0022-538X/86/080308-10$02.00/0
Copyright C) 1986, American Society forMicrobiology
Molecular
Basis for
Interference
of Defective Interfering Particles of
Pseudorabies Virus with Replication of
Standard Virus
CAROL ANN WU, LINDA HARPERANDTAMARBEN-PORAT*
Department ofMicrobiology, Vanderbilt University School of Medicine, Nashville, Tennessee 37232 Received 10February 1986/Accepted22April 1986
Serialpassage ofpseudorabiesvirus(PrV) at high multiplicity yields defective interfering particles (DIPs), butthe sharpcyclical increases and decreases in titer of infectious virus that areobserved upon continued passage at highmultiplicityof most DIPsof other viruses are not observed with DIPs of PrV (T. Ben-Porat and A. S. Kaplan, Virology 72:471479). Wehavestudied thedynamics of the interactions of the virions present inapopulation of
DIPs
toassessthecisfunctionsfor which thegenomes of the DIPs are enriched. The defective genomes present in onepopulationofDIPs, [PrV(1)42], replicatepreferentiallyoverthenondefective genomes present in that virion population at early stages of infection, indicating that the DIP DNA is enriched for sequences that can serve as origins of replication at early stages of infection. This replicative advantage of the DIPDNA is transientand disappears at later stages of infection. The defective DNA does not appear to be encapsidatedpreferentiallyover thenondefective DNA present in this virionpopulation, which might indicate that it is not enriched for cleavage-encapsidation sites. However, the nondefective DNA in the DIP virionpopulationhas become modified and has acquiredreiterations ofsequences originating from the end of the unique long (UL) region of the genome. Furthermore, both the infectious and defective genomes present in the DIPpopulation compete for encapsidation moreeffectivelythan do thegenomes of standard PrV. These results indicate that the defective genomes in the population of virionsstiudiedareenriched not only for an origin of replication but probably also forsequences necessary for efficient cleavage-encapsidation. Furthermore, the nondefective genomes present in this population of DIPs have also been modified and have acquired theability
tocompete with the defective genomes forcleavage-encapsidation. Serialpassage athighmultiplicity ofmanyviruses,
includ-ingtheherpesviruses, resultsintheemergence ofdefective
interfering
particles (DIPs) which contain altered genome structures. These DIPs, although unable to replicate in theabsence of standard helper virus, have agrowth advantage
over it. Theidentity of theDIPs is of interest because their
emergence hasobviousimplications concerningthemodeof replication andencapsidation of the virus genome.
That DIPsofherpesvirus containgenomes thatconsist,at
least inpart, of tandemrepeats of restricted regionsofthe
viralgenome inhead-to-tail alignment has beenrecognized for some time from theirpartial denaturation patterns (12, 24). Since populations ofDIPs
becomne
enrichedfor virionswhose genomes probably canreplicate and beencapsidated,
commonfeatures oftheserepeated sequences may, in
prin-ciple,
include
origins ofreplication, as well as recognition sites for cleavage and encapsidation. Indeed, it has beenestablished that the DIPs of herpes simplex virus (HSV) containgenomes composed of reiterated segments ofDNA thatinclude an origin ofreplication, as well as the signals required forefficient
cleavage-encapsidation
(1,26, 27,
29). Becausethese
DIPs have a clear replicative advantage,cyclical increases and decreases in
the
level of infectious virus areobserved as the populations of DIPs arepassaged athigh multiplicity.Thegenomes present in twoindependentlyderived popu-lations of DIPs of pseudorabies virus (PrV)
have
beenpartially characterized. The two
populations
differ in se-quence composition (22). PrV DIP DNA contains reitera-tions of a limited set ofsequences of the standard genome (2, 24) which are composed of segments of DNA originating from noncontiguous regions of the standard genome that* Correspondingauthor.
have become covalently linked (22). One of these popula-tions, [PrV(2)], consists ofDIPs withgenomesthat contain
an intact immediate-early gene that is expressed in the
absence of helper virus. Coinfection of cells with this
popu-lationofDIPsandhelper virus causestheoverproduction of
someotherviral
proteins
aswell(23). Infection of cells with the otherpopulation of DIPs, [PrV(1)], on the otherhand, does not detectably affect viralprotein
synthesis (23).Nev-ertheless, thebiological properties of thetwopopulations of
DIPs
appear to be quite similar (4). In both, there is areduction of approximately
90%
in the total number ofvirionsproduced. Both populations ofPrVDIPsbehavein a
significantly different manner from most DIPs of other herpesviruses (for example, HSV and equine herpesvirus [11, 13,
28]).
Thus, the DIPs of PrV accumulate ratherslowly; only after 25 passages at high multiplicity can a
significant dropin the titerof infectiousvirus beobserved
(2,
4). Furthermore, thesharpcyclical increasesand decreases in the titersof infectiousvirusobservedupon serialpassageofpopulations ofDIPsofHSVandequine herpesvirus (11, 13, 28), for example, do not occur when PrV DIPs are
passaged; after an initial drop in the titer ofabout
102
bypassage30, only slightfluctuations in the titerareobserved
upon continued passage (4). The studies presented in this paper were undertaken to
clarify
thedynamics of the inter-actions between thedefective
and nondefective virions present in thepopulations of DIPs.MATERIALS ANDMETHODS
Virus and cell culture. The preparation of PrV and culti-vation of primary rabbit kidney (RK) cells has been de-scribed previously (19). Two populations of PrV DIPs
[PrV(1)
and PrV(2)] were generated by independent serial passageathighmultiplicityof the standardPrV(Ka)strain in 308on November 10, 2019 by guest
http://jvi.asm.org/
RKcells (4).Populations ofDIPsatvariouslevelsofpassage were used, startingwith passage 33 [PrV(1)33] andgoingto passage64 [PrV(1)64].
Media and solutions. The following media were used: Eagle synthetic medium (9) plus 5% dialyzed calf serum (EDS); EDS withoutP04(EDS-PO4); EDS-PO4plus 20 ,ugof 5-fluorouracil (FU) and 5 F.g of thymidine per ml (EDS-P04+FU); EDS-PO4 with 20 ,ug ofFU, 10 ,ug of bromode-oxyuridine (BUdR), and5 ,ugofdeoxycytidine (CdR) perml
(BUdR medium); self-digested (nuclease-free) pronase (2 mg/ml) in 0.02 M NaCl-0.02 M Tris (pH 7.3) (pronase
solution); and 0.15 MNaCl-0.015 Msodium citrate (pH 7.2) plus 4% sodium lauryl sarkosinate-97 (lx SSC-4%
sarkosyl).
Enzymes and chemicals. All restriction enzymes were obtained from Bethesda Research Laboratories, and diges-tions were performed accordingto the specifications ofthe
supplier. Inorganic 32p (carrierfree) and [a-32P]dCTP were purchased from ICN Pharmaceuticals, Inc., FUand BUdR
wereobtained from Calbiochem-Behring, and
[3H]thymidine
and [3H]deoxycytidine(3H-CdR)
were obtained from Schwarz/Mann.Labeling and purification of replicating virus DNA. RK
cells were starved of phosphate by incubation in EDS-P04+FU for 48 h, a procedure which also inhibits cellular DNA synthesis(18). The cells werethen incubated for 24 h in the same mediumcontaining
32p
(100p.Ci/ml)
tolabel thenucleotide pools. The cultures were infected and further incubated in BUdRmediumcontaining
32p
(100 ,uCi/ml). At theindicated times, the cultureswereharvested by scrapingthe cells into lx SSC-4% sarkosyl. The samples were heated at 60°C for 15 min, an equal volume of pronase
solutionwasadded, and the sampleswere incubatedfor 2 h at 37°C. Viral DNA was separated from cellular DNA by isopycnic centrifugation in CsCl, with
[3H]thymidine-containing and[3H]BUdR-substituted
viral DNA as mark-ers, as describedpreviously
(6). DNA fractions were col-lecteddropwise (2 dropspertube),theradiolabeledmarkers werelocalized, theappropriate fractions werecollected anddialyzed,and the DNA was ethanol
precipitated.
Plaquepurification. Viruswas plaque assayedonRKcells grown on 90-mm petri plates. Plaques were
picked
fromplates containing not more than 20 plaques each, and the
virus wasamplified.
Purification of virionsandextraction of viralDNA. Virions were purified and viral DNA was extracted as described previously (2, 5).
RestrictionenzymedigestionandgelelectrophoresisofDNA fragments.Digestionand agarosegel
electrophoresis
ofvirus DNAwerecarriedout asdescribedby
Rixonand Ben-Porat (22). Filterstrips,towhichrestrictionfragmentsof PrVwere fixed, were prepared by the method of Southern (25).Nick translation of clonedPrV DNArestriction fragments.
PrV DNA restriction fragments cloned in pBR325 as
de-scribed previously (20) were nick translated by the method
ofRigby etal. (21).
RESULTS
Lack of DIP DNApreferential encapsidation. The genera-tion and amplification of DIP DNA may result from a replicative advantage of thisDNA, from a selective advan-tageof this DNAatthelevelofencapsidation, orfrom both. To determine whether preferential encapsidation of DIP DNA occurs (i.e., whether this DNA may be enriched for
cleavage-encapsidation signals), we
compared
therestrictionpatternsof the viral DNAthatis
packaged
into virionswiththoseoftheviral DNA that accumulateswithintheinfected cells.
PrV DNA is synthesized in excess, and only part of it becomes encapsidated (3, 17). Furthermore, restriction en-zyme digestion of PrV DIP DNA generates aberrantly
mi-grating DNA fragments thatare absent from the
digests
ofstandard PrV DNA (22). Preferential
encapsidation
ofDIP DNA should therefore bereadily detectable,
because the presence ofthese aberrant DNA bands isdiagnostic
ofthe presence ofDIP DNA. This procedurecanbe usedto study notonlytheevolution ofthe DIP upon passageofthevirionsbut also to compare the
efficiency
ofencapsidation
ofDIP DNA with that of standard DNA.Therestrictionpatternofvirus DNA that hadaccumulated
in cells infected with
populations
of PrV(1) at differentpassage levels was
compared
with that of DNA that had becomeencapsidated
and was present inpreparations
ofpurified
virions isolated from thesecells(Fig.
1). No signif-icantpreferentialencapsidation
ofDIP DNAat anylevel ofpassagewas
detectable;
the viral DNA that hadaccumulated within the infected cells during the infectious cycle had a restrictionpatternsimilartothatofthe viral DNA present in thepurified
virions isolatedfromthese cells. Therestrictionpatternof the viral DNA
changed, however,
uponcontinuedpassageof the virions. In
particular,
enrichmentforBamHIfragments
10aand 12b(generated
from defectiveDNA)
was observedduring early
passage levelsathigh multiplicity. By
passage 64, two defective DNAfragments,
lla and12b,
made up most of the DNA in the virion
population. Thus,
although the defective DNA (asexemplified by fragment
12b)didnotappeartohaveasignificant
selectiveadvantage
overnondefectiveDNA presentinthe DIP
population
atthe levelofencapsidation,
itwasneverthelessenriched foruponhigh-multiplicity
passage. Itappeared possible,
therefore,
thatenrichmentof the DIP DNA in the
population
occurs as aresult ofpreferential
replication.
Time course of synthesis of DIP DNA. To determine
whether DIP DNA hasa
replicative advantage
eitherduring
the first or
during
later rounds of DNAreplication,
wefollowed the time course of its
synthesis
relative to that ofthe nondefective DNA present in the
population
of DIPs.Phosphate-starved RKcellswere
pretreated
with FU andthymidine
(aprocedure
thatcompletely
inhibits cellular DNAsynthesis
withoutaffecting
viral DNAsynthesis [18])
and were
exposed
to32p
to prelabel the nucleotidepools.
The cellsweretheninfected with
PrV(1)33
and incubated inBUdR medium
containing
32P.(BUdR,
adensity label,
was addedtotheculturetoallow thedifferentiationbetween viral DNA that hadreplicated
and viral DNA that had become labeledby
repair synthesis only.)
At various times afterinfection,
someof the cultureswereharvested,
and the DNA was extracted and banded in CsClgradients along
with[3H]thymidine-containing
and[3H]BUdR-substituted
viral DNA as markers. DNA fractions werecollected,
and theposition
in thegradient
of32P-labeled
viral DNAwas deter-mined. The DNAbanding
with the[3H]thymidine-labeled
viral DNA and the DNA that had shifted to aposition
ofhigher density,
i.e., the DNA that hadreplicated,
were collectedseparately.
The DNApreparations
werepurified,
digested with BamHI restriction enzyme, and
electropho-resed in agarose
gels.
Figure
2 showsthatBamHIfragments
10aand 12b[which
arethe
fragments
characteristic ofPrV(1)33-defective
DNA]
hadreplicated
preferentially (lane 2) by
3 h 45 min postin-fection. Eventhough
these cells had been coinfected withon November 10, 2019 by guest
http://jvi.asm.org/
Pr(1)24
A B
Pr(1)33
A B
1
_u
1 _
2 _ 2 _f _
3 - _ 3 _ _0
4 =.-.;.
6 I4 mm 4 _
7 - -l ,o5 4tU
5 ,
tu:r
6~~._
8 ~ ~ ~ ~ m 6 Mo
9~~~~~~~~~~~~~~~~~~~~~~
10 ~ ~ ~
1~~~~~~~ ~0
~~~~~~~~
.d
loa
a1
t1 vY .). 1~~10 ; ]t 1 * 101
12
I
11I
.-:...;:.l* 12b
12b
FIG. 1. Comparison of the BamHl restrictionpatternsofvirion DNA with the total intracellular viral DNA. 32P-labeled virion DNA (A) andintracellular viral DNA (B)werepurified fromcells infected either with PrV(1)atpassagelevels24, 33, 43, and 64orwith standard PrV
[Pr(s)] asdescribed in Materials and Methods. An equal amountof32P-labeled DNA obtained from virions and from infected cellswas
digested with BamHI and electrophoresed inagarosegels, and autoradiogramswereprepared. The numbers onthe left side of each lane
designate the standard PrV BamHI fragments; the numbersontheright side designate the defective DNAfragments, i.e., the fragments that
arenotgeneratedby digestion of standardPrV DNA. Hindicatesaregion ofheterogeneity.
helper virus, fragments representative of the helper PrV
genome could barely be detected in the DNA that had
replicated (lane 2), i.e., had shifted to a position ofhigher
buoyant density as a result ofincorporation ofBUdR
(nor-mal thymidine-containing DIP DNA has a lower buoyant
density than doesstandard PrVDNA [4]). Inthis particular experimentasignificant amountof repair synthesishadalso occurred (lane 1);however, inotherexperiments much less repairsynthesiswas observed (data not shown). We donot know the reasonforthis variation.
Figure 3 shows the microdensitometer tracings of autora-diograms obtained from a similar experiment in which the
32P-labeled viral DNA that had accumulated in the infected cells at different times after infection was analyzed. The
results show that the defective DNA, exemplified by frag-ment 12b, replicated first and, in fact, was the prominent
species synthesized up to approximately 6 h postinfection. By 24 h postinfection, however, this band was much less
prominent,andtherestrictionpatternof the32P-labeled viral DNA that had accumulated in the cells resembled the restrictionpatternof the population of virionscontainingthe DIPs that had been usedtoinfectthe cells. We concludethat PrV(1)-defective DNA contains origins of replication that
are recognized at early times after infection by the DNA-synthesizing machinery of the infectedcells. Thereplicative advantage of the DIP DNA is,however, only transient.
The 32P-labeled DNAaccumulatingin cells infected with DIPs at various times after infection was also analyzed by
theSoutherntechnique(25).Themicrodensitometertracings of the hybridization pattern of this DNAtoKpnI fragments of standard PrV DNA fixed to nitrocellulose filters are
shown in Fig. 4.(See Fig. 6 formappositions ofrestriction
fragments.) Viral DNAsynthesized during the firstround(s) of replication hybridized exclusively to KpnI fragments B
and D (middle panel). (Further exposure did not reveal hybridization to any of the other KpnI bands.) By 24 h postinfection, the DNA synthesized by thecells hybridized toall KpnI fragments (toppanel), and the patternof hybrid-ization mimicked that of the DNA in the population of DIPs used for infection (see also reference 22). These results corroborate the conclusion that the DIP DNA replicates preferentially at early stages of infection in cells infected with a virion population which contains both DIPs and
helper virus [PrV(1)33].
Structural modification ofgenomesoftheinfectiousvirions
present in the populations of DIPs. The results described above indicate that although defective DNApresent in the population of DIPs has a replicative advantage (at early
stages ofinfection), itdoes not appearto posses an
advan-tagein encapsidation overthegenomesof the nondefective
virionspresentinthevirionpopulation (Fig. 1). The lack of preferential encapsidation of the defective DNA could be due to a lack of enrichment ofthis DNA in cis functions
required for encapsidation. On the other hand, it could be
dueto thepresence in thepopulation of DIPs of infectious
virus that has been modified and has acquired the abilityto compete effectively with DIPDNA, eventhough the latter has been enriched for cleavage-encapsidation sites. The
emergenceinapopulationofDIPs ofinfectious virionsthat,
as aresult ofsomemodification, haveacquiredtheabilityto
overcomethe selective advantages of the genomesofDIPs has been documented in other systems(10, 14-16).
To determine whether the genomes of the infectious
virions presentin thepopulation of DIPs ofPrVhave been modified, we analyzed (as a first step) the structure ofthe
genomes of individual plaque-purified infectious virions
ob-tained from the two independently derived populations of DIPs that we have been studying. Figure 5 illustrates the
Pr(1)43
A B Pr(1)64
A B
5.5
6 7
Pr(s)
A B
_UU
2 _&
-3_ _
4 -l _
56s l 7 _tWk
.8 _ _
98 _"f
9 _SO. _
10 w.
11 .,}- ---12
2. _
3 s m
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.138.483.74.321.2]w ..
-.
-U.~~.
7 . ,
8 0o
8
or
9
10 -.
IOIF M._
1 1
', 10a
12
*
~~~~~~I
1FIG. 2. Defective DNA replicates first in cells infected with a population ofPrV(1) DIPs.Cells wereincubatedin EDS-PO4+FU (toinhibit the synthesis of cellular DNA) and further incubated in EDS-PO4+FU containing32ptoequilibrate the nucleotide pools, as describedin Materials and Methods. The cells were then infected withstandard PrV or PrV(1)33 and were incubated in BUdR medium containing32pUp to3.75hpostinfection.The cells wereharvested, and the DNA waspurified by centrifugation inCsCI gradients, as described in Materials and Methods. The purified viral DNA was digested withBamHI and electrophoresed in agarose gels, and an autoradiogram wasprepared. Lanes: 1, viral DNAobtained from cells infected with DIPs that had banded in the characteristic position ofPrV DNA(6 days ofexposure); 2,viralDNAobtained from cells infected with DIPs that banded in a position of higher density (DIP DNA normally has a lower buoyant density than standard PrV DNA), i.e., that hadincorporated BUdR (6days of exposure); Pr, viralDNAobtainedfromcellsinfected with standard PrV that banded in a position of higher density, i.e., that had incorporated BUdR(2days ofexposure). M indicates the position of mitochondrial DNA, the synthesis of which is not completely inhibited by preincubation ofthe cells with FU.
Kpn digest of the DNA in the virions of10 representative
plaqueisolates obtained from PrV(1)42. Of interest was the
finding that in many cases a ladderlike series of submolar bands was observed; Kpnl end fragment D(Fig. 6), which normally ispresentinmolar amounts in standard DNA, was
in certain populations of virions either nondetectable or
considerably
reduced inintensity.
Severalrestrictiondigests of the DNA of individual plaque isolates were analyzed by the Southern technique (25) with
individual cloned nick-translated restriction fragments
en-compassing the entire PrV genome as probes, and the genomes of these plaque isolates were mapped. The results showed that although only occasional modifications were observed along various parts of the genome of theseplaque isolates, more than 75% had genomes in which sequences
fromthe left end of the genome had become reiterated. The
ladderlike arrays of bands derived from the left end of the genome that were observed in the restriction digests were
probably the result ofunequal (out-of-register)
recombina-tion, a process that occurs when sequences are tandemly
reiterated.
Examples of the hybridization pattern of the BamHI
digestsof the DNA of someplaque isolatewith someprobes
of interest areillustrated in Fig. 7. Digestion of the DNAof
plaque isolate B7 generated a series of bands which hybrid-izedtocloned BamHIfragment 14' only (i.e., the left end of the genome; Fig. 6), indicating that sequences from the left
end of the genome, which did notincludethe BamHIcutting
site, hadbecome reiterated. On the other hand, digestion of the DNAof plaque isolate All generated a multimolarband
at approximately the same molecular weight as that of
BamHI fragment 14' that was generated from the standard virus DNA. This fragment hybridized to sequences present in BamHI fragment 14' only, indicating that the segment of DNA thathad become reiterated consistedofthe sequences ofBamHI fragment 14' as wellas some adjacentsequences
DIP
12 b I 3.5h
Z3
4 5.5:7 8,8'9,10
11 12 13 14'6
FlG. 3. Preferential replication of defective DNA up to 6 h postinfection. Cells were incubated in EDS-PO4+FU and further incubated inEDS-PO4+ FU,asdescribedinthelegendtoFig.2.The cells were then infected with PrV(1)33 and further incubated in EDS-PO4+FU containing 32p. The cells were harvested at the indicatedtimes,and the DNAwaspurifiedanddigestedwith BamHI andelectrophoresedin agarosegels. Autoradiogramswereprepared after an appropriate exposure time and scanned to illustrate the relative abundance of the various restrictionfragments.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.326.515.293.645.2]including theBamHI cleavage site. In other plaque isolates, as, for example, isolate A9, the reiteration not only com-prised the extreme left of the genome(i.e.,BamHIfragment 14'), but also included significant stretches of sequences derived from the adjacent BamHI 5' fragment. For isolate A9, the BamHI cleavage sites between the sequences in BamHI fragments 14' and 5' were lost. A multimolar
frag-menthybridizing to both BamHI fragment 14' and BamHI
fragment 5' was consequently formed. (Isolate A9 also contains inverted repeats of unequal sizes, as indicated by
thehybridizationpatternof itsDNAtoBamHIfragment 8'.)
Thus, the size of thefragment of DNA originating from the left endofthe genome that isreiterateddiffers in thedifferent
virus isolates. This may also be deduced from the increments in the number of nucleotides between the rungs of the ladder observed in theKpnI digest (Fig. 5). Indeed, in some cases,
KpnI band D appeared only as adiffuse smear. Analysis of the genomes ofmorethan 100 individual plaque isolates of
PrV(1)42 showed that the reiterations at theleft end ofthe genome varied in size from less than 300 to at least 1,500 base pairs (data not shown). The reiterations always in-cluded sequences derived from the extreme left end of the
B
D
B
DIP 24h
DIP
A B C D E FGCA- I J K
FIG. 4. Microdensitometer tracings ofthehybridization pattern
of viralDNAsynthesized by cells infected with PrV(1)33. Cellswere
infected with PrV(1)33, as described in the legend to Fig. 3, and harvested at the indicated times. The DNA was purified and
hybridizedtoKpnl restriction fragmentsofstandardPrVDNA that
had been fixed to nitrocellulose filter strips. The autoradiograms
were scannedafter appropriate exposure times. The hybridization
[image:5.612.334.567.75.265.2] [image:5.612.110.277.317.645.2]patternof32P-labeled PrV DNAtotheKpnIfragmentsisalsoshown
[Pr(s)].
A21 A22 A23 A24 A25 A26 A27 A28 A29 A30
FIG. 5. KpnI restriction patterns of the genomes of different plaque isolates obtained from a population of virions containing DIPs. Individual plaques (A21 through A30) formed by infectious virions present in a population containing DIPs [PrV(1)33] were picked, and the virionswereamplified. ViralDNA waspurified and digested withKpnI. KpnIfragmentDisthefragment generatedfrom the left endofthegenome (see map in Fig. 6).
genome (BamHI fragment 14'). Similar results were also
obtainedwhenplaque isolates obtained fromPrV(2)42were analyzed (8).
The proportion of infectious virus genomes that had
reiterated ends in populations of DIPs was determined at several passage levels. Infectious virionspresentinPrV(1)at passages33, 43, and 53wereplaque purified,and 20plaques wereanalyzedin each case. No significant differences in the
relative numberof plaque isolates containinggenomeswith
reiterated ends were found; in all three cases, between 70 and85% ofthe plaques tested had genomes with reiterated
ends. Thus, the reiterations ofthe left end ofthe genome wereretained upon passageof the virions in the presenceof
DIPs. On the other hand, when individual plaque isolates
with reiterated end sequences were passaged in cell cultures in the absenceof DIPs, the genomes of some ofthe virions in the progenyacquired nonreiterated ends(Table 1). Thus,
it appears that the reiterated sequences atthe endoftheUL
donotconferonthe virionsanyreplicativeadvantage in the absence ofDIPs.
Underproduction of virions with nonreiterated end se-quencesby cellscoinfectedwith standardvirus and a popula-tion containing DIPs. Because
approximately
75% of the infectious virions present in the population of DIPs haverepetitive sequences at the left end of theirgenomes, these infectious virionscan be distinguished from standard infec-tious virions. It was possible, therefore, to determine whether standard virus would be amplified in unmodified
form as efficiently as was the endogenous infectious virus that was present in a population of DIPs when cells were coinfected with both.
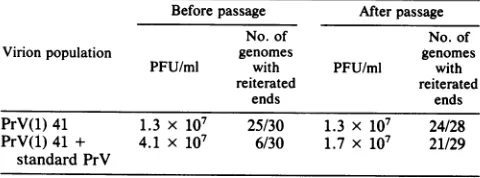
Table2summarizes the results of this experiment. In the
original population of DIPs, PrV(1)42, 25 of 30 plaque
isolates (83%) had genomeswith reiterated ends. After the
population of DIPs was mixed with standard virions, the relative number of plaque isolates with genomes that had reiterated ends decreased, as expected, and only 6 of 30
plaque isolates (20%) were found to have reiterated ends.
on November 10, 2019 by guest
http://jvi.asm.org/
15 14 12
* 5- * 2 *9,11* 4 * 3 * 6 #6*'S6 5 *10 77 * 5 IS 13
L m N M
D * C *A JIGG * f Er K # J KiIt1 H
UL IRs U3 TR5
I s s|I s l k s s s & | t | ffi | s s IAA A aIIIaa IIa I I I I III a I A I iIII.1II
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1.0
[image:6.612.66.528.75.186.2]MAP UNITS
FIG. 6. Restrictionmapsof the PrVgenome. Arrows indicate cleavage sites. Rectanglesrepresent theinvertedrepeats.
However,after thispopulation of virionswas passaged five
times at high multiplicity, most (21 of 29, or 72%) of the
plaque isolates hadreiterated ends, indicating that the stan-dard virus with thenonreiterated ends that had been added to the population of DIPs tended to be lost upon passage.
The disappearance of virus with genomes with standard nonreiterated ends observed in this experiment could be the result ofareplicative advantage of the virions with reiterated
ends thatareendogenouslypresentinthepopulationof DIPs
over standardvirions. On the otherhand, thestandard viral
genomes mayhaveacquired the reiterationsattheend ofthe
unique long(UL) segment as aresult ofrecombinationwith
thevirions presentin the population of DIPs.
To distinguish between these two possibilities, i.e., to ascertain whether in cellscoinfectedwithDIPs theDNA of standard viruscancompeteeffectively foramplification, we
compared (i) the relativeamounts of viral DNAsynthesized by cells either infected with standard virus alone or
coinfected with standard virus and DIPs that became
asso-ciated with mature virions (Table 3), and (ii) the relative
All
probes 5' 8 14' Pr
^.2
3
. 5'i
'I,j. 11 5
12
13
14'
87
5 8' 14 Pr
123
40
Ho
S& 0 Sisom
j 88.* .11
12
amountsofparental standard PrV DNA that, when addedto cells infected with these virion populations, replicated and becameassociated with mature virions (Table 4).
The amount of viral DNA synthesized by cells infected with standard PrV alone and by cells coinfected with stan-dard and defective virions was approximately the same
(Table 3). However, the number of virionsproducedby the cells coinfected with the DIPs(as determined bytheamount of labeled DNA that became associated withpreparationsof purified virions) was reduced by almost 90%. Thus, in this experiment, 22.6% of the total viral DNA synthesized by cells infected with standard virus alone butonly 3.1% of the viral DNA synthesized by cells coinfected with DIPs and standard viruswasfoundinthepurified virion preparations.
This finding confirms previously published reports (2, 4) which showed that the number of virions produced by the cells coinfected withDIPs ofPrV is reducedconsiderably.
To determine whether standard viral genomes can
com-pete at the level ofreplication and encapsidation with the viral genomes in the population of DIPs, the fate of
B5C 5' 8 8' 14 Pr
1
3
mm 4
*_ .6a7
0. 8
10
A9
5 8 8 2 14
a
4.
0
1112
to 4
06
A* 7
-wa
8
9 10
12
13
A* 14
.<+d 1 3
13
4
FIG. 7. Hybridizationofsomenick-translatedBamHI restrictionfragmentsof the PrV standardgenometotheBamHI restrictiondigests
of theDNA ofdifferentplaqueisolates. Thegenomesoftheindicatedplaqueisolates(All, B7, B5C,andA9)weredigestedwithBamHIand
electrophoresed,andtheDNAfragmentsweretransferredtonitrocellulosepaper.Stripswereprobedwithcloned,nick-translated restriction
fragments originating fromthefragmentencompassing theentirePrV genome;however,the results obtained withsome of the probes of
interestonly(BamHI fragments 5', 8',and 14') arepresented. Because of theheterogeneityofsomerestrictionfragments, allofthe viral
isolates inthisexperimentwereplaque purified again;the results obtained wereidentical.
14
Bus HI
Kpa
0 A& 14'+5'
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.112.492.432.675.2]TABLE 1. Reversion ofvirionscontaininggenomeswith
reiterated endsto virions withnonreiterated end fragmentsa No. of virions
withgenomes
Plaqueisolate containing
standard nonreiterated
ends
B1... 0/8"
B5... 6/9
B7... 2/9
B12... 0/3
All ... 9/12
aTheprogenyofsomeindividual plaque isolates obtained from populations
ofDIPs which containedgenomeswithreiterated ends (Fig. 7)waspassaged
fivetimes in RKcellsatlowmultiplicity andthenplaqueassayed.Individual
plaques were picked, the virions were amplified, and theirgenomes were
analyzed with restriction enzymes. The number ofplaque isolates which
contained DNAwith standard nonreiterated endswasdetermined.
bNumerator, number of plaques yielding virions with genomes with
standardends;denominator, number of plaque isolates analyzed.
[3H]thymidine-labeled parental standard viral DNA in cells coinfected witheither unlabeled standard virus orunlabeled
DIPs was determined (Table 4). This experiment was
per-formed underexperimental conditionsthatwereidentical to those used in the experiments summarized in Table 3. The results of a representative experiment are summarized in
Table 4. This experiment was repeated three times with
essentially similar results.
Both in cellsinfected with standard virus alone and in cells coinfected with DIPs, most of the parental standard viral DNA had replicated by 8 h postinfection. The amount of 3H-labeled parental standard virus DNA that became
asso-ciated with progeny virions produced by cells coinfected with DIPs was reduced approximately 10-fold compared
with that produced by cells infected with standard virus alone (Table 4, column 4). Sinc,e the total number of virions produced by these infected cells also differedapproximately
10-fold (Table3), onemight conclude that the standard and
defective genomes compete equally well forencapsidation.
However, a large proportion of the [3H]thymidine-labeled
parental virus DNA that was present in the population of
progeny virions obtained from cells coinfected with DIPs
was DNA that had notreplicated in the infected cells (Fig. 8). This DNA was probably derived from input parental virions that hadnotbeen uncoated and thatcopurified with
theprogeny virionsproduced by the cells. Ifonecompares
TABLE 2. Loss ofvirions withgenomeswithstandard ends from
apopulationcontaining DIPs'
Beforepassage Afterpassage
No. of No. of
Virionpopulation genomes genomes
PFU/ml with PFU/ml with reiterated reiterated
ends ends
PrV(1)41 1.3 x 107 25/30 1.3 x 107 24/28
PrV(1)41 + 4.1 x 107 6/30 1.7 x 107 21/29
standard PrV
ToapopulationofDIPs[PrV(1)411,sufficientstandard PrV wasaddedto
increase the titerofinfectious virusapproximatelythreefold.Part of thevirion
populationwasplaqueassayed,andpartwaspassagedfive times inundiluted
form andthenplaque assayed. Thirtyindividual plaquesfrom eachvirion
population were picked; the virions were amplified, and the DNA was extractedandanalyzedwithrestrictionenzymesto determine thetypeof end
[image:7.612.328.569.106.159.2]fragments (standardorreiterated)thatwerepresent.
TABLE 3. Synthesisandencapsidationintovirionsofviral DNA in cells infected with standard PrV aloneorin cells coinfected
withstandard PrV andDIPsa
3H-CdRin 3H-CdRin ViaDN
Cellsinfected with: viral DNA purifiedvirions al DNA
(103cpm) (103 cpm) invirion (%)
PrV 97.5 21.9 22.6
PrV + PrV(1)42 73.2 2.8 3.1
a RK cells (4 x 106persample) were incubated in EDS-FU for 16 h (to
inhibit cellular DNA synthesis) and were then infected with either PrV alone
(5PFU percell)or amixture of DIPs(approximately300particles;2PFU per
cell)and standardPrV(3 PFU percell).Unadsorbed virus was removed after
1h, and the cellswereincubated in BUdR mediumcontaining 3H-CdR(50
RCi/ml)for 48 h.The totalamountof3H-CdRincorporatedinto viralDNA,as
wellasthe amountof3H-CdR-labeledDNA that hadbecomeencapsidated
intomaturevirions, was determined as described in Materials and Methods.
the amount of3H-labeled parental standard DNA that had
replicated and become part of virions produced by cells coinfected with DIPs with the amount produced by cells infected with standard virus alone, one is led to conclude that thestandard DNAwasdiscriminatedagainstatthe level ofencapsidationin cellscoinfected with DIPs.Thus, incells infected with standard virus alone, the same proportion of the total viral DNA (22.6%) and of the 3H-labeled parental
viral DNA that hadreplicated (24.0%) becameencapsidated
into virions (see Tables 3 and 4). In cells coinfected with
DIPs, 3.1% of the total viral DNA but only 0.9% of the 3H-labeled parental viral DNA that had replicated became encapsidated. These results show that standard viral DNA is discriminated against at the level ofencapsidation in cells coinfected with DIPs.
Figure 1 shows that the defective DNA does not appearto be preferentially encapsidated overthe nondefective DNA endogenously present in the population of DIPs. On the otherhand,Tables 3 and 4 indicate thatexogenously added standard PrV DNA is discriminated against at the level of encapsidation in cells coinfected withamixture of DIPs and thestandard virus.Together, these findings indicate that the infectious virions present in the population of DIPs are modified forms of the standard PrV genome in thatthey have acquired characteristics which allow themtocompete
effec-tively with DIPs for encapsidation. Furthermore, these results indicate that the defective DNA isprobably enriched forcleavage-encapsidationsignals, despite the apparent lack ofpreferential encapsidation of the defective DNAoverthe nondefective DNA present in the virion population.
DISCUSSION
The salient features of the results presented in this paper can be summarized as follows. In a population containing DIPs ofPrV, the defective DNA has a replicativeadvantage atearly stages of DNAreplication. The defective DNA does not, however, appear to have an advantage at the level of
cleavage-encapsidation and is not preferentially encapsid-ated over nondefective viral DNA present in the virion population.Despite the lack ofpreferentialencapsidation of the DIP DNA, it appears that this DNA is nevertheless enriched forcleavage-encapsidation sites. This conclusion is based on thefollowing observations. (i) The genomes of the infectious virions present in the population of DIPs have beenaltered;notably reiterations of sequences derived from the left end of the genomearepresentinapproximately75% of the genomes of these virions. (ii) While standard viral genomes arediscriminated againstat the levelof
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.70.310.584.673.2]TABLE 4. Replication and encapsidation of13H]thymidine-labeledstandardparental viral DNA in cellscoinfected withDIPsa 3H-Pr(s)b 3H-Pr(s)DNAthat [3H]Pr(s)in progenyDNA [3H]PrV DNA inprogenyvirions [3H]PrVDNA in
Cellscoinfected with: DNA in cells had replicated by 8 h irons tahdepicated progenyvinons that
(103cpm) (103
cpm)
(13o
cpm)(103cpm)
hadreplicated (%)(3H)PrV(S) + PrV(S) 27.6 19.55 5.49 4.70 24.0
(3H)PrV(S)+ PrV(1)42 22.2 14.16 0.59 0.13 0.9
aCells were coinfected with purified[3H]thymidine-labeled virions(3 PFU per cell)and either standard or defective unlabeled virions (2 PFU per cell). After 1
h, unadsorbed virus was removed and the cells were further incubated in BUdR medium. At 8 h postinfection, a portion of the cultures was harvested and the
DNAwasextracted and centrifuged in aCsCI gradient with 32P-labeled thymidine-containing or BUdR-substituted DNA as markers to determine the amount of
parental virus DNAthathadreplicated (column 3). The remainder of the cultures was incubated up to 48 h postinfection, when the virions produced by the cells
were purified. The total amount of parental3H-labeledPrV DNA(column 4) as well as the amount of DNA that had replicated in the infected cells (column5; see
also Fig. 8) that were transferred to the progeny virions was deterined.
bPr(s), StandardPrV.
c P.i.,Postinfection.
tion in cells coinfected with DIPs, the nondefective and
defective genomes present in these populations of virions
compete equally well for encapsidation. It appears,
there-fore, that both the nondefective and defective genomes in these viral populations have an advantage over standard virus at the level of encapsidation and probably are enriched
for cleavage-encapsidation sites.
Our results showthat thedefective DNAreplicates firstin
the infected cells when the synthesis of the nondefective virus DNA present in the population of DIPs is barely detectable. Therefore, the defective DNA in thepopulation of virions studied is
probably
enriched for sequences that can serve asorigins
ofreplication,
sequences that canfunction at early stages of infection and compete
success-fully with the origins present in the nondefective DNA. Vlazny and Frenkel (29) showed that the defective DNA present in a population of DIPs of HSV is synthesized mainly at late times after infection, i.e., is enriched for origins of
replication
thatdonotfunction wellatearlystages12.8 Pr
I+H L-L
9.6-
6.4-CY 3.2
0
a.i 3
Pr(1)42
3-
2-20 30 40 50
[image:8.612.52.551.93.153.2]FRACTION NUMBER
FIG. 8. Analysis ofthe3Hparentalviral DNA strands thatwere transferredtothe virus progenyproduced by cells coinfected with DIPs. RK cells were coinfected with either standard PrV or PrV(1)42 and [3H]thymidine-labeled standard PrV, asdescribed in thefootnotetoTable 4. The DNAoftheprogeny virionsproduced bythe cells (Table4, column 4) was centrifugedtoequilibrium in CsCl, together with 32P-labeled thymidine-containing DNA and 32P-labeled BUdR-substituted viral DNA as markers. Fractions were collected, and the distribution of label in the gradient was determined.The bottomofthegradientis at the left.
of infection. The differences between our results with PrV DIPsandthose of Vlazny and Frenkel (29) with HSV DIPs are probably due to an enrichment of different types of
sequences that can serve as origins of replication in the genomes ofthe two populations ofDIPs; it is unlikely that they reflect intrinsic differences between these two herpesviruses.
Normally, during the first roundof replication, PrV DNA
synthesis is initiated mainly at a site near or within the
inverted repeats(7). Southern hybridization analysis ofthe DNAsynthesized duringthefirstround ofreplication in cells infected withDIPsindicates that thesequences thatreplicate firstdo notincludesequenceslocated intheinvertedrepeats
(Fig. 4). These results might indicate that an origin other
than that intheinverted repeatsispresent in the DIP DNA
and is recognized at early stages of infection. However, because recombinational events occurredduring the
gener-ation of theDIPDNA,wecannotexcludethepossibility that the
origin
normallypresentinthe invertedrepeatshas beenjuxtaposed (by a recombinational event) next to the se-quencesin theDIP DNA thatreplicate
preferentially
atearlystages of infection. The presence of a small stretch of
sequencesderivedfrom the invertedrepeatswouldnothave been detected inourexperiment. Thus, althoughoutresults show that defective DNA is enriched for an origin of replication that canbe usedat early stagesofinfection, this origincannotbeconclusively identified inthe absenceof the appropriate sequencing data. The reason for the transient preferential synthesis only ofthe DIP DNAis alsonotclear.
As mentioned above,although ourresults show that DIP DNA replicatespreferentially under certain conditions,it is not, as far as we can tell, preferentially encapsidated. However, the lack ofpreferential encapsidation of the DIP DNA does not necessarily reflect a lack ofenrichment for
thecleavage-encapsidation site. Ithas beenestablishedthat the dynamics of the interactions between the DIPs and
infectious virions present in a population containing DIPs areaffectednotonlybythesequencecomposition ofthe DIP DNA but alsoby mutational changes in the genomes ofthe
endogenous infectious virions (10,
14-16).
Indeed, our re-sults show that a selection for infectious virions that haveacquired
characteristics
that promote their ability to com-pete with the DIP DNA has occurredduringtheevolution of thepopulation ofDIPs. Mostoftheseinfectiousvirions haveacquiredreiterations of theleftendofthegenome
(Fig.
5and 7); these reiterations tend to be lost upon passage of the virions in the absence of DIPs (Table 1). Furthermore, infectious virions containing standard PrV DNA with nonreiterated ends are discriminated against in cells coinfected with DIPs(Table 2), andalthough the DIP DNA and the endogenousnondefective DNA present in the DIPon November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.64.276.437.642.2]population compete equally well (Fig. 1), standard PrV
genomes compete poorly at the level of encapsidation in cells coinfected with apopulationofDIPs (Tables 3 and 4). Taken together, these dataindicate that both the defective DNA and thegenomesof the infectious virionspresentin the population of DIPshaveanadvantage in encapsidationover
the standard viral genome.
The segments of DNA derived from the left end ofthe
standard viral genomethatare reiterated in thegenomes of infectious virionspresentin thepopulation ofDIPs contain someofthe cisfunctions thatare necessary forthe efficient
cleavage-encapsidation of PrV DNA (30; C. Wu, Ph.D thesis, Vanderbilt-University, Nashville, Tenn., 1986). The possibility that the reiteration of these sequencesis linkedto the advantage of these genomes in encapsidation is attrac-tive. However, conclusive evidence that these reiterations
are the basis for the selective advantage of the modified
virions is not available; other, undetected, modifications in thegenomes may play a role.
The cyclical changes in the level of infectious virus obtained upon passage of populations ofDIPs at high mul-tiplicity that are observed in many other viral systems are
not observed upon similar passage of PrV DIP. Further-more, the accumulation of the DIP DNA in this system is slow (4). The reason for these characteristics is probably
related to the fact that the DIPs only have a slight growth
advantageoverthe infectious viruspresentin thepopulation of DIPs that we have studied. Their genomes have only a
transient replicative advantage at early stages of infection (Fig. 2 and 3), and a significant advantage at the level of encapsidation of DIP genomes over the genomes of the
infectious virions present in the population of DIPs is also notobserved (Fig. 1). Asalready mentioned,this is probably because modifications of the genomes of the endogenous
infectious virions have occurred that allow them tocompete
forencapsidation.
Despite the fact that the DIP DNA has only a slight
replicative advantage, the populations ofDIPs do interfere significantly with the growth of standard virus (2). This
interference can be ascribed to apreferential encapsidation
of the genomes present in the DIP population over that of
standard virus. It is also due to an interference with the
process of cleavage and encapsidation of viral DNA
(stan-dardas wellasdefective) in DIP-infected cells (Tables 3 and
4). Thus, even though viral protein synthesis (22) and the
production of capsids(unpublishedresults)arenotmarkedly affected,thetotalnumberof viral particlesproducedand the cleavage of concatemericDNAis reduced byapproximately 90% in cells coinfected with DIPs (4). The reduction in cleavage and encapsidationof the virusmaybethe result of
an interference by the DIPs with the orderly expression of
the virus genome. On the other hand, it may be due to the direct inhibitory effect ofthe DIPgenomes on the
cleavage-encapsidation process as a consequence of the sequence
organization of the DIP DNA. This question remains to be clarified.
ACKNOWLEDGMENTS
This investigation was supported by Public Health Servicegrant
AI-10947from theNational InstitutesofHealth.
Weappreciate theexcellent technical assistance of PatComers.
LITERATURE CITED
1. Barnett, J. W., D. A. Eppstein, andH. W. Chan. 1983. Class I
defective herpes simplex virus DNA as a molecular cloning
vehicle in eucaryotic cells. J. Virol. 48:384-395.
2. Ben-Porat, T., J. M. DeMarchi, and A. S. Kaplan. 1974. Char-acterizationof defective interfering viral particles present ina population ofpseudorabies virions. Virology 61:29-37. 3. Ben-Porat, T., and A. S. Kaplan. 1963. Thesynthesisand fate of
pseudorabies virus DNA in infected mammalian cells in the stationary phaseof growth. Virology 20:310-317.
4. Ben-Porat, T., and A. S. Kaplan. 1976. A comparison of two populations of defective, interfering pseudorabies virus parti-cles.Virology 72:471-479.
5. Ben-Porat, T.,and F.J.Rixon.1979. Replication of herpesvirus DNA. IV. Analysis of concatemers. Virology 94:61-70. 6. Ben-Porat, T., B. Stehn, and A. S. Kaplan.1976. Fateof parental
herpesvirus DNA. Virology 71:412-422.
7. Ben-Porat, T., and R. A. Veach. 1980. Origin ofreplication of the DNA of a herpesvirus (pseudorabies). Proc. Natl. Acad. Sci. USA 77:172-175.
8. Ben-Porat, T., C. Wu, L. Harper, and B. Lomniczi. 1984. Biological significance of the alterations in restriction patterns of the genomes of different isolates of pseudorabies virus. UCLASymp. Mol. Cell. Biol.21:537-550.
9. Eagle, H. 1959. Amino acid metabolism in mammalian cell cultures. Science 130:432-437.
10. Enea, V., and N. D. Zinder. 1982. Interferenceresistantmutants of phage II. Virology 122:222-226.
11. Frenkel, N., R. J. Jacob, R. W. Honess, G. S. Hayward, H. Locker, and B. Roizman. 1975. Anatomyof herpessimplex virus DNA. III. Characterization ofdefective DNA molecules and biological properties of virus populations containing them. J. Virol. 16:153-167.
12. Frenkel, N., H. Locker, W. Batterson, G. S. Hayward, and B. Roizman. 1976. Anatomy ofherpes simplex virus DNA. VI. Defective DNA originates from the S component. J. Virol. 20:527-531.
13. Henry, B. E., W. W. Newcomb, and D. J. O'Callaghan. 1979. Biological and biochemical properties of defective interfering particles ofequine herpesvirus type 1.Virology 92:495-506. 14. Horiuchi, K. 1983. Co-evolutionof afilamentousbacteriophage
and its defectiveinterfering particles. J.Mol.Biol. 169:389-407. 15. Horodyski,F. M., and J. J. Holland. 1980. Virusesisolated from cells persistently infected with vesicular stomatitis virus show altered interactionswithdefective interfering particles.J. Virol. 36:627-631.
16. Jacobson, S., and C. J. Pfau. 1980. Viral pathogenesis and resistance to defective interfering particles. Nature (London) 283:311-313.
17. Kaplan, A.S.1964. Studies on thereplicatingpoolof viralDNA in cells infected with pseudorabies virus. Virology 24:19-25. 18. Kaplan, A. S., and T. Ben-Porat. 1961. The action of
5-fluorouracil on the nucleic acid metabolism of psedurabies virus-infected and non-infected rabbit kidney cells. Virology 13:78-92.
19. Kaplan, A. S., and A. E. Vatter. 1959. A comparison of herpes simplex and pseudorabies viruses. Virology 7:394-407. 20. Ladin, B. F., S. Ihara, H. Hampl, and T. Ben-Porat. 1982.
Pathway ofassembly ofherpesvirus capsids: ananalysis using DNA' temperature-sensitive mutants of pseudorabies virus. Virology 116:544-561.
21. Rigby, P. W. J.,M. Dieckmann, C.Rhodes, and P. Berg. 1977. Labelingdeoxyribonucleic acid tohighspecific activity in vitro by nick translation with DNA polymerase. J. Mol. Biol. 113:237-251.
22. Rixon, F. J., and T.Ben-Porat. 1979. Structural evolution ofthe DNA of pseudorabies defective viral particles. Virology 97:151-163.
23. Rixon, F. J., L. T.Feldman,and T.Ben-Porat. 1980.Expression of the genome of defective interfering pseudorabies virions in
the presence orabsence of helper functions provided by stan-dard virus. J. Gen. Virol. 46:119-138.
24. Rubenstein, A. S., and A. S. Kaplan. 1975. Electronmicroscopic studies of the DNA of defective and standard pseudorabies virions. Virology66:385-392.
25. Southern, E. M. 1975. Detection ofspecific sequences among DNAfragments separatedbygelelectrophoresis. J.Mol. Biol.
on November 10, 2019 by guest
http://jvi.asm.org/
98:503-517.
26. Spaete, R. R., and N. Frenkel. 1982. The herpes simplex virus amplicon: a neweucaryotic defective-virus cloning-amplifying
vector. Cell30:295-304.
27. Spaete,R. R., and N. Frenkel. 1985. The herpes simplex virus amplicon: analyses of cis-acting replication functions. Proc. Natl. Acad. Sci. USA82:694-698.
28. Stegmann, B.,H.Zentgraf, A.Ott, and C. H.Schroeder. 1978. Synthesis and packaging of herpes simplex virus DNA in the
course of virus passage at high multiplicity. Intervirology 10:228-240.
29. Vlazny, D. A., and N. Frenkel. 1981. Replication of herpes
simplex virus DNA: localization of replication recognition sig-nals within defective virus genomes. Proc. Natl. Acad. Sci. USA78:742-746.
30. Wu, C. A., L. Harper, and T. Ben-Porat. 1986. cis functions involved inreplication and cleavage-encapsidation of pseudora-bies virus. J. Virol. 59:318-327
![FIG.1.anddigesteddesignateare[Pr(s)] Comparison of the BamHl restriction patterns of virion DNA with the total intracellular viral DNA](https://thumb-us.123doks.com/thumbv2/123dok_us/1377342.90974/3.612.138.483.74.321/anddigesteddesignateare-comparison-bamhl-restriction-patterns-virion-total-intracellular.webp)



![TABLE 4. Replication and encapsidation of 13H] thymidine-labeled standard parental viral DNA in cells coinfected with DIPsa](https://thumb-us.123doks.com/thumbv2/123dok_us/1377342.90974/8.612.64.276.437.642/table-replication-encapsidation-thymidine-labeled-standard-parental-coinfected.webp)
