of the Phosphatidylinositol 3-Kinase/Akt/mTOR Pathway and
Inhibition of Autophagy
Zurab Surviladze, Rosa T. Sterk, Sergio A. DeHaro, Michelle A. Ozbun
Department of Molecular Genetics & Microbiology, University of New Mexico School of Medicine, Albuquerque, New Mexico, USA
The mammalian target of rapamycin (mTOR) downstream of phosphatidylinositol 3-kinase (PI3K) in the growth factor receptor (GFR) pathway is a crucial metabolic sensor that integrates growth factor signals in cells. We recently showed that human papil-lomavirus (HPV) type 16 exposure activates signaling from GFRs in human keratinocytes. Thus, we predicted that the virus would induce the PI3K/mTOR pathway upon interaction with host cells. We detected activation of Akt and mTOR several min-utes following exposure of human keratinocytes to HPV type 16 (HPV16) pseudovirions. Activated mTOR induced phosphoryla-tion of the mTOR complex 1 substrates 4E-BP1 and S6K, which led to inducphosphoryla-tion of the funcphosphoryla-tional protein translaphosphoryla-tional machin-ery. Blockade of epidermal GFR (EGFR) signaling revealed that each of these events is at least partially dependent upon EGFR activation. Importantly, activation of PI3K/Akt/mTOR signaling inhibited autophagy in the early stages of virus-host cell inter-action. Biochemical and genetic approaches revealed critical roles for mTOR activation and autophagy suppression in HPV16 early infection events. In summary, the HPV-host cell interaction stimulates the PI3K/Akt/mTOR pathway and inhibits au-tophagy, and in combination these events benefit virus infection.
L
ike many pathogens, human papillomavirus (HPV) entry into target cells is initiated by binding to cell surface heparan-sul-fonated proteoglycans (HSPGs). The virus must then move to secondary receptors, which are responsible for particle internal-ization. Recently we showed that after interaction with HSPGs, HPV in complex with HS and growth factors (GFs) interacts with GF receptors (GFRs) and induces rapid activation of their path-ways (1). Such receptors are often activated by viruses (2); the signals may be used to deceive the host’s defenses, allowing safe entrance into the cell.GFR activation triggers the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling cascade, which is involved in con-trolling cellular macromolecular synthesis, metabolism, growth, and survival. Activated PI3K induces the conversion of phospha-tidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), which recruits downstream factors to the cell membrane and regulates their activity (3). Akt is a key member of this pathway. PIP3anchors Akt to the plasma mem-brane, allowing its activation by phosphorylation. PIP3 concentra-tion is tightly controlled. The phosphatase PTEN negatively regu-lates PIP3 concentration, converting PIP3 to PIP2 and thereby inhibiting PIP3-mediated downstream signaling, including Akt activation. To prolong the infection cycle, viruses attempt to in-hibit apoptosis and have developed several ways to activate Akt by enhancing the functions of the PI3K upstream regulator or by inhibiting negative regulatory phosphatases, or both (4,5).
Akt’s downstream effector, mTOR, is a crucial metabolic sen-sor, integrating diverse cellular signals that play critical roles in regulating many pathophysiological processes. This evolution-arily conserved serine/threonine protein kinase functions as a component of two structurally and functionally distinct signaling complexes: mTORC1 and mTORC2 (6). mTORC1 is activated by GFs and nutrients, regulates protein translation and cell growth, and plays an important role in the control of lipid synthesis (7) and mitochondrial metabolism (8). The best-characterized targets
of mTORC1 are components of the translation machinery, in-cluding eukaryotic initiation factor (eIF)-4E-binding protein 1 (4E-BP1) and 40S ribosomal protein S6 kinase 1 (S6K1), both of which are important in the control of translation initiation (9). Sustaining the activity of mTORC1 is essential for continuing cap-dependent translation; therefore, viruses that rely on cap-depen-dent translation have acquired ways to prolong mTOR kinase ac-tivity (4). mTORC1 signaling can be potently inhibited by the naturally occurring antifungal macrolide rapamycin, which acts as an allosteric inhibitor (10) but does not completely inhibit mTORC1 activity (12). For this reason, PP242 and torin, recently discovered specific inhibitors capable of binding the catalytic site of mTORC1, are more widely used (14). The growth factor-sen-sitive but nutrient-insenfactor-sen-sitive mTORC2 phosphorylates Akt, SGK1, and PKC (12). These so-called “AGC group kinases” con-trol multiple cellular functions, such as the structure of the actin cytoskeleton and cell survival (13–15). In contrast to mTORC1, mTORC2 is resistant to acute rapamycin treatment. Recent stud-ies show that both mTORC1 and mTORC2 are involved in the regulation of autophagy (16,17).
Autophagy is a tightly regulated cellular process responsible for eliminating damaged organelles, cell membranes, and proteins via a lysosomal pathway. Cell stress and diseases can also trigger this process. Cell autophagic machinery is known to capture and de-grade intracellular pathogens (xenophagy in this case); this is an important component of the host response against viral infections
Received27 August 2012 Accepted7 December 2012 Published ahead of print19 December 2012
Address correspondence to Michelle A. Ozbun, mozbun@salud.unm.edu, or Zurab Surviladze, zsurviladze@salud.unm.edu.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02319-12
on November 7, 2019 by guest
http://jvi.asm.org/
(18). Therefore, many viruses have developed means to block au-tophagy or subvert this machinery (19).
HPVs regulate the PI3K/Akt/mTOR pathway, as do many other DNA viruses (5). HPV early proteins directly activate Akt and mTOR complexes (19–22). Here, we tested the activation of the PI3K/Akt/mTOR pathway upon keratinocyte exposure to HPV type 16 (HPV16) pseudovirions (PsVs), which deliver a re-porter plasmid (pseudogenome) to the cell but express no viral proteins. HPV contact induces rapid activation of several signal-ing pathways in host cells (23–25), including that of PI3K/Akt potentially via alpha-6 beta-4 integrins (26). We recently demon-strated activation of epidermal GFR (EGFR) and keratinocyte GFR (KGFR) pathways in HaCaT cells exposed to HPV16 and 31 PsVs (1). Here we show that upon HPV16 PsV exposure to kera-tinocytes, EGFR signals result in rapid Akt activation by phos-phorylation and by inactivation of PTEN. As a result, downstream events of this pathway were initiated: activation of mTOR and its substrates and suppression of autophagy. Biochemical and genetic approaches confirmed that these actions benefit virus infection.
MATERIALS AND METHODS
Cell culture, transfection, virus production, and infection quantifica-tion.HaCaT and 293T cells were maintained as reported previously (1). HPV PsVs encapsidating a luciferase reporter plasmid were generated via transfection of 293T cells, CsCl gradient purified, and quantified for viral genome equivalents (vge) as previously described (27). Specifically, PsV preparations were isolated from a discrete and visible buoyant density band following CsCl gradient centrifugation. PsVs were desalted into HSB buffer (25 mM HEPES [pH 7.5], 0.5 M NaCl, 1 mM MgCl2) using Amicon Ultra-4 100,000 MWCO centrifugation filter units (Millipore). PsV purity and L1 protein content were determined by SDS-PAGE and Coomassie brilliant blue staining against bovine serum albumin (BSA) standards (Fig. 1A). Virion morphology and quality were visualized by negative staining with 2% uranyl acetate and transmission electron microscopy (Hitachi 7500) at 80 kV following adsorption to a carbon-coated electron microscopy grid (Fig. 1B). Capsids reproducibly outnumber vge 2- to 10-fold (28). Infections were performed as reported earlier (see below) and allowed to proceed at 37°C, typically for 24 h before luciferase quan-tification (1). Luciferase quantities were normalized to protein content. For small interfering RNA (siRNA) depletions, HaCaT cells (20 to 25% confluence) were transfected with siRNA using Lipofectamine 2000 (In-vitrogen). siRNAs targeting mTOR (Cell Signaling) or Beclin 1 or Atg7 (Dharmacon) were used according to the manufacturer’s recommenda-tions. A nonspecific siRNA was used as a negative control (Dharmacon). All infections were initiated using HPV16 PsVs at 100 to 200 vge/cell.
Biochemical inhibition of infection and signaling.For detection of phosphorylation, subconfluent HaCaT cells were serum starved for 4 h in Tyrode’s buffer (10 mM HEPES [pH 7.4], 130 mM NaCl, 5 mM KCl, 1.4 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, and 0.05% BSA) and then were pretreated for 45 to 60 min with the indicated concentrations (see figure legends) of inhibitors in Tyrode’s buffer: PP2 (Tocris Bioscience), PP242 (Cayman Chemical), LY294004 (Cell Signaling), AG1478 and PD168393 (Calbiochem), wortmannin, genistein, and rapamycin (Sigma). In exper-iments for detection of infection, inhibitors were dissolved in complete medium (Dulbecco’s modified Eagle medium [DMEM] containing 10% fetal calf serum [FCS]) and incubated for 45 to 60 min with HaCaT cells grown in complete medium. HPV16 PsVs were added to inhibitor-pre-treated cells, and infections proceeded in the presence of the inhibitors at 37°C, 24 h before luciferase quantification or for the indicated times prior to harvesting. Cell viability was assessed with each treatment by trypan blue exclusion staining.
SDS-PAGE and immunoblotting.After addition of HPV16 PsVs, cells were incubated at 37°C for various times before being transferred to ice and solubilized with RIPA buffer (50 mM Tris [pH 7.5], 0.1% SDS, 1%
sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride [PMSF], 10 ng/ml leupeptin, 10 ng/ml aprotinin). Lysates were processed and ana-lyzed by SDS-PAGE and proteins transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were probed with various monoclonal and polyclonal antibodies purchased from Cell Signaling: p-Akt (Ser-473), p-Akt (Thr-308), p-mTOR (Ser-2448), p-mTOR (Ser2481), mTOR, eIF4G, p-p70 S6K (Thr-389), p-p70S6K (Ser-371), p-PTEN (Ser-380), p-4E-BP1 (Thr-37/46), 4E-BP1, and LC3B. Other antibodies included PTEN (Abcam) and actin (MP Biomedical). Horseradish peroxidase-la-beled secondary antibodies were used to detect antigen-antibody interac-tions (Pierce, Amersham Pharmacia Biotech). Immunoreactive bands were visualized by Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific). For loading control analysis, blots were stripped and incubated with antibodies to nonphosphorylated proteins or actin. The bands were scanned and quantified using AlphaEaseFC (Alpha Innotech).
Immunofluorescence staining and confocal microscopy. Immuno-fluorescence assays were performed essentially as described previously (1) using primary rabbit antibodies against p-PTEN (Ser-380), p-Akt (Thr-308), and p-Akt (Ser-473) (Cell Signaling) (1:150 dilution in 1% BSA– Tyrode) and secondary DyLight 488-conjugated Affinity Pure donkey an-ti-rabbit IgG (1:200; Jackson Immunochem.). For actin staining, phalloidin-rhodamine (1:500; Cytoskeleton, Inc.) was used. All images were acquired with a Zeiss LSM 510 META confocal system using appro-priate filters. Parameters of laser intensities were kept constant during the imaging. Full-projection (three-dimensional [3D]) cell images were gen-erated with Zen 2009 software (Zeiss), using z-stack confocal series. Flu-orescent images in this paper were generated in the University of New Mexico (UNM) Cancer Center Fluorescence Microscopy Facility, sup-ported as detailed on the webpagehttp://hsc.unm.edu/crtc/microscopy /index.html. Images were quantified with SlideBook 5.0.0.33 software and statistically analyzed using Prism software (GraphPad).
FIG 1Analysis of the purity of HPV16 PsV preparations. Following genera-tion of HPV16 PsVs via transfecgenera-tion in 293T cells, the particles were subjected to buoyant density centrifugation in CsCl or Optiprep (iodixanol) gradients. The visible bands were collected by side puncture and desalted into HSB buffer using Amicon Ultra-4 100,000 MWCO centrifugation filter units (Millipore). (A) PsV purity and L1 protein content were determined by SDS-10% PAGE and Coomassie brilliant blue staining against bovine serum albumin (BSA) standards. The asterisk (*) may represent an L1 cleavage product. MW, mo-lecular weight (in thousands). (B) Virion morphology and quality of PsVs purified by CsCl gradient visualized by negative staining and transmission electron microscopy. Bar, 100 nm.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.337.507.64.285.2]7-Methyl-GTP binding assays. Subconfluent HaCaT cells were starved 4 h at 37°C in Tyrode’s buffer prior to addition of HPV16 PsVs (200 vge/cell) for 1 h at 37°C. Cells were lysed (0.5% Triton X-100, 150 mM NaCl, 50 mM Tris [pH 7.5], 1 mM sodium orthovanadate; 1 mM PMSF; 10 ng/ml leupeptin). After centrifugation (10 min, 14,000⫻gat 4°C), supernatants were incubated with a 50% slurry of 7-methyl-GTP-Sepharose (GE Healthcare, United Kingdom) prewashed in lysis buffer, and incubated 1 h at 4°C. After washing the resin two times with lysis buffer and twice with phosphate-buffered saline (PBS), samples were an-alyzed by SDS-PAGE and immunoblotting using anti-eIF4G rabbit poly-clonal antibody (Cell Signaling). In experiments with inhibitors, cells were preincubated with 1M PD168393 (Calbiochem) or 5M PP242 (Cayman Chemical) before virus was added.
Cell cycle analyses.HaCaT cells were incubated with compounds (in-hibitors and siRNA) for 24 h. For detection of apoptotic cells, growing medium was harvested and floating cells were collected by centrifugation. The attached cells were washed with PBS and harvested using a trypsin-EDTA solution. Harvested cells were combined with apoptotic cells, washed 2 times with PBS, and fixed overnight in 70% ethanol. Cells were treated 30 min in the dark at room temperature with propidium iodide (PI) staining solution (0.1% TX100, 10g/ml PI, and 100g/ml RNase A in PBS). Samples were analyzed by flow cytometry at the UNM Center for Molecular Discovery.
RESULTS
HPV16 PsVs induce rapid activation of Akt in HaCaT cells.Full Akt activation requires its translocation to the plasma membrane and dual phosphorylation of two distant segments of the polypep-tide. Akt is phosphorylated at Thr-308 by PDPK1 and at Ser-473 by mTORC2 (13,29). We tested whether HPV PsVs induced Akt activation upon interaction with HaCaT cells. Serum-starved cells were briefly exposed to HPV16 PsVs, and we detected rapid phos-phorylation of Akt on both Thr-308 and Ser-473. Although
mock-exposed controls had basal Akt phosphorylation at both sites (Fig. 2A, lane 1), exposure to HPV16 PsVs led to increased Akt phos-phorylation, with levels reaching those achieved by EGF stimula-tion (Fig. 2A, lanes 2 and 6, respectively). The EGFR-specific in-hibitor AG1478 markedly reduced the Akt phosphorylation levels, indicating that Thr-308 and, to a lesser extent, Ser-473 phosphor-ylation were a consequence of EGFR pathway activation (Fig. 2A, lane 3). Specific inhibitors of PI3K (LY294002 and wortmannin) abolished phosphorylation of both sites on Akt (Fig. 2A, lanes 4 and 5). To verify that Akt activation by PsVs was due to interaction with the cells, we employed heparin as an agent that prevents HPV binding to HaCaT cells (25,30–32). When PsVs were saturated with heparin and unbound heparin was removed, Akt activation by HPV16 PsVs was blunted (data not shown). The well-charac-terized neutralizing antibodies H16.V5 and H16.U4 fail to avert HPV16 interaction with cells in culture (33) and also do not pre-vent Akt activation by HPV16 (data not shown). These findings provide further confirmation that HPV16 interaction with HaCaT cells triggers EGFR activation and also that HPV16 induces PI3K/ Akt signaling very quickly upon cell contact. Confocal microscopy corroborated the rapid phosphorylation of Akt on both residues upon HPV16 exposure (Fig. 2BandC). The intensity of phospho-AKT on images was quantified, andttest analysis of mean inten-sity confirmed that the differences were significant (Fig. 2F).
HPV16 stimulates PTEN phosphorylation.The PTEN phatase is a key inhibitor of the PI3K pathway, and PTEN phos-phorylation on Ser-380 impedes its activity (34). When serum-starved cells were exposed to HPV16, microscopy revealed a strong increase of p-PTEN (Ser-380) compared to that seen in mock-exposed cells (Fig. 2E), and quantification of confocal mi-croscopy images revealed a difference (Fig. 2F). Immunoblot
FIG 2Activation of Akt and inhibition of PTEN by HPV16 PsVs. HaCaT cells were serum starved for 4 h. (A) Cells were treated with inhibitors (1M AG1478, 25M LY294002, or 1M wortmannin) or DMSO alone (Ø) for 1 h at 37°C. Cells were then exposed to 100 vge/cell HPV16 PsV for 15 min at 37°C in the presence of inhibitors. Cell lysates were fractionated by SDS-PAGE. Immunoblotting was performed using antibodies to p-Akt (Ser-473) and p-Akt (Thr-308). Serum-starved (4 h) HaCaT cells were incubated with 100 vge/cell HPV16 PsV, and the distribution of p-Akt (B and C) or PTEN (E) was assessed by confocal microscopy at 25 min postexposure. (D) Cells were mock treated or exposed to HPV16 for 25 min or 5 ng/ml EGF for 5 min. Postnuclear supernatants were analyzed by SDS-PAGE and immunoblotting for PTEN or p-PTEN (Ser-380). Numbers at the left of the gel represent kDa. Panel A includes lanes spliced together from the same exposure of the same films. Data are representative of 3 independent assays. (F) Akt and PTEN phosphorylation from panels B, C, and E was quantified with SlideBook;Pvalues were determined by Student’sttest.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.114.475.65.278.2]analysis confirmed rapid Ser-380 phosphorylation of both 50-kDa and⬃100-kDa species of PTEN in response to HPV16 exposure. The higher-molecular-mass species specific to p-PTEN has not to our knowledge been described but was consistently detected as a dominant reactant even after samples were boiled 10 to 15 min under reducing conditions. We cannot exclude the possibility that the⬃100-kDa band is nonspecific. Nevertheless, levels of both the 50-kDa and⬃100-kDa p-PTEN forms following HPV16 expo-sure approached those induced by the Akt activator EGF (Fig. 2D, lane 3) (35). Densitometric analysis of the phospho-PTEN 50-kDa band in four independent experiments showed that phosphoryla-tion doubled after HPV incubaphosphoryla-tion with HaCaT cells. As Akt ac-tivation also could result from downregulated PTEN expression, we examined the total protein levels of PTEN in lysates from HPV-treated or unHPV-treated cells. The results showed a prominent PTEN species at 50 kDa and a minor form at⬃100 kDa (Fig. 2D); there was no significant difference in PTEN levels at either molecular mass in the presence of HPV16, indicating that the increase in Akt phosphorylation was not due to inhibited PTEN expression.
HPV16 rapidly activates mTOR. Inactivation of PTEN by phosphorylation constitutively activates Akt, which then can phosphorylate and subsequently trigger mTORC1 (36, 37). mTOR activity involves phosphorylation on Ser-2448 via PI3K/ Akt signaling and autophosphorylation of Ser-2481 (38,39). As amino acids play a crucial role in mTOR activation, cells were starved in Tyrode’s buffer to minimize nonspecific activation and then incubated with HPV16 for various times. mTOR was rapidly phosphorylated on both serine residues, and the phosphorylation levels were comparable to that observed upon cell stimulation by EGF or insulin (Fig. 3A, lanes 2 to 4 and lanes 5 and 6, respec-tively); Ser-2481 phosphorylation was not inhibited by rapamy-cin, even at high (M) concentration (Fig. 3B, lanes 3 to 5).
How-ever, PP242 effectively blocked HPV16-induced mTOR phosphorylation on Ser-2481 residue at nM concentrations (Fig. 3B, lanes 6 to 8), suggesting that the mTORC1 complex is involved in HPV16-induced signal transduction.
HPV16 triggers mTORC1 substrates.To more directly assess if HPV interaction with HaCaT cells increased mTORC1 activity, we evaluated the phosphorylation status of its two main sub-strates: S6K and 4E-BP1. Analysis of S6K revealed that HPV16 binding promotes rapid phosphorylation of both Thr-389 and Ser-371 residues, which are known to increase translational capac-ity by acting on translation initiation complex assembly (9,40). Further, phosphorylation levels of these residues reached those observed after EGF stimulation of cells (Fig. 3CandD, respec-tively). Similarly, the level of phosphorylation of 4E-BP1 was markedly increased on Thr-37/Thr-46 residues and approached that induced by EGF (Fig. 3E). 4E-BP1 is phosphorylated on mul-tiple sites, but phosphorylation of Thr-37/Thr-46 residues is a priming event catalyzed by mTOR (41,42).
We examined whether phosphorylation of these translational regulators induced by HPV16 PsVs was connected to activation of the EGFR pathway. Inhibitors of EGFR (AG1478 and PD168393) and PI3K (LY294002 and wortmannin) completely prevented vi-rus-induced 4E-BP1 phosphorylation, again demonstrating that HPV16 promotes phosphorylation of this protein as a result of EGFR pathway activation (Fig. 3F). Src kinases transduce signals from a variety of receptors (43,44) and regulate PI3K/Akt activity (45). Thus, it was not surprising that inhibitors of Src kinases (PP2) and tyrosine kinases (genistein) also substantially prevented HPV16-mediated 4E-BP1 phosphorylation (Fig. 3F, lanes 8 and 11, respectively). To determine if the observed effect on 4E-BP1 phosphorylation was a result of mTORC1 activation, we treated cells with mTOR inhibitors rapamycin and PP242. PP242
sub-FIG 3HPV16 PsVs induce phosphorylation of mTOR and activation of mTORC1 substrates S6K and 4E-BP1. HaCaT cells starved in Tyrode’s buffer for 4 h were incubated with HPV16 PsVs. (A) Cells were lysed at the indicated times, and postnuclear supernatants were analyzed by SDS-PAGE and immunoblotting for total mTOR and phosphorylation-mediated activation of mTOR on Ser-2448 and Ser-2481. (B) Cells were treated for 1 h with DMSO (Ø, lane 2) or inhibitors in DMSO (0.5, 1, and 5M rapamycin in lanes 3, 4, and 5, respectively; 50, 500, and 5,000 nM PP242 in lanes 6, 7, and 8, respectively) prior to exposure to virus for 30 min. Immunoblot analysis was performed for total mTOR and p-mTOR (Ser-2481). Panels include lanes spliced together from the same exposure of the same films. (C to F) Cells were exposed to HPV16 PsVs for the indicated times before lysis and SDS-PAGE and immunoblot analysis of mTORC1 substrate S6K phosphorylation on Thr-389 (C) and pS6K Ser-371 (D) and substrate 4E-BP1 phosphorylation on Thr-37/46 (E and F); actin (C and D) or total 4E-BP1 (E and F) was detected as loading control. (F) HaCaT cells were pretreated for 1 h with DMSO (Ø), EGFR inhibitors (1M AG1478; 0.5M PD168393), PI3K inhibitors (25M LY294002; 1M wortmannin), mTOR inhibitors (3M PP242; 1M and 5M rapamycin), Src inhibitor (5M PP2), and a paninhibitor of tyrosine kinases (100M genistein) prior to virus exposure for 30 min. (G and H) HPV16 PsV-exposed cell lysates were subjected to a 7-methyl-GTP cap-binding pulldown assay as described in Materials and Methods and analyzed by SDS-PAGE and immunoblotting. (G) Actin was detected as a loading control. (H) Starved cells were incubated with DMSO (Ø), 5M PP242, or 1M PD168393 for 1 h before addition of HPV16 PsVs and incubation for an additional 1 h in the presence of inhibitors. Immunoblot detection of eIF4G shows equal amounts of this protein in lysates (used for pulldown assay). Panels A, C, E, and F include lanes spliced together from the same exposure of the same films.
on November 7, 2019 by guest
http://jvi.asm.org/
stantially blocked phosphorylation of 4E-BP1 (Fig. 3F, lane 7). However, rapamycin, which is known to have little effect on the phosphorylation of 4E-BP1 (46,47), inhibited phosphorylation of 4E-BP1 only at a very high concentration (Fig. 3F, lanes 9 and 10). These results again support the conclusion that HPV16 activates the PI3K/Akt/mTOR signal cascade, primarily via EGFR, in hu-man keratinocytes.
HPV16 interaction with HaCaT cells leads to increased bind-ing of eIF4G to a 7-methyl-GTP mRNA cap.As phosphorylation of 4E-BP1 leads to the release of eIF4E to enable cap-dependent protein translation (48), we used anin vitrocap-binding assay to determine whether exposure to HPV16 PsV enhanced the assem-bly of the translation initiation complex at the mRNA cap. In three independent experiments, we observed increased eIF4G binding to the synthetic cap structure with lysates from HPV16 PsV-ex-posed cells compared to mock-exPsV-ex-posed cells (Fig. 3G). To deter-mine whether the increase of eIF4G binding to cap structures was caused by EGFR or mTORC1 activation, we performed similar pulldown experiments with cells treated with EGFR and mTOR inhibitors (PD168393 and PP242, respectively). As expected from the data above, both of these drugs inhibited eIF4G binding to the cap structure (Fig. 3H). Thus, it appears that the increased binding of translation initiation factors to the 7-methyl-cap is a result of EGFR/mTOR pathway activation catalyzed by HPV16 interac-tions with human keratinocytes.
HPV16 quickly suppresses autophagy.In addition to activat-ing biosynthetic processes, both mTORC1 and mTORC2 signal-ing paths are implicated in the inhibition of autophagy (49,50). The serum and amino acid starvation conditions that we em-ployed for mTOR phosphorylation detection induce autophago-some formation in HaCaT cells. Therefore, to determine whether HPV interaction with cells affects autophagy functions, we avoided starvation conditions and used HaCaT cells grown in complete medium (DMEM containing 10% FCS). HPV16 PsVs were incubated with cells for 1 h, and autophagy formation was evaluated by detecting the autophagosomal marker LC3. The lev-els of LC3-II, a form conjugated with phosphatidylethanolamine and present on membranes and autophagosomes, correlate with autophagy formation (51). As controls, cells were pretreated with 3-methyladenine (3MA), an inhibitor of autophagosome forma-tion, or with tamoxifen, an autophagy inducer (52). Six indepen-dent experiments showed significantly decreased ratios of LC3-II to LC3-I (Pvalue, 0.0145) in HPV16-exposed cell lysates com-pared to mock-exposed control cells (Fig. 4). This finding implies that HPV16 PsVs impair autophagosome formation early upon virus-host cell interaction.
Activation of PI3K/Akt/mTOR signaling is critical for HPV16 infection.The importance of the PI3K/Akt/mTOR path-way in HPV16 infection was assayed next. HaCaT cells were pre-treated with specific inhibitors of this pathway and then infected with HPV16 PsVs in complete medium (Fig. 5A). The PI3K-spe-cific inhibitor LY294002 reduced HPV16 pseudoinfection by
⬃75%, which is in agreement with a previous report from our lab showing strong inhibition of HPV31 infection with wortmannin (53). Although wortmannin is 500-fold more potent than LY294002, it lacks specificity and in vitrocan inhibit all three classes of the PI3K family, as well as mTOR, DNA-PK, some phos-phatidylinositol 4-kinases, myosin light-chain kinase, and mito-gen-activated protein kinase (MAPK) at high concentrations (54). Thus, the LY294002 data more specifically demonstrate the
in-volvement of PI3K in infection. As predicted from the data inFig. 3B, rapamycin had a negligible effect on HPV16 infection even at 1M, whereas the selective mTOR inhibitor PP242 significantly reduced virus infection with a 50% effective concentration (EC50)
of 2⫻10⫺6M (Fig. 5B). The Src family kinase inhibitor PP2 also
had a negative effect on HPV16 PsV infection (Fig. 5A), as antic-ipated from the fact that Src regulates PI3K/Akt and signal trans-duction from a variety of receptors (43–45). PP2 caused a dose-dependent reduction of infectivity with an EC50of 1.5⫻10⫺6M
(Fig. 5C). To verify that PP2 and PP242 did not inhibit HPV
infection by impeding HPV attachment, HaCaT cells were incu-bated with dimethyl sulfoxide (DMSO) or inhibitors for 1 h at 4°C. Unbound virus was washed out, lysates were fractionated by SDS-PAGE, and the amount of HPV16 bound to the cells was determined by immunoblotting.Figure 5Dshows that PP2 and PP242 did not alter the capacity of HPV to bind cells, indicating that the compounds do not inhibit HPV16 infection by impairing viral attachment to the cell surface. Similarly, the tyrosine kinase inhibitor genistein had no effect on HPV binding to cells. Again, these findings are consistent with the activation of GFRs by HPV16.
PI3K and Src activation are required at early steps of HPV infection.We next strove to identify the temporal window in which PI3K and Src signaling is important for infection and thus the times that PP242 and PP2, respectively, inhibit HPV infection. HaCaT cells were incubated in the presence of DMSO, PP2, or PP242 for 1 h prior to HPV16 PsV addition or for various intervals after virus exposure. Cell infectivity was scored after 24 h. Addi-tion of PP242 or PP2 at 4 to 5M concentrations prior to virus exposure inhibited infection 85% and 72%, respectively (Fig. 5E
andF, respectively). Supplying these compounds to cells at time intervals greater than or equal to 1 h after incubation with virus was increasingly less inhibitory, demonstrating that PI3K and Src signals are important very early in infection.
mTOR functions and autophagy are important for HPV in-fection.A genetic approach was used to interfere with mTOR and autophagosome functions to avoid possible off-target effects caused by biochemical inhibitors. First, HaCaT cells were trans-fected with either mTOR-specific or negative-control siRNAs. mTOR knockdown in three separate experiments ranged from
⬃20 to 50% compared to cells transfected with a nonspecific
con-FIG 4HPV16 interaction with keratinocytes suppresses autophagy. HaCaT cells grown in DMEM–10% FCS incubated with 100 vge/cell HPV16 PsVs and incubated at 37°C for 1 h. Controls included cells that were mock exposed to PsV, treated with 5M tamoxifen, or treated with 5 mM 3MA. (A) Cell lysates were analyzed by SDS-PAGE and immunoblotting for LC3, with actin detected as a loading control. Densities of LC3-I and LC3-II bands were analyzed with AlphaEaseFC software. (B) The graph represents data from six independent experiments quantifying the LC3-II/LC3-I ratio (two-tailedPvalue, 0.0145, indicating significance).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.302.543.67.163.2]trol siRNA (Fig. 6A). Remarkably, HPV16 PsV infection levels in three independent experiments were reduced 80 to 90% com-pared to negative-control-siRNA-transfected cells (Fig. 6B). As additional confirmation of mTOR knockdown and inhibited function, phosphorylation levels of 4E-BP1 were only⬃15% of those levels observed in nonspecific-siRNA-transfected cells (Fig. 6C). These data show that mTOR functions are needed for suc-cessful HPV16 infection of human keratinocytes. Inhibition of mTOR complexes is known to activate autophagy (55). Analysis of LC3-II/LC3-I levels confirmed the initiation of autophagy in
mTOR-depleted HaCaT cells (Fig. 6DandE). Consequently, the observed inhibition of infection upon mTOR depletion could re-sult from viral degradation by autophagy. To test this hypothesis, we activated or blocked autophagy in HaCaT cells using tamox-ifen or 3MA, respectively. Induction of autophagy with tamoxtamox-ifen completely abrogated HPV16 infection, whereas infection was en-hanced up to 6-fold when autophagy was inhibited with 3MA (Fig. 5A). As these widely used autophagy regulators are not exclusive to the autophagy machinery (56–61), we depleted key proteins of autophagosome formation, Beclin 1 and Atg7, using siRNAs.
Im-FIG 5Chemical inhibitors have varied effects on HPV16 infectivity. HaCaT cells were pretreated for 1 h with DMSO (Ø) or inhibitors at the indicated concentrations. Cells were exposed to HPV16 PsVs or mock exposed (M), and infectivity was scored after 24 h of incubation at 37°C. (A) Infection in the presence of 5M PP2, 50M LY294002, 3M PP242, 100M rapamycin, 5 mM 3MA, or 10M tamoxifen. (B and C) Dose-response curves of infection to mTOR inhibitor PP242 (B) or Src kinase inhibitor PP2 (C). The 50% infective concentrations (IC50s) were calculated using GraphPad Prism software. (D) Immunoblot
analysis of HPV16 L1 bound to HaCaT cells in the presence or absence of 5M PP2, 100M genistein, or 3M PP242. (E and F) Kinetics of HaCaT cell infection inhibition by PP242 and PP2. Cells were treated with DMSO (Ø), 4M PP242 (E), or 5M PP2 (F) for the indicated times pre- and postattachment of HPV16 PsVs (100 vge/cell), which was defined as 0 h; infection was analyzed 24 h following HPV exposure.
FIG 6Autophagy plays a negative role in HPV infection. HaCaT cells were transfected with a control siRNA or siRNAs specific to mTOR, Beclin 1, or Atg7. Downregulation of these proteins was determined in transfected cells by immunoblot analyses using antibodies against mTOR (A) and Beclin 1 and Atg7 (F). Immunoblotting was used to determine the effect of mTOR depletion on p-4E-BP1 (C) and autophagy formation detected by analysis of LC3 (D). The LC3-II/LC3-I ratio (E) was quantified from three independent experiments by densitometry, andPvalues were determined by Student’sttest. Actin (A, D, F) or 4E-BP1 (C) was detected as loading control. Infections were initiated 24 h posttransfection and proceeded for 24 h thereafter under normal conditions (B, G). Panel A includes lanes from the same exposure of the same films.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.116.473.63.261.2] [image:6.585.137.449.487.664.2]munoblot analysis revealed the successful knockdown of these proteins (Fig. 6F). Infection assays showed significant activation of infection compared to cells treated with nonspecific siRNA
(Fig. 6G). As mTOR signaling affects translation, we verified that
inhibitor-induced reductions in luciferase levels in the pseudoin-fection assays were not due to suppressed translational events. Assaying the levels of reporter gene RNAs by reverse transcrip-tion- and quantitative PCR demonstrated that the luciferase tran-script levels were consistent with luciferase protein levels (data not shown), suggesting that the inhibitors block infection prior to nuclear delivery and transcription of pseudogenomes. Thus, the genetic approach using siRNAs confirmed the results obtained from the biochemical analysis: autophagy inhibits HPV16 early infection, and interfering with autophagy results in enhanced HPV16 infection of host keratinocytes. Together with the data shown inFig. 4, these results suggest that autophagy plays an an-tiviral role in HPV infection and that HPVs have means of inter-fering with this cellular process to promote infection.
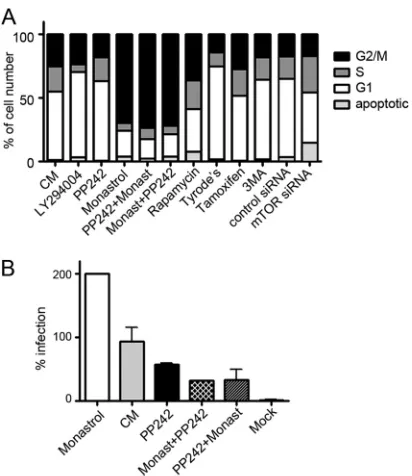
Cell cycle progression and mTOR inhibition.Because pro-gression into early M phase is needed for HPV infection (62), we tested whether the inhibitors prevented infection via cell cycle blockade. Therefore, we assayed the fraction of cells in each phase of the cell cycle during the inhibitor treatments under which in-fection levels were determined above. Although each inhibitor affected the cell cycle distribution, in no case did an inhibitor arrest the cells in any one phase of the cell cycle. No correlation was seen between infection inhibition and cell cycle distribution under the assay conditions employed (Fig. 7A). For example, the distributions of cells in the G1, S, or G2/M phases of the cell cycle were relatively similar whether cells were grown in complete me-dium and infected with HPV16 with no treatment or treated with 4M PP242 or 5M tamoxifen. However, infection levels ranged from 0% inhibition with no inhibitor to nearly 99% inhibition with tamoxifen (Fig. 5A). Specifically, the moderate changes ob-served in the number of cells in G2/M phase were not sufficient to account for the levels of infection inhibition demonstrated for a given inhibitor. To determine whether mTOR inhibitors were preventing infection by arresting the cell cycle in G1or S phase, PP242-treated cells were incubated with monastrol, which in-creases the number of cells in late M phase and promotes HPV16 infection (1,62). Cells were also pretreated with monastrol and then incubated with PP242 before exposure with HPV16 PsV. Treatment of cells with monastrol in all cases increased the num-ber of cells in G2/M phase (Fig. 7A). Monastrol alone significantly increased infection of cells with HPV16, and as expected we ob-served a 2-fold increase in infectivity (Fig. 7B). PP242 addition in the presence of monastrol did not alter the monastrol-character-ized cell cycle profile, but infection was dramatically inhibited, by
⬃85% (Fig. 7). These data indicate that cell cycle effects cannot account for the inhibition of infection by PP242 compounds. We expected to obtain an increase of G1 phase after depletion of mTOR by transfection of HaCaT cells with mTOR-specific siRNA, but interestingly two independent experiments showed an increase of S phase. It should be noted that the number of apop-totic cells after mTOR depletion increased and reached⬃15% of total cell number (Fig. 7A).
DISCUSSION
We recently reported that HPV16 PsVs induce rapid activation of GF-regulated pathways, particularly EGFR and KGFR (1). EGFR
activation by HPV16 PsVs was independently confirmed (25). EGFR signaling in most mammalian cells initiates activation of the PI3K/AKT/mTOR pathway; therefore, we examined if HPV trig-gers activation of these pathways downstream of EGFR. We found that HPV16 interaction with human keratinocytes initiates signal cascades in the PI3K/Akt/mTOR pathway to facilitate cap-depen-dent translation, inhibit autophagy, and thus bolster the early in-fection process.
Two important aspects of virus-induced PI3K/Akt/mTOR ac-tivation were revealed herein. First, HPV16 initiates PI3K/Akt/ mTOR activity prior to viral early protein expression, perhaps before virion entry. Many viruses pose as routine activating li-gands to induce normal cell signaling, avoid antiviral activity, and initiate safe uptake (2). However, activation of the PI3K/Akt path-way is typically associated withde novoviral protein expression aimed to hinder apoptosis and prolong infection (63). Most, if not all, mammalian DNA viruses modulate PI3K activity by changing the ratio of PIP2to PIP3. Some viruses encode proteins capable of activating PI3K, and others induce cellular proteins having similar functions. Repression of PTEN activity or its expression is another means whereby viruses modulate PIP3 concentration (4). HPV early proteins are well-known facilitators of PI3K-Akt signaling. HPV16 E5, E6, and E7 inhibit apoptosis by activating Akt (20–22).
FIG 7Cell cycle distribution fails to correlate with HPV16 infection inhibi-tion. (A) HaCaT cells were transfected with mTOR siRNA or treated with drugs (50M LY294004, 1M PP242, 100M monastrol, 1M rapamycin, 10M tamoxifen, or 5 mM 3MA). Some cells were first incubated for 6 h with 1M PP242 and then with 100M monastrol (PP242⫹Monast); other cells were first incubated for 6 h with 100M monastrol and then with 1M PP242 (Monast⫹PP242). After 24 h of incubation at 37°C, cells were assayed with propidium iodide and examined using flow cytometry. The fractions of cells in G1(1n), S (intermediate), and G2/M (2n) phases and apoptotic cells were
expressed as percentages of the total cells counted. (B) HPV16 PsV infection levels (24 h postinfection) in the presence of complete media (CM) or inhib-itors following pretreatment for 1 h with 100M monastrol, pretreatment with monastrol for 1 h plus 3M PP242 for the duration (Monast⫹PP242), or pretreatment with 3M PP242 for 1 h plus 100M monastrol for the duration (PP242⫹Monast).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.319.525.64.302.2]Interestingly, even after early HPV gene expression ensues, EGFR signaling plays a vital role in infection. For example, in human keratinocytes, HPV16 immortalization leads to heightened EGFR activity (64) and expression of HPV16 E5 induces dimerization of EGFR and subsequent upregulation of PI3K-Akt signaling (19).
We show that HPV interaction with host cells induced rapid phosphorylation of Akt and PTEN, leading to the activation of Akt by a two-pronged approach: phosphorylation of Akt kinase as well as phosphorylation of PTEN and, consequently, inactivation of this negative regulatory phosphatase. In addition, Akt activation is sensitive to EGFR-specific inhibition, indicating that HPV16 in-teraction with EGFR initiates this pathway. Our results are in good agreement with a publication from Fothergill and McMillan showing rapid activation of PI3K/Akt within several minutes of HPV or bovine PV virus-like particles (VLPs) binding to a serum-starved A431 epidermoid carcinoma cell line (26). Phosphoryla-tion of Akt on Ser-473 residue was markedly increased upon virus binding, while only minor phosphorylation of Akt on Thr-308 was observed (26). Phosphorylation of Akt on Ser-473 is an indi-cator of mTORC2 complex activation. Our data show that HPV interaction with host cells changes the functionality not only of mTORC2 but also of another mTOR-regulated complex, mTORC1. Fothergill and McMillan attributed PI3K/Akt signaling to VLP interaction with alpha-6 beta-4 integrin (26). Interest-ingly, there is signaling cross talk on epithelial cells among EGFR, alpha-6 beta-4 integrin, and tetraspanin CD151 (65–67), each of which has been shown to be important for mediating HPV early infection (1,68,69). Thus, these molecules may be cooperating during HPV entry into keratinocytes.
In the very early stages of HPV16 interaction with host cells, activation of mTORC1 was evidenced by phosphorylation of mTORC1 and its substrates, 4E-BP1 and S6K, both of which are important in the physiological control of translation initiation. A primary function of mTORC1 is regulating cap-dependent mRNA translation (70,71). Thus, it is not surprising that multiple DNA viruses, HPV among them, have evolved mechanisms to keep mTOR complexes active during infection. Phosphorylation of S6K increases the translational capacity by acting on translation initiation complex assembly (9,40), and phosphorylation of 4E-BP1 induces the dissociation of this protein from eIF4E, allowing eIF4G to bind and form the eIF4F complex, which enhances cap-dependent initiation of mRNA translation (9). We observed each of these phosphorylation events after HPV16 interaction with host cells under conditions of nutrient deprivation. The pulldown as-say with 7-methyl GTP cap analogue detected increased cap-de-pendent translation after only 1 h of virus binding to host cells, indicating changes in translation even before the bulk of virus internalization. Specific inhibitors of EGFR (PD168393) and mTOR (PP242) strongly inhibited eIF4G binding to 7-methyl GTP Sepharose 4B beads, demonstrating inhibition of translation and the crucial role of the EGFR/mTOR pathway in this process. The second remarkable finding of this work is that signaling downstream of HPV16-GFR contacts early after virus-host inter-actions serves to inhibit rather than activate autophagy to pro-mote more-efficient infection. HPV16 PsV exposure to HaCaT cells, by virtue of activation of mTOR complexes, opposes the formation of autophagosomes. This is significant, as autophagy is an important regulator of the host response against viral infec-tions, wherein cells capture and degrade intracellular pathogens (18). Many viruses have developed a variety of ways to block
au-tophagy or subvert this machinery for their own benefit (19). However, of those viruses that inhibit autophagy, all do so byde novoexpression of virulence factors that impair the function of autophagy proteins. For example, the DNA viruses of the Herpes-viridaefamily encode diverse proteins that inhibit autophagy by varied means (73,76–80).
Cell engagement by several viruses has been found to activate autophagy (19,72–74); however, our findings show that the initial HPV-cell interaction leads to autophagy inhibition to promote infection. Furthermore, HPV16 infectivity was directly affected by modulating keratinocyte autophagy. Infection levels were partic-ularly enhanced upon inhibiting autophagy with 3MA. This find-ing is in good agreement with work from the Pyeon laboratory, showing an⬃40-fold increase of HPV16 infectivity in primary keratinocytes in the presence of 3MA (81). Tamoxifen, an inducer of autophagy, practically abolished infection. Yet both tamoxifen and 3MA impact targets other than those related to autophagy (56–61). The specificity of genetic knockdowns of mTOR, Beclin 1, and Atg7 confirmed that HPV-induced restraint of autophagy is important for early infection events.
In summary, we report here that HPV16 PsV interaction with HaCaT human keratinocytes causes rapid activation of PI3K/Akt/ mTOR signaling and the impairment of autophagy and that both of these processes benefit HPV16 infection. This signaling cascade was primarily influenced by activation via the EGFR. Recently, we showed stimulation of the MAPK (ERK1/2) pathway after HPV16 interaction with EGFR and KGFR on HaCaT cells (1). Cross talk between ras/MAPK and PI3K/Akt effector paths generates con-text-dependent responses to growth factor stimulation, alters the dynamic topologies of signal propagation networks downstream of cell surface receptors, and leads to amplification or attenuation of key target protein activities (75). Thus, HPV interaction with cell surface GFRs is likely to trigger analogous communication between these pathways, resulting in activation of complementary signaling, cellular evasion of apoptosis, enhanced viral transcrip-tion and translatranscrip-tion, promoted proliferatranscrip-tion, and prolonged in-fection. As many other viruses are capable of interacting with EGFR at the cell surface, we propose that these viruses might also induce the PI3K/Akt/mTOR pathway via this GFR activation to inhibit autophagy upon viral uptake.
ACKNOWLEDGMENTS
This work was supported by NIH grants CA136132 (M.A.O) and K12GM088021 (S.A.D.).
The content is solely our responsibility and does not necessarily rep-resent the official views of the National Institutes of Health.
We thank N. Fusenig for HaCaT cells, M. Müller for the HPV16-L1/L2 plasmid, and C. Buck for 293TT cells. We are grateful to L. Laidler and N. Patterson for excellent technical assistance and to members of the Ozbun Lab for critical comments on the manuscript. We offer special apprecia-tion to T. Roitbak for insightful discussions and sharing of reagents.
REFERENCES
1.Surviladze Z, Dziduszko A, Ozbun MA.2012. Essential roles for soluble virion-associated heparan sulfonated proteoglycans and growth factors in human papillomavirus infections. PLoS Pathog.8:e1002519. doi:10.1371 /journal.ppat.1002519.
2.Marsh M, Helenius A.2006. Virus entry: open Sesame. Cell124:729 –740. 3.Andjelkovic M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA.1997. Role of transloca-tion in the activatransloca-tion and functransloca-tion of protein kinase B. J. Biol. Chem. 272:31515–31524.
on November 7, 2019 by guest
http://jvi.asm.org/
4.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat. Rev. Microbiol.6:266 –275.
5.Cooray S.2004. The pivotal role of phosphatidylinositol 3-kinase-Akt signal transduction in virus survival. J. Gen. Virol.85:1065–1076. 6.Pópulo H, Lopes JM, Soares P.2012. The mTOR signalling pathway in
human cancer. Int. J. Mol. Sci.13:1886 –1918.
7.Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, Schulze A.2008. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8:224 –236.
8.Schieke SM, Phillips D, McCoy JP, Jr, Aponte AM, Shen RF, Balaban RS, Finkel T.2006. The mammalian target of rapamycin (mTOR) path-way regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem.281:27643–27652.
9.Hay N, Sonenberg N.2004. Upstream and downstream of mTOR. Genes Dev.18:1926 –1945.
10. Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. 2006. mTOR signaling: implications for cancer and anticancer therapy. Br. J. Cancer94:195–199.
11. Crusius K, Auvinen E, Steuer B, Gaissert H, Alonso A.1998. The human papillomavirus type 16 E5-protein modulates ligand-dependent activa-tion of the EGF receptor family in the human epithelial cell line HaCaT. Exp. Cell Res.241:76 – 83.
12. Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. 2004. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol.6:1122–1128.
13. Alessi DR, Pearce LR, Garcia-Martinez JM. 2009. New insights into mTOR signaling: mTORC2 and beyond. Sci. Signal.2:pe27. doi:10.1126 /scisignal.267pe27.
14. Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B.2006. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell127:125–137. 15. Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated
translational control. Nat. Rev. Mol. Cell Biol.10:307–318.
16. Fan QW, Weiss WA. 2011. Autophagy and Akt promote survival in glioma. Autophagy7:536 –538.
17. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M.2007. FoxO3 controls autophagy in skeletal mus-cle in vivo. Cell Metab.6:458 – 471.
18. Deretic V, Levine B.2009. Autophagy, immunity, and microbial adapta-tions. Cell Host Microbe5:527–549.
19. Jordan TX, Randall G.2012. Manipulation or capitulation: virus inter-actions with autophagy. Microbes Infect.14:126 –139.
20. Menges CW, Baglia LA, Lapoint R, McCance DJ.2006. Human papil-lomavirus type 16 E7 up-regulates AKT activity through the retinoblas-toma protein. Cancer Res.66:5555–5559.
21. Pim D, Massimi P, Dilworth SM, Banks L. 2005. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene24:7830 –7838. 22. Spangle JM, Munger K.2010. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol.84:9398 –9407.
23. Abban CY, Meneses PI.2010. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology403:1–16.
24. Payne E, Bowles MR, Don A, Hancock JF, McMillan NAJ.2001. Human papillomavirus type 6b virus-like particles are able to activate the Ras-MAP kinase pathway and induce cell proliferation. J. Virol.75:4150 – 4157.
25. Schelhaas M, Shah B, Holzer M, Blattmann P, Kühling L, Day PM, Schiller JT, Helenius A.2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog.8:e1002657. doi:10.1371/journal.ppat.1002657.
26. Fothergill T, McMillan NAJ.2006. Papillomavirus virus-like particles activate the PI3-kinase pathway via alpha-6 beta-4 integrin upon binding. Virology352:319 –328.
27. Campos SK, Ozbun MA.2009. Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for in-fectivity in human keratinocytes. PLoS One4:e4463. doi:10.1371/journal .pone.0004463.
28. Ozbun MA, Kivitz MP.2011. The art and science of obtaining virion stocks for experimental human papillomavirus infections, p 19 –35.In
Gaston K (ed), Small DNA tumor viruses. Horizon Scientific and Caister Academic Press, Norfolk, United Kingdom.
29. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM.2005. Phosphoryla-tion and regulaPhosphoryla-tion of Akt/PKB by the rictor-mTOR complex. Science 307:1098 –1101.
30. Culp TD, Budgeon LR, Christensen ND.2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology347: 147–159.
31. Joyce JG, Tung J-S, Przysiecki CT, Cook JC, Lehman ED, Sands JA, Jansen KU, Keller PM.1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem.274:5810 –5822.
32. Shafti-Keramat S, Handisurya A, Kriehuber E, Meneguzzi G, Slupetzky K, Kirnbauer R.2003. Different heparan sulfate proteoglycans serve as cellular receptors for human papillomaviruses. J. Virol.77:13125–13135. 33. Day PM, Gambhira R, Roden R, Lowy DR, Schiller JT.2008. Mecha-nisms of human papillomavirus type 16 neutralization by L2 cross-neutralizing and L1 type-specific antibodies. J. Virol.82:4638 – 4646. 34. Vazquez F, Ramaswamy S, Nakamura N, Sellers WR.2000.
Phosphor-ylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol.20:5010 –5018.
35. Ono M, Kuwano M.2006. Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin. Cancer Res.12:7242–7251.
36. Guertin DA, Sabatini DM.2007. Defining the role of mTOR in cancer. Cancer Cell12:9 –22.
37. Manning BD, Cantley LC.2007. AKT/PKB signaling: navigating down-stream. Cell129:1261–1274.
38. Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. 1999. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J.344(Part 2): 427– 431.
39. Peterson RT, Beal PA, Comb MJ, Schreiber SL. 2000. FKBP12-rapamycin-associated protein (FRAP) autophosphorylates at serine 2481 under translationally repressive conditions. J. Biol. Chem.275:7416 – 7423.
40. Holz MK, Ballif BA, Gygi SP, Blenis J.2005. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic pro-tein interchange and ordered phosphorylation events. Cell123:569 –580. 41. Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM.1998. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. U. S. A.95:1432–1437.
42. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoek-stra MF, Aebersold R, Sonenberg N.1999. Regulation of 4E-BP1 phos-phorylation: a novel two-step mechanism. Genes Dev.13:1422–1437. 43. Donepudi M, Resh MD.2008. c-Src trafficking and co-localization with
the EGF receptor promotes EGF ligand-independent EGF receptor acti-vation and signaling. Cell. Signal.20:1359 –1367.
44. Sandilands E, Akbarzadeh S, Vecchione A, McEwan DG, Frame MC, Heath JK.2007. Src kinase modulates the activation, transport and sig-nalling dynamics of fibroblast growth factor receptors. EMBO Rep. 8:1162–1169.
45. Nijhuis E, Lammers JW, Koenderman L, Coffer PJ.2002. Src kinases regulate PKB activation and modulate cytokine and chemoattractant-controlled neutrophil functioning. J. Leukoc. Biol.71:115–124. 46. Choo AY, Blenis J.2009. Not all substrates are treated equally:
implica-tions for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle 8:567–572.
47. Vu C, Fruman DA.2010. Target of rapamycin signaling in leukemia and lymphoma. Clin. Cancer Res.16:5374 –5380.
48. Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev.15:2852–2864.
49. Jung CH, Ro SH, Cao J, Otto NM, Kim DH.2010. mTOR regulation of autophagy. FEBS Lett.584:1287–1295.
50. Wang X, Proud CG.2011. mTORC1 signaling: what we still don’t know. J. Mol. Cell. Biol.3:206 –220.
51. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T.2000. LC3, a mammalian
on November 7, 2019 by guest
http://jvi.asm.org/
logue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J.19:5720 –5728.
52. Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P.2004. Ceramide-mediated macroautophagy in-volves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem.279:18384 –18391.
53. Smith JL, Lidke DS, Ozbun MA.2008. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Vi-rology381:16 –21.
54. Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD. 2001. Synthesis and function of 3-phosphorylated inositol lipids. Annu. Rev. Biochem.70: 535– 602.
55. Raught B, Gingras AC, Sonenberg N.2001. The target of rapamycin (TOR) proteins. Proc. Natl. Acad. Sci. U. S. A.98:7037–7044.
56. Altan N, Chen Y, Schindler M, Simon SM.1999. Tamoxifen inhibits acidification in cells independent of the estrogen receptor. Proc. Natl. Acad. Sci. U. S. A.96:4432– 4437.
57. Caro LH, Plomp PJ, Wolvetang EJ, Kerkhof C, Meijer AJ. 1988. 3-Methyladenine, an inhibitor of autophagy, has multiple effects on me-tabolism. Eur. J. Biochem.175:325–329.
58. Lopes MC, Vale MG, Carvalho AP.1990. Ca2(⫹)-dependent binding of tamoxifen to calmodulin isolated from bovine brain. Cancer Res.50: 2753–2758.
59. O’Brian CA, Housey GM, Weinstein IB.1988. Specific and direct bind-ing of protein kinase C to an immobilized tamoxifen analogue. Cancer Res.48:3626 –3629.
60. Wiseman H, Cannon M, Arnstein HR, Halliwell B.1990. Mechanism of inhibition of lipid peroxidation by tamoxifen and 4-hydroxytamoxifen introduced into liposomes. Similarity to cholesterol and ergosterol. FEBS Lett.274:107–110.
61. Wu Y-T, Tan H-L, Shui G, Bauvy C, Huang Q, Wenk MR, Ong C-N, Codogno P, Shen H-M.2010. Dual role of 3-methyladenine in modula-tion of autophagy via different temporal patterns of inhibimodula-tion on class I and III phosphoinositide 3-kinase. J. Biol. Chem.285:10850 –10861. 62. Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF.2009.
Estab-lishment of human papillomavirus infection requires cell cycle progres-sion. PLoS Pathog.5:e1000318. doi:10.1371/journal.ppat.1000318. 63. Ji WT, Liu HJ.2008. PI3K-Akt signaling and viral infection. Recent Pat.
Biotechnol.2:218 –226.
64. Zyzak LL, MacDonald LM, Batova A, Forand R, Creek KE, Pirisi L. 1994. Increased levels and constitutive tyrosine phosphorylation of the epidermal growth factor receptor contribute to autonomous growth of human papillomavirus type 16 immortalized human keratinocytes. Cell Growth Differ.5:537–547.
65. Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG.2001. EGF-R signaling through Fyn kinase disrupts the function of integrin alpha6beta4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J. Cell Biol.155:447– 458.
66. Sterk LM, Geuijen CA, Oomen LC, Calafat J, Janssen H, Sonnenberg A. 2000. The tetraspan molecule CD151, a novel constituent of hemidesmo-somes, associates with the integrin alpha6beta4 and may regulate the spa-tial organization of hemidesmosomes. J. Cell Biol.149:969 –982. 67. Yang XH, Richardson AL, Torres-Arzayus MI, Zhou P, Sharma C,
Kazarov AR, Andzelm MM, Strominger JL, Brown M, Hemler ME. 2008. CD151 accelerates breast cancer by regulating alpha 6 integrin func-tion, signaling, and molecular organization. Cancer Res.68:3204 –3213. 68. Evander M, Frazer IH, Payne E, Qi YM, Hengst K, McMillan NAJ.
1997. Identification of the a6integrin as a candidate receptor for
papillo-maviruses. J. Virol.71:2449 –2456.
69. Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L.2008. Clathrin- and caveolin-independent entry of human papilloma-virus type 16 —involvement of tetraspanin-enriched microdomains (TEMs). PLoS One3:e3313. doi:10.1371/journal.pone.0003313. 70. Sarbassov DD, Ali SM, Sabatini DM.2005. Growing roles for the mTOR
pathway. Curr. Opin. Cell Biol.17:596 – 603.
71. Wullschleger S, Loewith R, Hall MN.2006. TOR signaling in growth and metabolism. Cell124:471– 484.
72. Joubert PE, Meiffren G, Gregoire IP, Pontini G, Richetta C, Flacher M, Azocar O, Vidalain PO, Vidal M, Lotteau V, Codogno P, Rabourdin-Combe C, Faure M.2009. Autophagy induction by the pathogen receptor CD46. Cell Host. Microbe6:354 –366.
73. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B.2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell122:927–939.
74. Shelly S, Lukinova N, Bambina S, Berman A, Cherry S.2009. Autophagy is an essential component of Drosophila immunity against vesicular sto-matitis virus. Immunity30:588 –598.
75. Aksamitiene E, Kiyatkin A, Kholodenko BN.2012. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: a fine balance. Biochem. Soc. Trans.40:139 –146.
76. Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A.2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its in-teraction with Beclin 1. J. Virol.86:2571–2584.
77. Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B.2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe1:23–35. 78. Shoji-Kawata S, Levine B.2009. Autophagy, antiviral immunity, and viral
countermeasures. Biochim. Biophys. Acta1793:1478 –1484.
79. Sinha S, Colbert CL, Becker N, Wei Y, Levine B.2008. Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy4:989 –997.
80. Tallóczy Z, Virgin HW, IV, Levine B.2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy2:24 –29. 81. Griffin LM, Cicchini L, Pyeon D.Human papillomavirus infection is
inhibited by host autophagy in primary human keratinocytes. Virology, in press.