three copies are present in lineage
1.3.46). Since approximately 20 to 30% of the hepatocytes
sustain high-level HBV replication in these animals (see
be-low) and since the hepatocytes represent about 80% of total
liver cells, by comparing the signal intensity of the integrated
transgene with those for HBV replicative forms (Fig. 2B), we
estimated that 100 to 200 copies of HBV replicative
interme-diates are present in each HBV replicating hepatocyte. This
estimate is consistent with the results of an independent
ex-periment in which HBV replicative intermediates were
quan-titated by a comparison with serial dilutions of a purified 3.2-kb
HBV DNA fragment (not shown).
[image:4.612.316.551.76.268.2]Total hepatic DNA from the transgenic lineages was
com-pared with DNAs from previously described transgenic mice
produced by other investigators to evaluate the relative
effi-ciency of HBV replication in the various strains of mice. As
shown in Fig. 2, HBV replication in lineages 1.3.32 and 1.3.46
was 10- to 100-fold greater than that in lineage PC21, described
by Farza et al. (9) and that in lineage 1.2HB-BS, produced by
FIG. 2. HBV replication in lineages 1.3.32 and 1.3.46 compared with that in previously reported transgenic lineages and that in a chronically HBV-infected human liver. (A) Southern blot analysis of 20mg of total DNA extracted from transgenic-mouse liver and kidney tissues of lineages 1.3.32 (lanes 1 and 2) and 1.3.46 (lanes 3 and 4); (B) Southern blot analysis of 20mg of total hepatic DNA extracted from chronically HBV-infected human liver (lane 1), transgenic-mouse lineage PC21 (9) (lane 2), transgenic-mouse lineage 1.2HB-BS (1) (lane 3), and transgenic-mouse lineage 1.3.32 (lane 4). All DNA samples were RNase treated before gel electrophoresis. The filters were hybridized with a32P-labeled HBV-specific DNA probe. Bands corresponding to the integrated transgenes can be used to normalize the amount of DNA bound to the membrane. Bands corre-sponding to the expected size of the integrated transgene, relaxed circular (RC), double-stranded linear (DS), and single-stranded (SS) HBV DNAs are indi-cated.
on November 9, 2019 by guest
http://jvi.asm.org/
Araki et al. (1). We have also compared the level of HBV
replication in our transgenic mice with those in six chronically
infected human livers. Both lineages replicate HBV at a level
comparable to that in the human liver that showed the highest
intrahepatic concentration of HBV DNA replicative
interme-diates in terms of total cellular DNA per microgram (Fig. 2B).
Developmental expression of viral RNA.
The kinetics of
hepatic expression of HBV mRNA was examined for lineages
1.3.32 and 1.3.46. As shown in Fig. 3, viral RNA is not
detect-able in the liver tissue in newborn mice from lineage 1.3.32. In
contrast, the 3.5- and 2.1-kb transcripts are already detectable
in the liver tissue in fetal or newborn mice from lineage 1.3.46
(not shown). In both lineages, the level of expression increases
steadily during the first several weeks of life, reaching a stable
plateau at approximately 4 weeks of age. The 2.4-kb transcript,
which is easily detectable at 2 to 3 weeks of age (Fig. 3),
appears to become obscured by the 2.1-kb band as it and the
3.5-kb mRNA increase as the mice reach maturity.
Tissue distribution of viral RNA in adult mice.
Northern
blot analysis of tissues derived from 8-week-old mice from
lineages 1.3.32 and 1.3.46 revealed high-level expression of the
3.5-kb pregenomic RNA and the 2.1-kb envelope mRNA
spe-cies in liver and kidney tissues in both lineages, with lower-level
expression in a variety of other tissues (Fig. 4).
As stated above, a 2.4-kb mRNA was easily detectable by
Northern analysis of liver RNA from 2- to 3-week-old
trans-genic mice (Fig. 3), but it became obscured as the abundance
of the 2.1-kb transcript increased with time (Fig. 3 and 4).
Nonetheless, transcripts consistent with this species were
de-tectable by primer extension analysis of liver RNA in the adult
animals (not shown). This is consistent with the presence of
pre-S1 antigen in the serum (see Table 1), since pre-S1 is a
unique marker for the large envelope polypeptide which is
translated from the 2.4-kb viral mRNA (10).
A 0.7-kb viral mRNA was never detectable by Northern blot
analysis in any tissue in these animals, a finding similar to its
low-level expression in the liver during natural infection (19);
however, multiple bands consistent with this species were
de-tected in the liver by primer extension analysis (not shown),
suggesting that all four viral promoters are probably active in
these animals.
One additional, currently undefined
;
3.2-kb transcript was
also observed in the kidney of lineage 1.3.32 and in all positive
tissues in lineage 1.3.46 (Fig. 4).
Expression of viral proteins.
At the protein level, HBsAg
(product of the 2.1-kb mRNA), pre-S1 (product of the 2.4-kb
mRNA), and HBeAg (product of the 3.5-kb mRNA) were
easily detectable in the serum and urine in both lineages, as
shown in Table 1. As expected, HBcAg (also a product of the
3.5-kb mRNA) was easily detectable in the liver and kidney by
immunohistochemical staining (Fig. 5), and it was also present
at lower levels in other tissues (Table 2). The 17-kDa precore
protein (HBeAg) was not detectable in total protein extracts
from transgenic-mouse liver or kidney by Western blot analysis
(data not shown), indicating that intracellular retention of the
precore protein is not detectable at this level in the transgenic
mouse. Only small amounts of HBsAg were detectable in the
cytoplasm of centrilobular hepatocytes (Table 2), consistent
with production of the rapidly secretable, HBsAg-containing
middle and major envelope proteins by translation of the
abun-dant 2.1-kb mRNA. Immunoreactive X protein was not
detect-able in liver or kidney by Western blot analysis (not shown).
FIG. 3. Developmental regulation of hepatic HBV gene expression. North-ern blot analysis of 20mg of total hepatic RNA extracted from lineage 1.3.32. The filters were hybridized with HBV- and GAPDH-specific probes. The results obtained from a representative transgenic mouse at each time point are indi-cated. The housekeeping GAPDH gene was used to normalize the amount of RNA bound to the membrane.
[image:5.612.314.554.511.685.2]FIG. 4. Tissue distribution of HBV gene expression. (A) Northern blot anal-ysis of 20mg of total RNA extracted from the liver (lane 1), kidney (lane 2), stomach (lane 3), and small intestine (lane 4) of lineage 1.3.32; (B) Northern blot analysis of 20mg of total RNA extracted from the liver (lane 1), kidney (lane 2), pancreas (lane 3), heart (lane 4), lung (lane 5), stomach (lane 6), and small intestine (lane 7) of lineage 1.3.46. The filters were hybridized with HBV- and GAPDH-specific probes.
TABLE 1. HBV antigen levels in serum and urine
Samplea Pre-S1
(cpm)
HBsAg (ng/ml)
HBe/cAg (ng/ml)
Normal mouse
Serum 80 ,0.1 ,0.1
Urine 80 ,0.1 ,0.1
1.3.32
Serum 2,532 700 50
Urine 2,822 900 100
1.3.46
Serum 1,923 700 50
Urine 2,830 900 100
pFC80-219 serum 205 6,000 ,0.1
Human serum 6,779 800 NT
Small envelopeb,c 100 600 ,0.1
Large envelopeb,d 6,441 35 ,0.1
a
Except for the human serum, which was tested at a 1/5 dilution, all serum and urine samples were undiluted.
b
Recombinant antigen provided by Ron Ellis (Merck).
c
20mg of protein per ml.
d
255mg of protein per ml.
on November 9, 2019 by guest
http://jvi.asm.org/
Distribution of HBcAg in the liver and kidney.
Both lineages
displayed the same subcellular and architectural distribution
of HBcAg in the liver and kidney. As illustrated in Fig. 5A
for lineage 1.3.32, HBcAg is present in the nuclei of almost all
of the hepatocytes distributed widely throughout the hepatic
lobule from the centrilobular region around central veins to
the periportal region around the portal tracts. At the
cytoplas-mic level, however, HBcAg is detectable mainly in the
centri-lobular hepatocytes surrounding the central veins, despite the
fact that all of the hepatocytes contain abundant HBcAg in
their nuclei. As illustrated in the inset in Fig. 5, the cellular
HBcAg appears to be particulate since typical 25- to 30-nm
HBcAg particles (core particles and nucleocapsid particles)
were detected in both the cytoplasm and the nucleus in the
centrilobular hepatocytes. HBcAg is also present in the
nu-cleus and the cytoplasm of the proximal convoluted tubules of
the kidney, especially the deep cortical tubules immediately
adjacent to the renal medulla, indicating the existence of
im-portant host influences on viral gene expression in these
ani-mals (Fig. 5B).
[image:6.612.88.524.71.391.2]Differential expression and replication of HBV in the
cyto-plasm in centrilobular hepatocytes.
The basis for the
prefer-ential expression of cytoplasmic HBcAg in the centrilobular
hepatocytes was examined by in situ hybridization analysis
us-ing antisense probes that are specific for either all HBV
mRNAs (Fig. 6A) or the 3.5-kb viral mRNA (Fig. 6B). As
shown in Fig. 6A, HBV RNA is detectable in virtually all of the
hepatocytes when a probe that spans sequences on the
non-coding strand of the X region of HBV (between residues 1243
and 1948) was used. When a probe specific for the 3.5-kb viral
mRNA (between residues 2139 and 2800) was used (Fig. 6B),
we observed a stronger expression of this transcript in the
centrilobular region, in keeping with the presence of HBcAg in
the cytoplasm in these cells (Fig. 5A) and suggesting that
higher-level expression is required for HBcAg to accumulate
FIG. 5. Immunohistochemical and ultrastructural analysis of HBcAg in the liver and kidney. (A) Immunohistochemical analysis of the liver of an 8-week-old male mouse shows that nuclear HBcAg is present in almost all of the hepatocytes while cytoplasmic HBcAg is detectable mainly in centrilobular hepatocytes surrounding the central veins. (B) HBcAg is also present in the nucleus and the cytoplasm in the proximal convoluted tubules of the kidney, especially the deep cortical tubules immediately adjacent to the renal medulla. An immunoperoxidase stain was used for HBcAg. Magnification,3170. Electron microscopic analysis of hepatic nuclei and cytoplasm from lineage 1.3.32 (inset) revealed abundant 25- to 30-nm particles that were not present in nontransgenic controls. Bar, 50 nm.
TABLE 2. Tissue distribution of HBV envelope and nucleocapsid antigens
Tissue
Distribution
1.3.32 1.3.46
HBsAg HBeAg and
HBcAg HBsAg
HBeAg and HBcAg
Liver 1 1111 1 1111
Kidney 1 1111 1 1111
Pancreas 2 2 2 11a
Stomach 2 2 2 11b
Lung 2 2 2 1c
a
Pancreatic acinar, ductal, and islet cells were positive.
b
Gastric parietal cells were positive.
c
Bronchiolar columnar epithelial cells were positive.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.58.296.598.703.2]FIG. 6. Detection of HBV mRNA and HBV DNA sequences by in situ hybridization. A33
P-labeled antisense probe that covers the X region of HBV was used on transgenic-mouse (A) and nontransgenic-mouse (D) liver tissues. A33
P-labeled antisense probe specific for the 3.5-kb mRNA was used on transgenic-mouse (B) and nontransgenic-mouse (E) liver tissues. A33
P-labeled sense probe specific for the minus-strand DNA that covers the X region of HBV was used on RNase-treated transgenic-mouse (C) and nontransgenic-mouse (F) liver sections. PV and CV, portal and central veins, respectively. Magnification,3320.
on November 9, 2019 by guest
http://jvi.asm.org/
to detectable levels in the cytoplasm than that required in the
nucleus.
When the same tissue was RNase treated and probed by an
HBV-specific sense probe that can detect the viral
minus-strand DNA, the signal was also stronger in the cytoplasm in
the centrilobular hepatocytes (Fig. 6C). The cytoplasmic
com-partmentalization of minus-strand viral DNA was confirmed
and extended by Southern blot analysis of DNA prepared from
8
3
10
6isolated nuclei and the cytoplasmic fraction derived
from 4
3
10
5liver cells from these animals. In this study, using
a total HBV DNA probe capable of detecting viral plus and
minus strands, DNA species corresponding in size to the linear
single-stranded, linear double-stranded, and relaxed circular
double-stranded forms of the HBV genome were detectable in
the total liver extract (Fig. 7, lanes 1 and 2) and in the
cyto-plasmic compartment of the liver, where they were shown to be
DNase resistant (Fig. 7, lanes 5 and 6), but not in the nucleus
(Fig. 7, lanes 3 and 4). These results suggest that viral
replica-tion is restricted to a DNase-resistant compartment in the
cytoplasm in these animals and that viral DNA is not
detect-able in the nucleus by these techniques.
Structure of the replicative intermediates.
To examine the
structure of the DNA replicative intermediates, cytoplasmic
DNA was extracted from the livers of both transgenic lineages
and from the liver of an infected patient and either heated at
100
8
C for 5 min or treated with Klenow polymerase before gel
electrophoresis. Southern blot analysis was carried out with
HBV strand-specific probes. In all instances, HBV replicative
intermediates in the transgenic-mouse and human livers were
identical in structure (not shown).
Detection of cccDNA.
The presence of the cccDNA species,
which normally serves as the intranuclear transcriptional
tem-plate for viral gene expression during HBV infection (33), was
tested in the transgenic-mouse liver by Southern blot analysis
of non-protein-bound nucleic acid extracts. We compared the
contents of cccDNA and replicative intermediates in
trans-genic-mouse and infected human livers. In this experiment the
cccDNA and replicative intermediate preparations were
de-rived from the same amount of liver tissue, and equal amounts
of human and mouse livers were used to prepare these extracts.
As shown in Fig. 8, cccDNA was not detectable in the
trans-genic-mouse liver, while it was readily detectable in the human
liver by Southern blot analysis, despite the fact that the levels
of viral replication were equivalent in these tissues. The reason
for this important difference is not clear at this time.
Circulating viral particles.
To determine whether
poten-tially infectious viral particles are formed and secreted into the
blood of these mice, serum was analyzed for the presence of
HBV DNA, endogenous polymerase activity, and
sediment-able particles displaying the ultrastructural characteristics of
complete virions (Dane particles).
Serum HBV DNA content was analyzed by two different dot
blot experiments. In the first instance, 300
m
l of filtered serum
[image:8.612.108.246.71.229.2]was treated with DNase prior to dot blot hybridization with an
HBV-specific probe in order to eliminate any transgene DNA
contaminating the specimen because of normal cell turnover or
tissue damage during phlebotomy, and the results were
com-pared with a serially diluted HBV DNA standard. As shown in
Fig. 9A, this analysis yielded
;
9
3
10
7HBV genome eq/ml in
lineage 1.3.46 (
;
300 pg of HBV DNA per ml) and
;
3
3
10
7HBV genome eq/ml in lineage 1.3.32 (
;
100 pg of HBV DNA
per ml). HBV DNA was undetectable in the serum of an HBV
transgenic-mouse lineage (pFC80-219) that does not replicate
the viral genome, even though this serum was not treated with
DNase. This illustrates that the signal in lineages 1.3.32 and
1.3.46 was not derived from contaminating genomic DNA. In
the second instance, HBV DNA quantitation yielded an
esti-mate of
;
100 pg of HBV DNA per ml in lineage 1.3.32. This
is approximately 20% of the HBV DNA concentration in the
serum of a chimpanzee at the peak of infection with HBV
produced from a cloned HBV DNA genome identical to the
one used here as transgene (Fig. 9B). The association of DNA
polymerase activity with HBV genomes was demonstrated by
restriction endonuclease digestion of the
32P-labeled reaction
product. As shown in Fig. 9C, digestion of the
transgenic-mouse-derived reaction product with EcoRI, BglII, and BamHI
yielded labeled fragments displaying the same mobility as a
similarly treated reaction product derived from the serum of
the chimpanzee infected with HBV with an identical sequence
(35).
FIG. 7. Cytoplasmic localization of HBV DNA replicative intermediates. Southern blot analysis of 20mg of uncut DNA extracted from total liver of lineage 1.3.32 (lane 1) or lineage 1.3.46 (lane 2), DNase-resistant nuclear DNA extracted from 83106purified liver nuclei of lineage 1.3.32 (lane 3) or lineage 1.3.46 (lane 4), and DNase-resistant cytoplasmic DNA extracted from 43105 liver cells of lineage 1.3.32 (lane 5) or lineage 1.3.46 (lane 6). All DNA samples were RNase treated before gel electrophoresis. The filters were hybridized with a32P-labeled HBV-specific DNA probe. Bands corresponding to the expected size of the integrated transgene, relaxed circular (RC), double-stranded linear (DS), and single-stranded (SS) HBV DNAs are indicated.
FIG. 8. Detection of HBV cccDNA. Identical amounts of chronically in-fected human liver (lanes 1) and transgenic-mouse livers from lineages 1.3.32 (lanes 2) and 1.3.46 (lanes 3) were extracted to produce DNase-resistant cyto-plasmic DNA (A) and non-protein-bound nucleic acids (cccDNA) (B). The DNA samples were RNase treated, and identical quantities of the two types of extract were analyzed by Southern blot using a32
P-labeled HBV-specific DNA probe. Bands corresponding to the expected size of relaxed circular DNA (RC), double-stranded linear DNA (DS), cccDNA, and single-stranded DNA (SS) are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
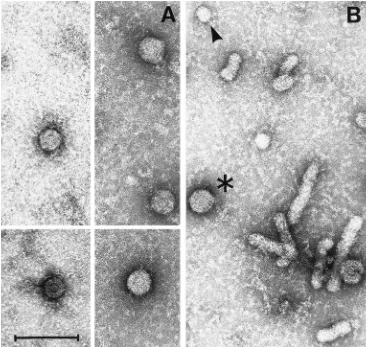
[image:8.612.377.493.506.652.2]Ultrastructural analysis of endogenous polymerase-positive
fractions of ultracentrifuged transgenic-mouse serum captured
on anti-pre-S1 coated grids revealed the presence of
charac-teristic 42-nm particles (Fig. 10A) that are morphologically
indistinguishable from Dane particles purified from infected
human serum (Fig. 10B). These particles were not detectable
in the same fractions applied to grids that had not been coated
with anti-pre-S1 antibody, nor were they detected in the
cor-responding endogenous polymerase-negative fractions from
nontransgenic-mouse serum applied to antibody-coated grids
(not shown).
Histopathological findings.
Animals from lineages 1.3.32
and 1.3.46 have been monitored histologically for over 1 year
without evidence of pathological changes in any organ,
espe-cially the liver and kidney (not shown).
DISCUSSION
HBV transgenic mice whose hepatocytes replicate the virus
at high levels without any evidence of cytopathology have been
produced. Approximately 100 to 200 copies of HBV replicative
intermediates were detectable in each hepatocyte. This
com-pares favorably with the estimate of 100 to 700 copies per
infected hepatocyte during acute woodchuck hepatitis virus
infection (18). Furthermore, the estimated 10
7to 10
8viral
genomes detectable per ml of serum in these animals compares
favorably with the concentration of virus present in the sera of
chronically infected humans (28). These results confirm the
noncytopathic behavior of HBV in healthy carriers (16).
High-level viral gene expression and replication were
ob-served in the liver and kidney in three independent lineages
produced with a terminally redundant, 1.3
3
genome length
transgene that starts just upstream of the X promoter and
enhancer I and ends just downstream of the unique HBV
polyadenylation site. Slightly shorter constructs lacking these
upstream elements, including lineage 1.2HB-BS produced by
Araki et al. (1), consistently yielded lower-level expression and
much lower levels of replication, which were preferentially
detectable in the kidney tissue instead of the liver tissue in the
corresponding animals. These data suggest that the transgene
structure may contribute to the level of liver-specific HBV
gene expression in these animals.
The above finding is interesting because the extra sequences
that are uniquely present at the 5
9
end of the 1.3 genome
construct integrated in the mouse genome encompass the
sig-nals needed for expression of the X gene. Since the X gene is
known to be important for woodchuck hepatitis virus
replica-tion in the liver in vivo (36), the level of X gene expression
might also influence the level of viral replication in the liver in
these mice. Although low-level X mRNA is detectable in the
liver in the 1.3 series of animals by primer extension analysis,
we do not have direct evidence to prove that this notion is
correct at this time, since we have been unable to detect X
protein in the liver in these animals. Additionally, the 1.3, but
not the 1.1 or 1.2, series of animals contains an extra copy of
the entire HBV enhancer I at the 5
9
end of the integrated
transgene. This factor could be responsible for higher-level
liver gene expression in these animals.
The family of 3.5-kb HBV transcripts normally serves as the
mRNA for the viral nucleocapsid proteins (HBcAg and
HBeAg) and for the viral polymerase protein, and it also serves
as the viral pregenome which is reverse transcribed by the
polymerase to produce the first strand of viral DNA during
viral replication (30). The 2.4-, 2.1-, and 0.7-kb transcripts
normally serve as the mRNAs for the viral large and middle/
major envelope proteins and the X protein, respectively.
Dur-FIG. 9. Quantitation and characterization of circulating viral particles. (A) Dot blot analysis of HBV DNA extracted from transgenic-mouse sera of lineages 1.3.46, 1.3.32, and pFC80-219. The signals for serial dilutions of a plasmid of known HBV DNA concentration (expressed in picograms) are shown at the top. DNase-resistant HBV DNAs extracted from 300-ml serum samples from lineages 1.3.46 and 1.3.32 are shown below. As a negative control, 300ml of untreated serum from a lineage (pFC80-219) that does not replicate the viral genome was also included. (B) In an independent experiment, the HBV DNA content of 100
ml of transgenic-mouse serum (lineage 1.3.32) was compared with the HBV DNA content of 100ml of serum from a chimpanzee acutely infected with HBV (35). The signals for serial dilutions of a plasmid of known HBV DNA concen-tration (expressed in picograms) are shown above. (C) The endogenous poly-merase activity present in the ultracentrifuged pellet derived from 1 ml of transgenic-mouse serum (lineage 1.3.32) was compared with that in 100ml of chimpanzee serum. The32P-radiolabeled endogenously elongated HBV DNA products were analyzed by electrophoresis in a 1.5% agarose gel containing 1% SDS before and after digestion with selected restriction enzymes. Lane M, molecular weight markers.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.57.301.95.558.2]ing normal viral replication, the 3.5- and 2.1-kb transcripts are
most abundant and present in roughly equimolar quantities,
while the 2.4-kb transcript is a minor species and the 0.7-kb
mRNA is not detectable (10).
All of these transcripts are expressed in the liver in the
current lineages in the proportions and abundance typical of
natural HBV infection (Fig. 3 and 4), and they are also
abun-dant in the subset of proximal convoluted renal tubules that
are located deep in the renal cortex immediately adjacent to
the medulla (Fig. 4). In addition, viral RNA is present in trace
amounts in scattered tissues and absent from other tissues (Fig.
4), presumably because of the influence of host factors (e.g.,
transcription factors) that regulate viral gene expression.
Along these lines, it may be germane that hepatocyte nuclear
factor 1 alpha (HNF 1
a
) is known to be preferentially
ex-pressed in hepatocytes and in the juxtamedullary proximal
convoluted tubules of the kidney (29).
Presumably for similar reasons, the 3.5-kb HBV mRNA is
more abundant in the centrilobular hepatocytes than
else-where in the hepatic lobule in these transgenic mice (Fig. 6B).
This is reflected in the expression of HBcAg particles in the
cytoplasm of the centrilobular hepatocytes, i.e., the same cells
that contain large amounts of replicative DNA forms in the
liver lobule (Fig. 6C). Collectively, these results strongly
sug-gest that viral replication is occurring in the cytoplasmic
nu-cleocapsid particles in the centrilobular hepatocytes of these
animals and, by inference, in the HBcAg-positive cytoplasm of
the juxtamedullary proximal convoluted tubules of the kidney.
The data also indicate that the viral polymerase protein is
expressed in these mice and that it is packaged in the
nucleo-capsid particles together with the 3.5-kb pregenomic RNA,
since the minus-strand DNA is known to be produced by
re-verse transcription of the 3.5-kb pregenomic RNA by the viral
polymerase protein inside nucleocapsid particles (30).
Addi-tionally, the data indicate that an expression threshold for
HBcAg, viral polymerase, and 3.5-kb pregenomic RNA must
be reached before these events can occur.
[image:10.612.123.491.71.418.2]Finally, the results suggest that the intranuclear
nucleocap-sid particles present in the liver and kidney in these animals
(Fig. 7) do not contain viral DNA (i.e., they are empty). When
considered together with the fact that most of the hepatocytes
contain HBcAg in their nuclei in these and other transgenic
mice that we have previously described (13), the current
ob-servations indicate that the intranuclear HBcAg particles form
de novo in the nucleus and that they are not transported there
from the cytoplasm, even in cells that also contain cytoplasmic
HBcAg. This conclusion confirms our previous evidence (13)
that HBcAg particles do not cross the nuclear membrane in
either direction in transgenic mice, and it suggests that HBcAg
particles do not carry the viral DNA into the nucleus during
FIG. 10. Ultrastructure of circulating viral particles. Ultrastructural analysis of endogenous polymerase-positive fractions of ultracentrifuged transgenic-mouse serum captured on anti-pre-S1-coated grids revealed the presence of negatively stained characteristic 42-nm particles (A) that are morphologically indistinguishable from Dane particles (asterisk) and larger in diameter than subviral HBsAg particles (arrowhead) and HBsAg filaments purified from infected human serum (B) for comparison. Bar, 100 nm.
on November 9, 2019 by guest
http://jvi.asm.org/
the viral life cycle. If this finding extends to infected human
liver, it implies that nucleocapsid disassembly must occur at the
cytoplasmic face of the nuclear membrane to allow the viral
DNA to enter the nucleus. If this notion is correct, it raises
serious questions about the significance of the intranuclear
HBcAg particles during the viral life cycle.
As a consequence of efficient viral replication, it is not
sur-prising that ultrastructurally complete viral particles
morpho-logically indistinguishable from human Dane particles were
detected in the transgenic mouse serum (between 3
3
10
7and
9
3
10
7viral particles per ml). This finding further indicates
that the HBV life cycle can be efficiently completed in the
transgenic-mouse hepatocyte.
Surprisingly, HBV cccDNA, the normal viral transcriptional
template during natural infection, is not readily detectable by
direct Southern blot analysis in the hepatocyte nucleus in these
animals (Fig. 8). In preliminary experiments, however, using a
PCR-based technique that has been previously shown to
selec-tively amplify cccDNA (21), HBV cccDNA was detectable in
total hepatic DNAs from both transgenic lineages (not shown).
The results of these studies will be reported elsewhere.
It is important to reiterate that similar transgenic mice have
been produced by other investigators in recent years (1, 9).
While those animals clearly demonstrated that HBV
replica-tion can occur in the transgenic murine hepatocyte, their
ex-perimental value has been limited because they replicate the
virus at a very low level. The high level of HBV replication in
the new lineages now creates the opportunity to examine the
influence of viral and host factors on HBV replication and
pathogenesis and to assess the potential of pharmacological
agents and physiological processes, including the immune
re-sponse, to control HBV replication in a genetically defined
small-animal system.
ACKNOWLEDGMENTS
We thank John Brems, Michael Gerber, and David Shafritz for providing human liver specimens; Ken-ichi Yamamura for providing 1.2HB-BS transgenic mice; Christine Pourcel for providing PC21-de-rived transgenic tissue specimens; Laura Runkel for producing HBV constructs used for microinjection; Christa Kuhn for performing dot blot assays and endogenous polymerase assays on mouse and chim-panzee sera; Hans-Ju¨rgen Schilcht and Joseph Kock for performing PCR-based detection of HBV cccDNA; David Bylund, Dwight Dun-can, and the Scripps Immunology Reference Laboratory for perform-ing serum HBV DNA quantitation; John Gerin for providperform-ing purified Dane particles; Wolfram Gerlich for providing the mouse monoclonal anti-pre-S1 antibody MA 18/7; Claus Schro¨der and Hanswalter Zent-graf for providing the mouse monoclonal anti-X antibodies 11/S1/G5 and 11/4/80; Jesse Summers for providing protocols for liver extraction of HBV cccDNA and HBV replicative intermediates; Ron Ellis for providing recombinant small and large envelope proteins; and Alan McLachlan for consultation and advice. We also thank Jenny Price and the Scripps Transgenic Mouse Facility for embryo microinjections; Cheng-Ming Chang for assistance with ultrastructural analysis; Margie Pagels for preparation and staining of tissue sections; Patricia Fowler, Josan Chung, and Jan Shoenberger for excellent technical assistance; and Bonnie Weier for help with manuscript preparation.
This work was supported by National Institutes of Health Public Health Service merit award R37-CA40489 and by Deutsche For-schungsgemeinschaft grant SFB229.
REFERENCES
1. Araki, K., J.-I. Miyazaki, O. Hino, N. Tomita, O. Chisaka, K. Matsubara,
and K.-I. Yamamura.1989. Expression and replication of hepatitis B virus genome in transgenic mice. Proc. Natl. Acad. Sci. USA 86:207–211. 2. Babinet, C., H. Farza, D. Morello, M. Hadchouel, and C. Pourcel. 1985.
Specific expression of hepatitis B surface antigen (HBsAg) in transgenic mice. Science 230:1160–1163.
3. Burk, R. D., J. A. DeLoia, M. K. ElAwady, and J. D. Gearhart. 1988. Tissue
preferential expression of the hepatitis B virus (HBV) surface antigen gene in two lines of HBV transgenic mice. J. Virol. 62:649–654.
4. Calvert, J., and J. Summers. 1994. Two regions of an avian hepadnavirus RNA pregenome are required in cis for encapsidation. J. Virol. 68:2084– 2090.
5. Chisari, F. V., and C. Ferrari. 1995. Immunopathogenesis of hepatitis B virus. Annu. Rev. Immunol. 13:29–60.
6. Chisari, F. V., P. Filippi, A. McLachlan, D. R. Milich, M. Riggs, S. Lee, R. D.
Palmiter, C. A. Pinkert, and R. L. Brinster.1986. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secre-tion in transgenic mice. J. Virol. 60:880–887.
7. Chisari, F. V., C. A. Pinkert, D. R. Milich, P. Filippi, A. McLachlan, R. D.
Palmiter, and R. L. Brinster.1985. A transgenic mouse model of the chronic hepatitis B surface antigen carrier state. Science 230:1157–1160. 8. Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation
by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Bio-chem. 162:156–159.
9. Farza, H., M. Hadchouel, J. Scotto, P. Tiollais, C. Babinet, and C. Pourcel. 1988. Replication and gene expression of hepatitis B virus in a transgenic mouse that contains the complete viral genome. J. Virol. 62:4144–4152. 10. Ganem, D., and H. E. Varmus. 1987. The molecular biology of the hepatitis
B virus. Annu. Rev. Biochem. 56:651–693.
11. Gilles, P. N., G. Fey, and F. V. Chisari. 1992. Tumor necrosis factor alpha negatively regulates hepatitis B virus gene expression in transgenic mice. J. Virol. 66:3955–3960.
12. Guidotti, L. G., K. Ando, M. V. Hobbs, T. Ishikawa, R. D. Runkel, R. D.
Schreiber, and F. V. Chisari.1994. Cytotoxic T lymphocytes inhibit hepatitis B virus gene expression by a noncytolytic mechanism in transgenic mice. Proc. Natl. Acad. Sci. USA 91:3764–3768.
13. Guidotti, L. G., V. Martinez, Y. T. Loh, C. E. Rogler, and F. V. Chisari. 1994. Hepatitis B virus nucleocapsid particles do not cross the hepatocyte nuclear membrane in transgenic mice. J. Virol. 68:5469–5475.
14. Guilhot, S., P. Fowler, G. Portillo, R. F. Margolskee, C. Ferrari, A. Bertoletti,
and F. V. Chisari.1992. Hepatitis B virus (HBV) specific cytolytic T cell response in humans: production of target cells by stable expression of HBV-encoded proteins in immortalized human B cell lines. J. Virol. 66:2670–2678. 15. Guilhot, S., L. G. Guidotti, and F. V. Chisari. 1993. Interleukin-2 downregu-lates hepatitis B virus gene expression in transgenic mice by a post-transcrip-tional mechanism. J. Virol. 67:7444–7449.
16. Hoofnagle, J. H., D. A. Shafritz, and H. Popper. 1987. Chronic type B hepatitis and the ‘‘healthy’’ HBsAg carrier state. Hepatology 7:758–763. 17. Junker-Niepmann, M., R. Bartenschlager, and H. Schaller. 1990. A short
cis-acting sequence is required for hepatitis B virus pregenome encapsida-tion and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396. 18. Kajino, K., A. R. Jilbert, J. Saputelli, C. Aldrich, J. Cullen, and W. S. Mason.
1994. Woodchuck hepatitis virus infections: very rapid recovery after a pro-longed viremia and infection of virtually every hepatocyte. J. Virol. 68:5792– 5803.
19. Katayama, K., N. Hayashi, Y. Sasaki, A. Kasahara, K. Ueda, H. Fusamoto,
N. Sato, O. Chisaka, K. Matsubara, and T. Kamada.1989. Detection of hepatitis B virus X gene protein and antibody in type B chronic liver disease. Gastroenterology 97:990–998.
20. Kim, C.-M., K. Koike, I. Saito, T. Miyamura, and G. Jay. 1991. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature (London)
351:317–320.
21. Kock, J., and H.-J. Schlicht. 1993. Analysis of the earliest steps of hepad-navirus replication: genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J. Virol. 67:4867–4874. 22. Lee, T. H., M. J. Finegold, S. Rong-Fong, J. L. DeMayo, S. L. C. Woo, and
J. S. Butel.1990. Hepatitis B virus transactivator X protein is not tumori-genic in transtumori-genic mice. J. Virol. 64:5939–5947.
23. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Har-bor, N.Y.
24. McLachlan, A., D. R. Milich, A. K. Raney, M. G. Riggs, J. L. Hughes, J.
Sorge, and F. V. Chisari.1987. Expression of hepatitis B virus surface and core antigens: influences of pre-S and precore sequences. J. Virol. 61:683– 692.
25. Milich, D. R., J. E. Jones, J. L. Hughes, T. Maruyama, J. Price, I. Melhado,
and F. Jirik.1994. Extrathymic expression of the intracellular hepatitis B core antigen results in T cell tolerance in transgenic mice. J. Immunol.
152:455–466.
26. Milich, D. R., J. E. Jones, J. L. Hughes, J. Price, A. K. Raney, and A.
McLachlan.1990. Is a function of the secreted hepatitis B e antigen to induce immunologic tolerance in utero? Proc. Natl. Acad. Sci. USA 87:6599– 6603.
27. Radziwill, G., W. Tucker, and H. Schaller. 1990. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J. Virol. 64:613–620.
28. Ranki, M., H. M. Scha¨tzl, R. Zachoval, M. Uusi-Oukari, and P. Lehtovaara.
1995. Quantification of hepatitis B virus DNA over a wide range from serum for studying viral replicative activity in response to treatment and in
on November 9, 2019 by guest
http://jvi.asm.org/
rent infection. Hepatology 21:1492–1499.
29. Sladek, F. M., and J. E. Darnell, Jr. 1992. Mechanisms of liver-specific gene expression. Curr. Opin. Genet. Dev. 2:256–259.
30. Summers, J., and W. S. Mason. 1982. Replication of the genome of a hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell
29:403–415.
31. Summers, J., P. M. Smith, and A. L. Horwich. 1990. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol.
64:2819–2824.
32. Treinin, M., and O. Laub. 1987. Identification of a promoter element located upstream from the hepatitis B virus X gene. Mol. Cell. Biol. 7:545–548.
33. Tuttleman, J. S., C. Pourcel, and J. Summers. 1986. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell
47:451–460.
34. Tuttleman, J. S., J. C. Pugh, and J. W. Summers. 1986. In vitro experimental infection of primary duck hepatocyte cultures with duck hepatitis B virus. J. Virol. 58:17–25.
35. Will, H., R. Cattaneo, G. Darai, F. Deinhardt, H. Schellekens, and H.
Schaller.1985. Infectious hepatitis B virus from cloned DNA of known nucleotide sequence. Proc. Natl. Acad. Sci. USA 82:891–895.
36. Zoulim, F., J. Saputelli, and C. Seeger. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 68:2026–2030.