Copyright © 1998, American Society for Microbiology
Characterization of Replication-Competent Hepatitis A Virus
Constructs Containing Insertions at the N Terminus
of the Polyprotein
YUAN ZHANGANDGERARDO G. KAPLAN*
Laboratory of Hepatitis Research, Division of Viral Products, Center for Biologics Evaluation and Research, Food and Drug Administration, Bethesda, Maryland 20892
Received 25 July 1997/Accepted 17 September 1997
To determine whether hepatitis A virus (HAV) could tolerate the insertion of exogenous sequences, we constructed full-length HAV cDNAs containing in-frame insertions at the N terminus of the polyprotein and transfected the derived T7 RNA polymerase in vitro transcripts into FRhK-4 cells. Replication of HAVvec1, a construct containing an insertion of 60 nucleotides coding for a polylinker, a 2B/2C cleavage site for HAV protease 3Cpro, and two initiation codons that restored the sequence of the N terminus of the polyprotein, was
detected 2 weeks after transfection by indirect immunofluorescence analysis using anti-HAV monoclonal antibodies. Western blot analysis of HAVvec1-infected cells using anti-VP2 and anti-VP4 antibodies failed to detect the expression of the inserted sequences. Insertion of a 24-mer oligonucleotide coding for a FLAG epitope into HAVvec1 resulted in its HAV-mediated expression which was retained upon deletion of a Gln residue from the inserted 2B/2C cleavage site. Western blot analysis using anti-FLAG and anti-VP2 antibodies showed that the FLAG epitope accumulated in infected cells fused to VP0. Replacement of the FLAG epitope with an epitope of the circumsporozoite protein (CSP) of Plasmodium falciparum resulted in its stable HAV-mediated expression for at least six serial passages in FRhK-4 cells. Sedimentation analysis in sucrose density gradients showed that the CSP epitope accumulated in infected cells fused to VP0, forming 80S empty capsids which also contained native VP0. Our data suggest that the HAV internal ribosome entry site can efficiently direct dual initiation of translation of the polyprotein from AUG codons separated by 66 to 78 nucleotides and show that HAV can tolerate insertions at the N terminus of the polyprotein.
Hepatitis A virus (HAV), the causative agent of acute hep-atitis in humans (23), is the single member of the Hepatovirus genus of the Picornaviridae (21). This family of small, nonen-veloped, positive-strand RNA viruses include human patho-gens such as poliovirus (PV) and rhinovirus and animal pathogens such as foot-and-mouth disease virus and encepha-lomyocarditis virus, which are grouped within the Enterovirus,
Rhinovirus, Aphthovirus, and Cardiovirus genera, respectively.
HAV is transmitted via the oral-fecal route and can be pre-vented with formalin-inactivated vaccines containing HAV propagated in cell culture (24, 47).
The RNA genome of HAV is about 7,500 bases long and has a small virus-encoded VPg protein covalently linked to its 59
end (46) and a poly(A) tail at its 39 end. After binding to a cellular receptor identified in African green monkey kidney cells as HAVcr-1 (28), the HAV genome is delivered to the cytoplasm by an unknown mechanism. The 59 nontranslated region of the HAV genome, which is a long and complex structure containing an internal ribosome entry site (IRES), directs the cap-independent translation of the viral message (reference 26 and references therein). A single long open read-ing frame codes for a polyprotein from which structural pro-teins VP0, VP3, and VP1 and nonstructural propro-teins are cleaved by 3Cpro, the only HAV-encoded protease (41). Sixty copies of each of the structural proteins assemble into viral capsids, which in association with the HAV genome form pro-virions that undergo a slow RNA-dependent maturation
cleav-age of VP0 into VP4 and VP2 (13). Downstream from the IRES there are two in-frame AUG codons at nucleotides (nt) 735 to 737 and 741 to 743; although both are used, translation of the polyprotein preferentially starts from the second AUG, which has a less favorable initiation context (43). Cardioviruses and aphthoviruses also have two in-frame AUG initiation codons separated by 9 and 84 nt, respectively, and both are used to initiate translation of the polyprotein producing two forms of the leader (L) protein, a nonstructural protein which precedes the structural proteins. HAV, on the other hand, does not code for an L protein, and its first translated protein is an unusually small VP4 protein of 21 to 23 amino acids that has not yet been found in the viral capsid (22, 29, 44). Although the N-terminal amino acid of VP4 of other picornaviruses is a myristylated Gly residue essential for virus viability (9, 16), there is no evidence that internal residues of VP4 of HAV are exposed and myristylated (44). Furthermore, it has recently been shown that addition of a Pro residue preceding the N-terminal Gly residue, which blocks its myristylation, is lethal for PV (35).
To further study the mechanism of initiation of translation in HAV and to determine whether this virus could tolerate the addition of sequences immediately downstream of the polypro-tein initiation codons, we made in-frame insertions at the N terminus of the polyprotein. Infection of FRhK-4 cells with HAV constructs coding for a FLAG peptide or an NANP immunodominant epitope of the circumsporozoite protein (CSP) of Plasmodium falciparum inserted at the N terminus of the polyprotein resulted in the HAV-mediated expression of the tag epitopes fused to VP0 and termed ExtVP0. The simul-taneous expression of ExtVP0 and a protein that comigrated with native VP0 in cells infected with our constructs pointed to
* Corresponding author. Mailing address: Division of Viral Prod-ucts, CBER-FDA, 8800 Rockville Pike, Bldg. 29A-NIH, Rm. 1D10, HFM-448, Bethesda, MD 20892. Phone: (301) 827-1870. Fax: (301) 480-5326. E-mail: [email protected].
349
on November 9, 2019 by guest
http://jvi.asm.org/
a model of dual initiation of translation of the polyprotein. Our data suggested that HAV can initiate translation of the polyprotein from two AUG codons separated by 66 to 78 nt and that the larger-than-VP0 proteins containing insertions in VP4 accumulated as empty capsids in infected cells. The HAV-mediated expression of exogenous sequences is a tool to study poorly understood mechanisms of HAV such as the fate of VP4.
MATERIALS AND METHODS
Cells and viruses.Fetal rhesus monkey kidney (FRhK-4) cells were a gift of S. Emerson, National Institutes of Health. Cells were grown in monolayer cultures in Eagle minimal essential medium supplemented with 10% horse serum.
The stocks of tissue culture adapted HM175 strain of HAV (HAV HM175) were derived from infectious cDNA (17) and grown in FRhK-4 cell monolayers. Nucleotide positions of the HAV genome are according to the published HAV HM175 cDNA sequence (17).
PV type 1 Mahoney strain (PV1/M) was obtained from the American Type Culture Collection and grown in HeLa S3 cell monolayers (27).
Antisera.Anti-VP4 antiserum was obtained from rabbits immunized with a synthetic 23-mer full-length VP4 peptide conjugated to keyhole limpet hemocy-anin (KLH). Anti-VP2 antiserum was obtained from rabbits immunized with synthetic peptide CTDLELHGLTPLSTQ corresponding to the C terminus of VP2 fused to KLH. Peptides conjugated to KLH were emulsified in Freund’s complete adjuvant and inoculated into female New Zealand White rabbits. Animals were boosted several times; sera were collected and stored at270°C. Western blot analysis showed that these anti-VP4 and anti-VP2 antisera reacted specifically against HAV VP0 (Fig. 3; compare lanes 9 and 10) and VP0 and VP2 (Fig. 4; compare lanes 5 and 6), respectively. Unlabeled and125I-labeled human
anti-HAV polyclonal antisera were from a HAVAB kit (Abbott Laboratories). Murine anti-FLAG monoclonal antibody (MAb) M2, which reacts against the FLAG epitope (DTKDDDDK) irrespective of its location, was used as suggested by the manufacturer (IBI, Inc.). Alkaline phosphatase-labeled goat anti-rabbit and goat anti-human antibodies (Abs) were used as suggested by the manufac-turer (Kirkegaard-Perry Laboratories, Inc.). Fluorescein isothiocyanate (FITC)-labeled goat anti-human immunoglobulin G and immunoglobulin M Ab (Accu-rate), HAV-neutralizing murine MAbs K2-4F2, K3-4C8, and K3-2F2 (32), biotinylated affinity-purified horse anti-mouse Ab (Vector Laboratories), and FITC-labeled avidin (Vector Laboratories) were used for indirect immunofluo-rescence (IF) analysis. Purified murine MAb 2A10 raised against the
immuno-dominant NANP region of the CSP of the human malaria parasite P. falciparum (15) was obtained from Robert T. Wirtz (Centers for Disease Control and Prevention, Atlanta, Ga.).
Indirect IF analysis.Growth of HAV and expression of the FLAG epitope were assessed by indirect IF analysis. Monolayers of FRhK-4 cells grown in eight-well Permanox culture slides (Nunc, Inc.) were fixed with cold acetone for 30 min, treated (1mg/ml) with a mix of HAV-neutralizing murine MAbs K2-4F2, K3-4C8, and K3-2F2 (19) or anti-FLAG MAb M2 for 1 h at room temperature, and stained with biotinylated affinity-purified horse anti-mouse Ab (1.5mg/ml) and FITC-labeled avidin (40 mg/ml). Immunofluorescent micrographs were taken with a Zeiss Axioscope microscope at a magnification of3400 with an oil immersion objective.
Plasmid constructions.Recombinant DNA manipulations were done by stan-dard methods (34). Constructions were verified by nucleotide sequence analysis using a PCR-based dideoxynucleotide termination method (fmol sequencing system kit; Promega Co.). All plasmids were grown in Escherichia coli DH5aand purified by equilibrium centrifugation in CsCl2in the presence of ethidium
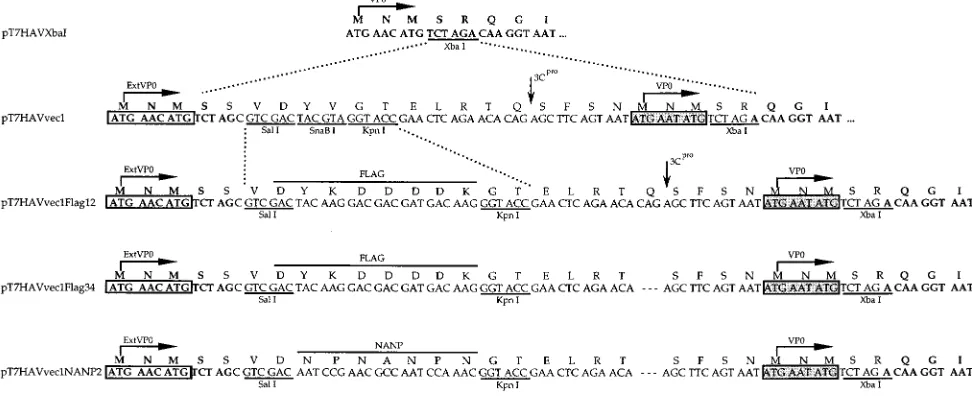
bromide (34). Figure 1 shows the nucleotide and deduced amino acid sequences of the N terminus of the polyprotein of the following plasmids constructed for this work.
(i) pT7HAV.The NheI-BspEI DNA fragment of pHAV/7 (17), which contains the bacteriophage SP6 RNA polymerase promoter and nt 1 to 23 of the HAV cDNA, was replaced with annealed synthetic oligonucleotides 59-CTAGCTAA TACGACTCACTATAGTTCAAGAGGGGTCTCCGGGAATTT-39 and 59-C CGGAAATTCCCGGAGACCCCTCTTGAACTATAGTGAGTCGTATTAG C-39, coding for a bacteriophage T7 RNA polymerase promoter followed by a G residue and the first 23 nt of the HAV cDNA. T7 RNA polymerase in vitro transcripts of the resulting plasmid, pT7HAV, linearized with HaeII were infec-tious upon transfection into FRhK-4 cells.
(ii) pT7HAVXbaI.The XbaI site immediately downstream of the poly(A) tail in pT7HAV was eliminated by filling in with DNA polymerase I Klenow enzyme. The resulting plasmid, pT7HAVXbaI, contains a unique XbaI site at position 744 of the HAV cDNA. T7 RNA polymerase in vitro transcripts of HaeII-linearized pT7HAVXbaI were infectious upon transfection into FRhK-4 cells.
(iii) pT7HAVvec1.Annealed synthetic oligonucleotides 59-CTAGCGTCGAC TACGTAGGTACCGAACTCAGAACACAGAGCTTCAGTAATATGAATA TGT-39 and 59-CTAGACATATTCATATTACTGAAGCTCTGTGTTCTGA GTTCGGTACCTACGTAGTCGACG-39, coding for SalI, SnaBI, and KpnI re-striction sites (polylinker), a 2B/2C cleavage site for HAV protease 3Cpro, and
[image:2.612.52.538.74.274.2]two adjacent initiation codons which restored the native VP0 sequence, were cloned into the unique XbaI site of pT7HAVXbaI. The resulting plasmid, pT7HAVvec1, contains an in-frame insertion of 20 codons at the N terminus of FIG. 1. Nucleotide and amino acid sequences of constructs containing insertions in the unique XbaI site at nt 744 of the HAV cDNA. Synthetic oligonucleotides coding for a polylinker containing SalI, SnaBI, and KpnI restriction sites, a 2B/2C cleavage site for the HAV 3Cpro, and peptide MNMSR containing two initiation
codons which restore the native sequence of VP0 were cloned into the unique XbaI site of pT7HAVXbaI and termed pT7HAVvec1. Synthetic oligonucleotides coding for the DYKDDDDK FLAG peptide were inserted into the SalI and KpnI sites of pT7HAVvec1, and the resulting construct was termed pT7HAVvec1Flag12. Deletion of the Gln residue required for the 3Cpro-mediated cleavage of the inserted 2B/2C cleavage site was introduced into pT7HAVvec1Flag12 by replacing the KpnI-XbaI
DNA fragment with synthetic oligonucleotides, and the construct was termed pT7HAVvec1Flag34. The SalI-KpnI DNA fragment coding for the FLAG peptide in pT7HAVvec1Flag34 was replaced with synthetic oligonucleotides coding for an immunodominant NANP epitope of the CSP of P. falciparum. Horizontal arrows indicate the initiation sites for translation of ExtVP0 and VP0. Unique restriction sites are underlined; HAV HM175 nucleotides and amino acids of the N terminus of the polyprotein are boldfaced; inserted sequences are shown in plain text. Sequences of the FLAG and NANP epitopes are marked. Dotted lines indicate the insertion of synthetic nucleotides between two restriction sites; dashes indicate deletion of inserted nucleotides. The putative cleavage of the inserted 2B/2C site by 3Cpro
is indicated with a vertical arrow. The first (or native) set of initiation codons is boxed in white, and the second (or inserted) set of initiation codons is boxed in grey.
on November 9, 2019 by guest
http://jvi.asm.org/
the HAV polyprotein. The synthetic oligonucleotides restored only the XbaI site within the VP0 coding sequence.
(iv) pT7HAVvec1Flag12.Annealed synthetic oligonucleotides 59-TCGACTA CAAGGACGACGATGACAAGG-39and 59-GTACCCTTGTCATCGTCGTC CTTGTAG-39, coding for the FLAG octapeptide DTKDDDDK, were cloned into the SalI and KpnI sites in the polylinker of pT7HAVvec1. The resulting plasmid, pT7HAVvec1Flag12, contains an in-frame insertion of 25 codons at the N terminus of the HAV polyprotein.
(v) pT7HAVvec1Flag34.The KpnI-XbaI fragment of pT7HAVvec1Flag12 was exchanged with annealed synthetic oligonucleotides 59-GTACCGAACTCAGA ACAAGCTTCAGTAATATGAATATGT-39and 59-CTAGACATATTCATAT TACTGAAGCTTGTTCTGAGTTCG-39. The resulting plasmid, pT7HAVvec1 Flag34, contains a deletion of a Gln residue from the inserted 2B/2C cleavage site and has an in-frame insertion of 24 codons at the N terminus of the HAV polyprotein.
(vi) pT7HAVvec1NANP2. The SalI-XbaI fragment of pT7HAVvec1Flag34 coding for the FLAG peptide was replaced with annealed synthetic oligonucle-otides 59-GACAATCCGAACGCCAATCCAAACG-39and 59-GTACCGTTTG GATTGGCGTTCGGATTG-39coding for the CSP epitope NPNANPN of P. falciparum (42). The resulting plasmid, pT7HAVvec1NANP2, contains an in-frame insertion of 24 codons into the N terminus of the HAV polyprotein.
In vitro RNA synthesis and transfection. In vitro synthesis of RNA from cDNA templates was performed with T7 RNA polymerase (Pharmacia-LKB). Reaction mixtures of 50ml contained 2mg of plasmid DNA digested with HaeII, which cuts immediately downstream of the HAV poly(A), 1 mM each nucleoside triphosphate, RNasin (1 U/ml; Promega), bovine serum albumin (RNase and DNase free; 10.5 mg/ml; Boehringer Mannheim Inc.), 5 mM dithiothreitol, 40 mM Tris-HCl (pH 8.0), 15 mM MgCl2, and 30 U of T7 RNA polymerase. After
incubation for 30 min at 37°C, the RNA was examined by electrophoresis in a 1% agarose gel (17). Similar amounts of RNA (approximately 5 to 10mg) were synthesized in each reaction regardless of which DNA template was used. The in vitro RNA synthesis reaction without any further purification was transfected by using DEAE-dextran as a facilitator (45) into 60 to 80% confluent monolayers of FRhK-4 cells grown in 25-cm2flasks. Transfected cells were incubated at 35°C,
and every 2 weeks cells were split 1:10 and grown in 25-cm2flasks and eight-well
slides (Nunc). HAV growth was monitored in the eight-well slides by IF staining with anti-HAV MAbs.
Viruses produced by transcription of pT7HAV, pT7HAVvec, pT7HAVvec1 Flag12, pT7HAVvec1Flag34, pT7HAVvec1Flag01, and pT7HAVvec1NANP2 and transfection into FRhK-4 cells were named HAV HM175, HAVvec1, HAVvec1Flag12, HAVvec1Flag34, HAVvec1Flag01, and HAVvec1NANP2, re-spectively.
Infections and preparation of cytoplasmic extracts.Growth of HAV in trans-fected and intrans-fected FRhK-4 cells was assessed each week by IF analysis. After 100% of the cells became positive for HAV antigen, viral stocks were prepared by three freezing-thawing cycles and stored at270°C. Fresh FRhK-4 cells grown in 25-cm2flasks were infected with a multiplicity of infection of 1 to 10 50%
tissue culture infective doses/cell; after 1 week of incubation at 35°C under 5% CO2, cells were trypsinized, transferred to two 225-cm2flasks, and grown for 2
additional weeks in a 35°C CO2incubator. Cells were scrapped into 1 ml of
phosphate-buffered saline, pelleted, resuspended in 1 ml of reticulocyte standard buffer (RSB; 10 mM NaCl, 10 mM Tris-HCl [pH 7.2]) containing 1% Nonidet P-40 (NP-40), incubated 2 min at room temperature, and centrifuged at 12,0003 g for 1 min to remove nuclei. Supernatants containing cytoplasmic extracts were used immediately or stored at270°C.
Viruses were cloned by limiting dilution in 96-well plates containing confluent FRhK-4 cell monolayers. Cells were inoculated with 200ml of 10-fold serial dilutions of viral stocks and incubated at 35°C under 5% CO2. After 2 weeks,
supernatants were transferred to new 96-well plates, and cells were assayed for HAV replication by using125I-labeled human anti-HAV antisera (19). Wells
from the highest dilution that reacted with the125I-labeled human anti-HAV
antisera were selected, and half of the supernatant was used to inoculate con-fluent FRhK-4 cell monolayers in 25-cm2flasks. After 1 week incubation at 35°C
under 5% CO2, cytoplasmic extracts were prepared and stored as described
above.
Nucleotide sequence analysis.Cytoplasmic extracts of four 225-cm2flasks
containing confluent monolayers of FRhK-4 cells were treated for 2 h at room temperature with a mixture of detergents (0.5% sodium dodecyl sulfate [SDS], 1% sodium deoxycholate, 1% NP-40) and extracted with chloroform until there was no interface. HAV contained in the cytoplasmic extracts was pelleted through a 40% sucrose–0.5% Sarkosyl-NTE (150 mM NaCl, 10 mM Tris-HCl [pH 7.4], 1 mM EDTA [pH 8.0]) cushion by centrifugation at 40,000 rpm for 4 h at 4°C in a Beckman SW40 rotor, and genomic RNA was extracted with phenol– chloroform–1% SDS, precipitated with ethanol, and resuspended in 100ml of water. Reverse transcription with random hexamer primers was followed by PCR (Perkin-Elmer Cetus, Inc.) using synthetic oligonucleotide 1 (59-TCAAG AGGGGTCTCCGGGAAT T TCCGGAGTCCC TCT TGGAAG TCCATGG-39), a positive-sense primer corresponding to nt 1 to 50 of HAV, and 2 (59-AG CACCAGTCACTGCAGTCCTATC-39), a negative-sense primer correspond-ing to nt 837 to 860 of HAV. Amplified PCR fragments were purified by TAE–1% agarose gel electrophoresis and sequenced by using a PCR-based
dideoxynucleotide termination method (fmol sequencing system kit; Promega) priming with oligonucleotide 2.
The nucleotide sequence of the N terminus of the polyprotein of HAV clones was obtained by automatic sequencing using an ABI Prism model 377 automatic sequencer. Briefly, cytoplasmic extracts from 25-cm2confluent monolayer of
FRhK-4 cells were extracted with phenol-chloroform, and nucleic acids were ethanol precipitated. One-twentieth of the nucleic acids extracted was used as the template for reverse transcription-PCR (RT-PCR) using synthetic oligonu-cleotides 1 and 2 as described above. The cDNA fragments were amplified further by nested PCR using synthetic oligonucleotides 3 (59-ATGATTAGCA TGGAGCTGTAGGAG-39), a positive-sense primer corresponding to nt 286 to 309 of HAV, and 4 (59-GGTCAAGACCACTCCCAACAGTCTGG-39), a neg-ative-sense primer corresponding to nucleotides 761 to 786 of HAV. Amplified cDNA was analyzed by TAE–1.2% low-melting-point agarose gel electrophore-sis, and bands of expected molecular weights were cut and purified. Amplified bands were sequenced by using an ABI PRISM Dye terminator cycle sequencing ready reaction kit (Perkin-Elmer Cetus, Inc.) and synthetic oligonucleotide 5 (59-TGGCCTTAAATGGGATTCTGTGAG-39), a positive-sense primer corre-sponding to nucleotides 606 to 629 of HAV.
Western blot analysis.Cytoplasmic extracts of 107FRhK-4 cells were
frac-tionated by polyacrylamide gel electrophoresis (PAGE) in SDS–12.5% polyacryl-amide gels (30), and proteins were transferred to nylon (Zeta-probe; Bio-Rad, Inc.) or polyvinylidene difluoride (Immobilon-P; Millipore, Inc.) membranes, probed with 1:5,000 dilution of rabbit VP4, rabbit VP2, human anti-HAV, mouse anti-CSP MAb 2A10, or mouse anti-FLAG MAb M2, and stained with 1:1,000 dilution of alkaline phosphatase-labeled goat anti-rabbit, anti-hu-man, or anti-mouse antibody, respectively. 5-Bromo-4-chloro-3-indolylphos-phate–nitroblue tetrazolium was used as a substrate as recommended by the manufacturer (Kirkegaard & Perry Laboratories). Alternatively, 200ml of cyto-plasmic extracts obtained from 23107to 33107cells was immunoprecipitated
with 5ml of human anti-HAV antibody and 50ml of 10% (wt/vol) formaldehyde-denatured staphylococcus protein A (Sigma Co.) in 700ml of PLB buffer (0.1% SDS, 1% sodium deoxycholate, 1% Triton X-100, 0.05% sodium azide) at 4°C for 2 h. Western blot analysis of immunoprecipitated proteins was done as described above.
Sedimentation analysis of HAV constructs.Cytoplasmic extracts of 23108
cells were prepared in 2 ml of RSB–1% NP-40 as described above and treated with 0.4% sodium deoxycholate–0.4% Sarkosyl–0.2% NP-40 for 2 h at room temperature. Cellular debris were pelleted in a microcentrifuge at 14,0003g for 15 min, and the supernatants were loaded onto continuous 15 to 30% sucrose gradients in NTE buffer containing 0.25% NP-40. Gradients were centrifuged for 2 h at 40,000 rpm in a Beckman SW41 rotor at 4°C and collected from the bottom into 20 fractions.35S-labeled poliovirus 160S and 80S particles (27) were run in
similar 15 to 30% sucrose gradients as sedimentation markers, and peaks were identified by scintillation counting. The first 16 fractions of each gradient were immunoprecipitated with human anti-HAV antisera as described above, and half of the immunoprecipitated proteins from each fraction were subjected to West-ern blot analysis probing with either rabbit anti-VP2 Ab or anti-NANP 2A10 MAb.
RESULTS
Insertion of a multiple cloning site immediately downstream of the polyprotein initiation codons.To test whether HAV can tolerate the insertion of exogenous sequences at the N termi-nus of the polyprotein, we cloned synthetic oligonucleotides immediately downstream of the AUG initiation triplets at the unique XbaI site (nt 744) of pT7HAVXbaI. Preliminary results indicated that cloning of oligonucleotides coding for a poly-linker and a tag epitope at this XbaI site resulted in highly unstable HAV constructs which deleted the inserted sequences upon few passages in FRhK-4 cells. To circumvent this stability problem, we cloned into the unique XbaI site of pT7HAVXbaI complementary 60-mer synthetic oligonucleotides coding for a polylinker containing SalI, SnaBI, and KpnI restriction sites, followed by the 2B/2C cleavage site ELRTQ/SFSN for HAV protease 3Cpro(33) and peptide MNMS, which restored the native VP4 sequence (Fig. 1). The resulting construct, pT7HAVvec1, contains two in-frame sets of adjacent AUG initiation codons immediately downstream of the HAV IRES. Consequently, ribosomes could initiate translation of the polyprotein at (i) the first set of AUG codons, giving rise to a protein larger-than-VP0 that we named ExtVP0, (ii) the sec-ond set of AUG codons, giving rise to native VP0, or (iii) both sets of AUG codons, giving rise simultaneously to ExtVP0 and VP0 (Fig. 1). It is also possible that 3Cprocould recognize the
on November 9, 2019 by guest
http://jvi.asm.org/
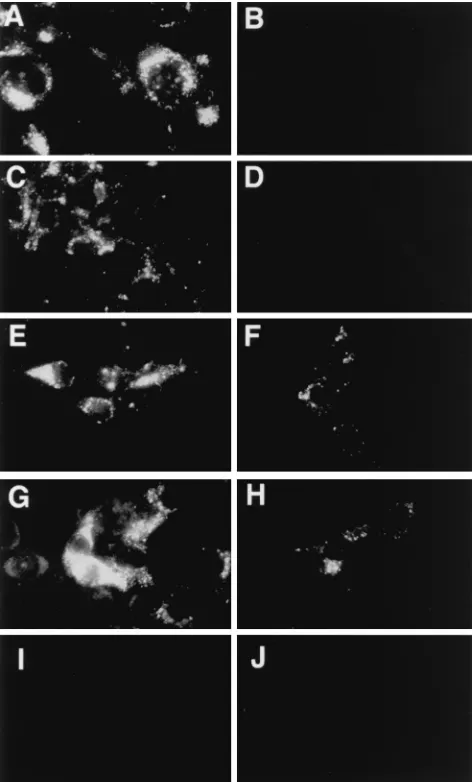
2B/2C inserted site cleaving the putative ExtVP0 protein. A similar strategy has been successfully used in PV expression vectors (7). T7 RNA polymerase in vitro transcripts of pT7HAVvec1 were transfected into FRhK-4 cells, and repli-cation of HAVvec1 was detected 14 days posttransfection by indirect IF using anti-HAV MAbs. Under similar conditions, replication of HAV HM175 was detected 5 to 7 days after transfection of in vitro transcripts of pT7HAV into FRhK-4, which suggested that replication of HAVvec1 was delayed due to the insertions of sequences at the N terminus of the polypro-tein. Stocks of HAV HM175 and HAVvec1 were prepared and passaged twice in FRhK-4 cells, and infected cells were ana-lyzed by indirect IF using anti-HAV MAbs (Fig. 2), which revealed that HAV HM175-infected (Fig. 2A) and HAVvec1-infected (Fig. 2C) cells had the characteristic granular cyto-plasmic fluorescence of HAV-infected cells, whereas no fluo-rescence was detected in cells stained with control anti-FLAG MAb M2 (Fig. 2B and D). It should be pointed out that
mock-infected FRhK-4 cells stained with anti-HAV MAbs (Fig. 2I) did not fluoresce, confirming that the staining was HAV specific. RT-PCR and nucleotide sequence analysis of the amplified cDNA revealed that HAVvec1 coded for the expected 60 nt inserted at the N terminus of the polyprotein (Fig. 1) and that these sequences were stably maintained for at least six serial passages in FRhK-4 cells.
HAV-mediated expression of a FLAG epitope. Since our data indicated that HAV could tolerate insertions at the N terminus of the polyprotein, it was of interest to determine whether the inserted sequences were expressed in infected cells. To do so, we cloned complementary synthetic oligonu-cleotides coding for the FLAG octapeptide DTKDDDDK (IBI) into the SalI and KpnI sites of the polylinker of pHAV-vec1. The resulting plasmid, pHAVvec1Flag12 (Fig. 1), was transcribed in vitro and transfected into FRhK-4 cells. Repli-cation of HAVvec1Flag12 was detected 25 days posttransfec-tion by indirect IF analysis using anti-HAV MAbs. A stock of HAVvec1Flag12 was prepared and passaged twice in FRhK-4 cells, and infected cells were analyzed by indirect IF using anti-Flag M2 and anti-HAV MAbs (Fig. 2), which showed the presence of FLAG (Fig. 2F) and HAV epitopes (Fig. 2E), respectively. Mock-infected (Fig. 2J), HAV HM175-infected (Fig. 2B), and HAVvec1-infected (Fig. 2D) FRhK-4 cells stained with anti-FLAG MAb M2 did not fluoresce, indicating that the staining was FLAG specific. These data confirmed that HAV can mediate the expression of an epitope tag inserted at the N terminus of the polyprotein.
To analyze whether the inserted 2B/2C cleavage site was necessary for the HAV-mediated expression of the FLAG epitope, we deleted the Gln residue required for the 3Cpro -mediated cleavage of this site (2). The resulting plasmid, pT7HAVvec1Flag34, was transcribed in vitro and transfected into FRhK-4 cells. After 20 days posttransfection, indirect IF analysis using anti-HAV MAbs revealed the characteristic granular fluorescence of HAV-infected cells. A stock of HAVvec1Flag34 virus was prepared and passaged twice in FRhK-4 cells, and infected cells were analyzed by indirect IF using anti-FLAG M2 and anti-HAV MAbs (Fig. 2), which showed the presence of FLAG (Fig. 2H) and HAV (Fig. 2G) epitopes, indicating that the 3Cpro-mediated cleavage of the inserted 2B/2C site was not required for the HAV-mediated expression of the tag epitope. It is possible that the lower level of the FLAG-specific fluorescence (Fig. 2F and H) compared to the HAV-specific fluorescence (Fig. 2E and G) was due to the intrinsic characteristics of the MAbs used in the staining. However, the preferential initiation of translation from the second set of initiation codons, the possible deletion of the inserted sequences, and/or the proteolytic degradation of ExtVP0 could also play a role in the lower level of FLAG-specific fluorescence.
The FLAG peptide accumulates as ExtVP0 in infected cells.
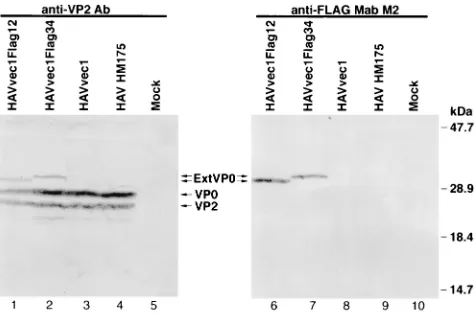
[image:4.612.52.288.69.460.2]To further analyze the HAV-mediated expression of the tag epitope, we studied cytoplasmic extracts of infected and mock-infected FRhK-4 cells by Western blot analysis probing with anti-FLAG MAb M2, rabbit anti-VP4, and human anti-HAV antibodies (Fig. 3). Anti-FLAG MAb M2 recognized a 31-kDa band in HAVvec1Flag12-infected cells (closed arrowhead, lane 1) and a 32-kDa band in HAVvec1Flag34-infected cells (open arrowhead, lane 2), but it did not react with HAVvec1-, HAV-, and mock-infected cells (lanes 3 to 5). The anti-VP4 antibody also recognized a 31-kDa band in HAVvec1Flag12-infected cells (closed arrowhead, lane 6) and a 32-kDa band in HAVvec1Flag34-infected cells (open arrowhead, lane 7) which comigrated with similar bands detected with MAb M2 (com-pare lanes 1 and 2 with lanes 6 and 7). As expected, the FIG. 2. IF analysis of HAV-infected cells. Monolayers of FRhK-4 cells grown
in eight-well slides were infected with HAV HM175 (A and B), HAVvec1 (C and D), HAVvec1Flag12 (E and F), or HAVvec1Flag34 (G and H) or were mock infected (I and J), acetone fixed, treated with a mixture of HAV-neutralizing MAbs K2-4F2, K3-4C8, and K3-2F2 (A, C, E, G, and I) or anti-FLAG MAb M2 (B, D, F, H, and J), and stained with biotinylated anti-mouse antibodies and FITC-labeled avidin.
on November 9, 2019 by guest
http://jvi.asm.org/
anti-VP4 Ab reacted specifically against a 29-kDa VP0 band present in HAV-infected (lanes 6 to 9) but not in mock-in-fected cells (lane 10) (23); this Ab also detected a faint cellular band of approximately 20 kDa in HAV-infected (lanes 6 to 9) and mock-infected (lane 10) cells. Surprisingly, the anti-VP4 Ab did not detect larger-than-VP0 proteins in HAVvec1-in-fected cells (lane 8), indicating that the sequences inserted at the N terminus of the polyprotein were poorly translated, not translated at all, or degraded. The expression of larger-than-VP0 proteins in HAVvec1Flag12- and HAVvec1Flag34-in-fected cells that reacted with anti-VP4 Ab and anti-FLAG MAb M2 indicated that the FLAG epitope accumulated fused to VP0, forming a novel set of proteins that we defined as ExtVP0 proteins. The putative 3Cpro-mediated cleavage of ExtVP0 of HAVvec1Flag12 should have yielded a FLAG-con-taining peptide of 19 to 21 residues and VP0 conFLAG-con-taining 4 residues more than native VP0; however, we did not de-tect such protein species by Western blot analysis. On the other hand, the presence of native VP0 and ExtVP0 in HAVvec1Flag12- and HAVvec1Flag34-infected cells sug-gested the possibility that there was a dual initiation of trans-lation of the polyprotein starting from two sets of AUG initi-ation codons: the first set located immediately downstream of the 39end of the IRES that initiates translation of ExtVP0, and the second set located 24 to 25 codons downstream that ini-tiates translation of native VP0. Our results did not rule out the unlikely possibility that ExtVP0 was digested by cellular proteases, giving rise to a single VP0 molecule that comigrated with native VP0. It is also interesting that we did not detect larger-than-VP4 proteins corresponding to VP4 fused to the FLAG epitope. Such proteins would result from the putative maturation cleavage of ExtVP0 to form VP2 and a VP4-FLAG fusion peptide ranging in size from the 21- to 23-residue VP4 to the 46- to 48-residue VP4-FLAG fusion peptide in HAVvec1Flag12. The human anti-HAV polyclonal antisera reacted strongly against a 33-kDa protein present in HAV-infected cells but not in mock-HAV-infected cells that probably cor-responded to VP1 (Fig. 3; compare lanes 11 to 14 with lane 15), which suggested that, besides VP0, migration of other viral
proteins was not affected by the insertions at the N terminus of the polyprotein. It should be pointed out that cytoplasmic extracts from similar amounts of cells were loaded in each well; therefore, the weak reaction of the anti-VP4 Ab with the HAVvec1Flag34-infected cell extract (lane 7) reflects a lower amount of viral proteins present in that particular extract.
To improve the specificity and sensitivity of the detection of HAV proteins in infected cells, cytoplasmic extracts were im-munoprecipitated with human anti-HAV antiserum prior to Western blot analysis, using anti-VP2 Ab and anti-FLAG MAb M2 (Fig. 4). The anti-VP2 Ab detected a 27-kDa VP2 band in HAV-infected (lanes 1 to 4) but not mock-infected (lane 5) cells. A faint band migrating faster than VP2 was also detected by the anti-VP2 Ab in infected (lanes 1 to 4) but not mock-infected (lane 5) cells, indicating that it was of viral origin and typically present in HAV-infected cells. The anti-VP2 Ab also reacted with a 29-kDa VP0 band in HAV HM175-, HAVvec1-, HAVvec1Flag12-, and HAVvec1Flag34-infected cells (lanes 1 to 4). ExtVP0 bands of 31 and 32 kDa, which were previously recognized by the anti-VP4 Ab (Fig. 3), also reacted with the anti-VP2 Ab in HAVvec1Flag12-infected (lane 1) and HAVvec1Flag34-infected (lane 2) cells, respectively. The anti-FLAG MAb M2 also recognized ExtVP0 bands of 31 and 32 kDa in HAVvec1Flag12-infected (lane 6) and HAVvec1 Flag34-infected (lane 7) cells, but it did not react with HAV-vec1-, HAV HM175-, and mock-infected cells (lanes 8 to 10), indicating that the reaction was FLAG specific. Although the immunoprecipitation step prior to the Western blot analysis resulted in a stronger signal of the viral proteins and a lower background of the assay, we were unable to detect bands cor-responding to (i) VP4 and VP4-FLAG fusion proteins coming from the maturation cleavage of VP0 and ExtVP0, respec-tively, and (ii) FLAG peptides and VP0 containing four extra residues coming from the 3Cpromediated cleavage of the in-serted 2B/C site.
ExtVP0 in HAVvec1Flag12-infected cells should have con-tained one Gln residue more than in pHAVvec1Flag34-in-FIG. 3. Western blot analysis of cell extracts of HAV-infected cells.
[image:5.612.53.290.69.204.2]Cyto-plasmic extracts of HAVvec1Flag12-infected (lanes 1, 6, and 11), HAVvec1 Flag34-infected (lanes 2, 7, and 12), HAVvec1-infected (lanes 3, 8, and 13), HM175 HAV-infected (lanes 4, 9, and 14), and mock-infected (lanes 5, 10, and 15) FRhK-4 cells were prepared in RSB–1% NP-40. Proteins were separated by SDS-PAGE (12.5% gel), transferred to nylon membranes, and probed with anti-FLAG MAb M2 (lanes 1 to 5), rabbit anti-VP4 Ab (lanes 6 to 10), or human anti-HAV Ab (lanes 11 to 15). The closed arrowheads point to a 31-kDa ExtVP0 band in HAVvec1Flag12-infected cells; the open arrowheads point to a 32-kDa ExtVP0 band in HAVvec1Flag34-infected cells. The positions and sizes of prestained molecular weight markers are shown on the left. The migration of VP0 and VP1 is indicated with arrows.
FIG. 4. Western blot analysis of immunoprecipitated cytoplasmic extracts of HAV-infected cells. Cytoplasmic extracts of HAVvec1Flag12-infected (lanes 1 and 6), HAVvec1Flag34-infected (lanes 2 and 7), HAVvec1-infected (lanes 3 and 8), HAV HM175-infected (lanes 4 and 9), and mock-infected (lanes 5 and 10) FRhK-4 cells were prepared in RSB–1% NP-40. Cytoplasmic extracts were immunoprecipitated with human anti-HAV antisera and pelleted proteins were separated by SDS-PAGE (12.5% gel), transferred to nylon membranes, and probed with rabbit anti-VP2 antibody or anti-FLAG MAb M2. The migration of ExtVP0, VP0, and VP2 is indicated with arrows. The closed arrowhead points to a smaller-than-VP2 band detected only in HAVvec1Flag01-infected cells. The positions and sizes of prestained molecular weight markers are shown on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.311.549.470.627.2]fected cells (Fig. 1); however, Western blot analysis showed that it migrated faster than ExtVP0 of HAVvec1Flag34 (Fig. 3 and 4). This discrepancy in the migration of the ExtVP0 pro-teins suggested that deletions occurred during viral replication of the FLAG-containing constructs. Since HAVvec1Flag12-and HAVvec1Flag34-infected cells contained similar amounts of ExtVP0 (Fig. 3, lanes 6 and 7), it is likely that the higher level of VP0 in the HAVvec1Flag12-infected cells was due to a greater proportion of revertant viruses that deleted the in-serted sequences.
Stability of the FLAG-containing HAV constructs.The sta-bility of the HAV constructs was tested by reverse transcription of viral RNA, PCR amplification using HAV primers flanking the inserted sequences, and nucleotide sequence analysis of the amplified cDNA. While HAVvec1 had the expected se-quence (Fig. 1) and remained stable for at least six serial passages, the RT-PCR analysis of stocks of HAVvec1Flag12 and HAVvec1Flag34 showed several bands that migrated at and below the expected size. Nucleotide sequence analysis of the amplified HAVvec1Flag12 and HAVvec1Flag34 cDNAs was ambiguous due to the superimposition of deleted se-quences accumulated during viral replication. These data agree well with the data obtained by Western blot analysis and indicate that the FLAG-containing constructs are unstable. To analyze individual sequences, viral stocks were cloned by lim-iting dilution in 96-well plates containing confluent FRhK-4 cell monolayers. Expression of the FLAG epitope was deter-mined in 16 randomly selected clones of each construct by Western blot analysis using MAb M2. Western blot analysis of the eight HAVvec1Flag12 clones that expressed the FLAG epitope showed that the anti-FLAG MAb M2 reacted very strongly with the 31-kDa ExtVP0 band and the anti-VP2 Ab reacted with the 31-kDa ExtVP0, 29-kDa VP0, and 27-kDa VP2 bands, which further suggested that the FLAG epitope was expressed by replication-competent HAV recombinants.
[image:6.612.57.546.64.276.2]Two clones that did not react (clones 3 and 7) and one that did react (clone 10) strongly with anti-FLAG MAb M2 were sub-jected to nucleotide sequence analysis (Fig. 5A). Clones 3 and 7 deleted 43 and 71 nt of the inserted sequences, respectively, resulting in smaller out-of-frame insertions, whereas clone 10 deleted only 38 nt of the inserted sequences and still coded for the DYKXXD core epitope recognized by the anti-FLAG MAb M2 (38) inserted in frame with the polyprotein. The nucleotide sequence of an additional HAVvec1Flag12 clone that reacted with the anti-FLAG MAb M2 (clone 6) was iden-tical to that of clone 10, which further confirmed that the M2 core epitope in the context of the HAV recombinants was recognized by the anti-FLAG MAb. It should be pointed out that in clone 10, the A residue of the first AUG triplet from the second set of initiation codons was also deleted. A similar analysis showed that only 3 of 16 randomly selected HAVvec1Flag34 clones strongly expressed the FLAG epitope and produced mature VP0 and ExtVP0, as assessed by West-ern blot analysis probing with anti-VP2 Ab. Nucleotide se-quence analysis (Fig. 5B) revealed that while clones 8 and 9, which did not express the FLAG epitope, deleted all inserted sequences, clone 10, which expressed the FLAG epitope, re-tained all 72 inserted nucleotides. In addition, an A residue preceding the AUG initiation codons of the polyprotein was deleted in clone 8. These results showed that stocks of HAVvec1Flag12 and HAVvec1Flag34 are heterogeneous and contain viruses with different deletions at the N terminus of the polyprotein. Moreover, these experiments allowed us to esti-mate the proportion of revertants that deleted the inserted sequences in the stocks of HAV recombinants. Since 50% of the HAVvec1Flag12 clones expressed ExtVP0 with a molecu-lar weight smaller than expected (due to the deletion of part of the inserted sequences [Fig. 5A]) and none of the isolated clones contained the full-length inserted sequence, we con-cluded that 100% of the viruses of the HAVvec1Flag12 stock FIG. 5. Nucleotide sequence alignment of the N termini of the polyproteins of cloned viruses. Stocks of FLAG-containing HAV were cloned by limiting dilution. Sequencing of three representative clones of each construct was done by RT-PCR. The nucleotide sequence of the N terminus of the polyprotein of each viral clone was compared with the sequence of the originating construct. (A) Clones 3, 7, and 10 were isolated from the HAVvec1Flag12 viral stock. Only clone 10 reacted with anti-FLAG MAb M2. (B) Clones 8, 9, and 10 were isolated from the HAVvec1Flag34 viral stock. Only clone 10 reacted with anti-FLAG MAb M2. Horizontal arrows indicate the initiation sites for translation of ExtVP0 and VP0. Unique restriction sites are underlined; the sequence of the FLAG epitope is marked with an horizontal line; a dash indicates the deletion of a nucleotide; three dots indicate the continuation of the HAV HM175 polyprotein sequence. The putative cleavage of the inserted 2B/2C site by 3Cprois indicated with a vertical arrow.
on November 9, 2019 by guest
http://jvi.asm.org/
contained deletions in the inserted sequences. On the other hand, 3 of 16 HAVvec1Flag34 clones expressed full-length ExtVP0 (Fig. 5B, clone 10), which indicated that approximately 80% of the viruses of the HAVvec1Flag34 stock contained deletions in the inserted sequences.
Stable HAV-mediated expression of an NANP epitope.To determine whether the instability of the recombinant viruses was due to the composition of the FLAG epitope, we ex-changed it with an immunodominant NANP epitope of the CSP of P. falciparum. To do so, synthetic oligonucleotides coding for the NPNANPN peptide were cloned into the SalI and KpnI sites of pT7HAVvec1Flag34, and the resulting plas-mid, pT7HAVvec1NANP2 (Fig. 1), maintained the same num-ber of amino acids inserted at the N terminus of the polypro-tein. In vitro T7 RNA polymerase transcripts of pT7HAVvec1 NANP2 were transfected into FRhK-4 cells, and replication of HAVvec1NANP2 was detected 3 weeks posttransfection by indirect IF using anti-HAV MAbs. A stock of HAVvec1NAN P2 was prepared, passaged twice in FRhK-4 cells, and studied by Western blot analysis (Fig. 6). Anti-VP2 Ab recognized a 33-kDa ExtVP0, a 29-kDa VP0, and a 27-kDa VP2 band in HAVvec1NANP2-infected cells (lane 1). The latter two bands comigrated with the VP0 and VP2 bands detected in HAV HM175-infected cells (lane 2), whereas no virus-specific bands were recognized in mock-infected cells (lane 3). Anti-CSP MAb 2A10 recognized only a 33-kDa ExtVP0 protein in HAVvec1NANP2-infected cells (lane 4) and did not react with any viral protein in HAV HM175-infected (lane 5) and mock-infected (lane 6) cells. These data indicated that the HAV vec1NANP2 ExtVP0 protein had the expected molecular weight and contained CSP and VP2 epitopes. Western blot analysis using anti-VP2 Ab and anti-CSP MAb 2A10 indicated that HAVvec1NANP2 was stable for at least six serial passages in FRhK-4 cells, which was further confirmed by RT-PCR and nucleotide sequence analysis. Moreover, HAVvec1NANP2 grew to similar titers in FRhK-4 cells as HAV HM175 and HAVvec1 and produced similar amounts of viral proteins and RNA. The stability of HAVvec1NANP2 suggested that the instability of HAVvec1Flag34, which differ only in the quence of the epitope tag, was mainly due to the FLAG se-quence itself. We did not detect deletions in the inserted sequences of HAVvec1NANP2 during six serial passages, as assessed by RT-PCR, nucleotide sequence analysis, and West-ern blot analysis, which indicated that the stock of this virus is composed of a homogeneous population and not a mixture of HAV mutants containing deletions in the inserted sequences
as found in the FLAG-containing HAV recombinant stocks (Fig. 5). The lack of deletions in the inserted sequences and the similar levels of ExtVP0 and VP0 proteins in HAVvec1NAN P2-infected cells (Fig. 6, lane 1) further supported the model of a dual initiation of translation of its polyprotein and confirmed that peptides fused to the N terminus of the polyprotein can be expressed by replication-competent HAV recombinants.
[image:7.612.61.283.69.162.2]The NANP epitope accumulates in HAVvec1NANP2-infect-ed cells as 80S empty particles. To determine whether the NANP epitope was present in HAV particles, cytoplasmic ex-tracts of HAV HM175-infected and HAVvec1NANP2-in-fected cells were sedimented over 15 to 30% sucrose density gradients. Fractions of each gradient were analyzed by West-ern blotting using anti-VP2 Ab and anti-CSP MAb 2A10 (Fig. 7). Cytoplasmic extracts of FRhK-4 cells infected with PV1/M and labeled with [35S]Met were sedimented in similar gradi-ents, and the positions of the 160S and 80S particles were determined by scintillation counting and used as sedimentation markers for HAV virions/provirions and empty capsids (5), respectively. In HAV HM175-infected cells (Fig. 7A), the anti-VP2 Ab detected a peak of virions and provirions (fractions 1 to 4) containing VP2 and traces of VP0 which comigrated with PV1/M 160S particles. The same Ab detected a peak of empty capsids (fractions 8 to 13) containing mainly VP0 which comi-grated with PV1/M 80S particles. In HAVvec1NANP2-in-fected cells, the anti-VP2 Ab (Fig. 7B) detected a peak of virions (fractions 1 to 4) containing VP2 and a peak of empty capsids (fractions 8 to 13) containing VP0 and ExtVP0 which also comigrated with PV1/M 160S and 80S markers, respec-tively. MAb 2A10, on the other hand, detected only ExtVP0 in HAVvec1NANP2-infected cells forming a peak of empty cap-sids (Fig. 7C, fractions 8 to 13). We do not know whether ExtVP0 could form provirions and undergo maturation cleav-age into a NANP-VP4 fusion protein and VP2, and we are currently performing experiments trying to address this impor-tant question. We were unable to detect NANP-VP4 fusion protein in HAVvec1NANP2-infected cells by Western blot analysis, suggesting that this peptide is never formed, is pro-duced after the maturation cleavage of ExtVP0 but is unstable, or is technically difficult to detect. However, our results clearly FIG. 6. Western blot analysis of the HAV-mediated expression of the CSP
epitope. Cytoplasmic extracts of HAVvec1NANP2-infected (lanes 1 and 4), HAV HM175-infected (lanes 2 and 5), or mock-infected (lanes 3 and 6) FRhK-4 cells were prepared in RSB–1% NP-40. Extracts were immunoprecipitated with human anti-HAV antisera, and pelleted proteins were separated by SDS-PAGE (12% gel), transferred to nylon membranes, and probed with rabbit anti-VP2 Ab or anti-CSP MAb 2A10. Arrows point to ExtVP0, VP0, and VP2 as revealed by the antibodies. The position and size of the prestained molecular weight marker are shown on the right.
FIG. 7. Sucrose density gradient ultracentrifugation of HAV particles. Cyto-plasmic extracts of FRhK-4 cells infected with HAV HM175 (A) or HAV vec1NANP2 (B) were ultracentrifuged on 15 to 30% sucrose gradients. Twenty fractions of 0.5 ml were collected from the bottom of the gradient, and 0.25 ml of each of the first 16 fractions was immunoprecipitated with human anti-HAV antisera. Pelleted proteins were separated by SDS-PAGE (12% gel), transferred to nylon membranes, and probed with rabbit anti-VP2 Ab (A and B) or anti-CSP MAb 2A10 (C). Arrows point to proteins recognized by the antibodies. Positions of the prestained 28.9-kDa molecular weight marker are indicated to the right. Sucrose gradient fractions 1 (bottom of the gradient) to 16 were analyzed. Migration in similar sucrose gradients of35S-labeled PV 160S and 80S particles
is indicated by vertical arrows. Fractions containing the peaks of HAV virions/ provirions (1 to 4) and empty capsids (8 to 13) are indicated with a line.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.309.545.500.618.2]showed that the NANP epitope contained in ExtVP0 accumu-lates in infected cells as empty capsids. Similar results were obtained with HAVvec1Flag12 and HAVvec1Flag34 where, despite the instability of the constructs, the FLAG peptide also accumulated in infected cells as empty capsids.
DISCUSSION
Although IRES elements of the different picornaviruses show little sequence homology, computer-assisted RNA fold-ing predictions showed three distinct groups (for a review, see reference 25): (i) enteroviruses and rhinoviruses, (ii) cardiovi-ruses and aphthovicardiovi-ruses, and (iii) hepatovirus. The 39 end of the IRES element in cardioviruses, aphthoviruses, and hepa-toviruses is adjacent to the authentic initiation site of the polyprotein, whereas in polioviruses and rhinoviruses, it is far upstream and initiation probably occurs following internal en-try and scanning of ribosomes (for a review, see reference 12). In the present study, the sequences inserted at the N terminus of the polyprotein were placed in a location analogous to that of the nonstructural L protein of cardioviruses and aphthovi-ruses. In some cardioviruses, the dual initiation of translation of the polyprotein proceeds from two AUG triplets separated by only 9 nt. In aphthoviruses, two AUG triplets located 84 bases apart initiate translation of two forms of the polyprotein, producing the Lab and Lb forms of the L protein (40) through a leaky scanning mechanism (11). Some of the viruses con-structed in this work, such as HAVvec1Flag12, HAVvec1Flag 34, and HAVvec1NANP2, mimic the aphthovirus dual initia-tion configurainitia-tion. Infecinitia-tion with HAVvec1NANP2, which is the most stable of our HAV constructs expressing a tag epi-tope, resulted in the expression of similar levels of ExtVP0 and VP0 (Fig. 6), indicating that the HAV IRES can most likely direct a dual initiation of translation. Since it has been sug-gested that the cardiovirus, aphthovirus, and hepatovirus IRES elements are related (14, 31), it is also interesting to speculate that their capability to direct the dual initiation of translation of the polyprotein is also a characteristic of this type of IRES. HAVvec1 contains two sets of AUG triplets separated by 17 codons, but initiation of translation of the polyprotein was detected only from the second set, resulting in the production native VP0 (Fig. 3 and 4). If translation of the polyprotein had started from the first set of initiation codons in HAVvec1, Western blot analysis using anti-VP2 and anti-VP4 Abs most likely would have detected a shift in the migration of VP0 due to the additional sequences inserted at the N terminus of the polyprotein. Although initiation of translation from the second set of AUG triplets in HAVvec1 seems the most likely sce-nario, we cannot completely rule out the possibility that pro-teolytic degradation of the inserted sequences resulted in a protein that comigrated with native VP0. Unfortunately, dele-tion of the second set of AUG triplets in the FLAG-containing HAV recombinants resulted in highly unstable viruses which rapidly deleted the inserted sequences (data not shown); there-fore, we could not use such constructs to determine whether proteases can mediate the unlikely cleavage of ExtVP0 into mature VP0. Dual initiation of translation of the polyprotein from both sets of initiation codons was detected only after a tag epitope was inserted into the polylinker of HAVvec1. Since sequences at and upstream of the first set of initiation codons were not modified, our data suggested that determinants downstream from the native initiation codons could play a role in regulating initiation of translation of HAV. Therefore, the insertion of foreign epitopes in HAV could be a useful tool to further understand the mechanism initiation of translation in HAV.
The rationale for introducing a 3Cprocleavage site at the N terminus of the polyprotein was to determine whether the viral protease could recognize it cutting some of the inserted se-quences, thus increasing the possibility of obtaining an infec-tious HAV cDNA construct. A similar approach was success-fully used with PV expression vectors by engineering a PV 3Cprocleavage site at the N terminus of the polyprotein, which, after being cleaved, restored the native sequence of VP4 (7). An identical approach of restoring the native VP4 sequence of HAV could not have been undertaken because, unlike the case for PV, the initiation AUGs of the HAV polyprotein are not posttranslationally cleaved, exposing a Gly residue (44). Since it was shown that purified 3Cpro efficiently cleaved synthetic peptide ELRTQ/SFSN coding for the 2B/2C junction (33), we inserted such sequences in HAVvec1 (Fig. 1). Unfortunately, due to the instability of the HAVvec1Flag12 construct, we were unable to determine whether 3Cpro recognized the in-serted 2B/2C cleavage site in the context of the expression vector. Moreover, it is possible that deletion of a Gln residue from the inserted 2B/2C cleavage site in HAVvec1Flag12 to generate HAVvec1Flag34 increased the stability of this con-struct and allowed the isolation of clones containing all the inserted sequences (Fig. 2, 3, and 5). It has recently been shown that the 2A/2B and 2C/3A junctions are the preferred HAV 3Cprocleavage sites and that longer periods are required for cleavage of the 2B/2C junction (41). Therefore, it is possi-ble that efficient cleavage of the inserted sequences can be achieved by exchanging the 2B/2C for the 2A/2B or 2C/3A site. Modification of the HAVvec1NANP2 construct to include other protease cleavage sites will help us to understand and possibly improve the proteolytic processing in the context of the recombinant HAV vectors.
Since the HAV constructs containing the FLAG epitope tended to be unstable, we constructed HAVvec1NANP2, in which the FLAG epitope was replaced by the CSP NPNANPN peptide. HAVvec1NANP2 was stable for more than six pas-sages as assessed by RT-PCR and nucleotide sequence analy-sis, suggesting that the amino acid composition of the FLAG peptide contributed to the instability of the construct. The stability of HAVvec1NANP2 allowed us to determine that the tag epitope accumulated in infected cells as empty capsids (Fig. 7). Although we did not detect ExtVP0 and VP0 forming HAVvec1NANP2 provirions and virions (Fig. 7B and C), we cannot rule out the possibility that ExtVP0 undergoes matu-ration cleavage into an NANP-VP4 fusion protein and VP2. More detailed experiments will be required to analyze the possible formation of HAVvec1NANP2 provirions and their maturation cleavage into virions (13).
Picornaviruses have been used as expression vectors to pro-duce live recombinant vaccines (1, 3, 4, 6–8, 10, 18, 20, 35–37, 39, 48). Although construction of HAV expression vectors has been difficult due to the poor growth characteristics of the virus, it is possible that such vectors can also be used to develop recombinant vaccines. In this study, we clearly showed that exogenous sequences can be cloned into the N terminus of the HAV polyprotein, resulting in replication-competent viruses. We also showed that the inserted sequences are expressed as ExtVP0 proteins and accumulate in infected cells as 80S empty particles. Our data, therefore, support the development of HAV expression vectors as a tool to study this poorly under-stood picornavirus and to produce recombinant vaccines.
ACKNOWLEDGMENTS
We thank Jean Paterson for constructing pT7HAV and pT7HAV XbaI and Kathleen Mihalik for producing anti-VP4 and anti-VP2 Abs. We also thank Stephen Feinstone for helpful discussions and Sara
on November 9, 2019 by guest
http://jvi.asm.org/
Gagneten, Barry Falgout, and Hira Nakhasi for comments on the manuscript.
REFERENCES
1. Alexander, L., H. H. Lu, M. Gromeier, and E. Wimmer. 1994. Dicistronic polioviruses as expression vectors for foreign genes. AIDS Res. Hum. Ret-roviruses 10(Suppl. 2):S57–S60.
2. Allaire, M., M. M. Chernaia, B. A. Malcolm, and M. N. James. 1994. Picornaviral 3C cysteine proteinases have a fold similar to chymotrypsin-like serine proteinases. Nature 369:72–76.
3. Altmeyer, R., N. Escriou, M. Girard, A. Palmenberg, and S. van der Werf. 1994. Attenuated Mengo virus as a vector for immunogenic human immu-nodeficiency virus type 1 glycoprotein 120. Proc. Natl. Acad. Sci. USA 91: 9775–9779.
4. Altmeyer, R., M. Girard, S. van der Werf, V. Mimic, L. Seigneur, and M. F.
Saron.1995. Attenuated mengovirus: a new vector for live recombinant vaccines. J. Virol. 69:3193–3196.
5. Anderson, D. A., and B. Ross. 1990. Morphogenesis of hepatitis A virus: isolation and characterization of subviral particles. J. Virol. 64:5284–5289. 6. Anderson, M. J., D. C. Porter, P. N. Fultz, and C. D. Morrow. 1996.
Polio-virus replicons that express the gag or the envelope surface protein of simian immunodeficiency virus SIV(smm) PBj14. Virology 219:140–149. 7. Andino, R., D. Silvera, S. D. Suggett, P. L. Achacoso, C. J. Miller, D.
Baltimore, and M. B. Feinberg.1994. Engineering poliovirus as a vaccine vector for the expression of diverse antigens. Science 265:1448–1451. 8. Ansardi, D. C., Z. Moldoveanu, D. C. Porter, D. E. Walker, R. M. Conry,
A. F. LoBuglio, S. McPherson, and C. D. Morrow.1994. Characterization of poliovirus replicons encoding carcinoembryonic antigen. Cancer Res. 54: 6359–6364.
9. Ansardi, D. C., D. C. Porter, and C. D. Morrow. 1992. Myristylation of poliovirus capsid precursor P1 is required for assembly of subviral particles. J. Virol. 66:4556–4563.
10. Arnold, G. F., D. A. Resnick, A. D. Smith, S. C. Geisler, A. K. Holmes, and
E. Arnold.1996. Chimeric rhinoviruses as tools for vaccine development and characterization of protein epitopes. Intervirology 39:72–78.
11. Belsham, G. J. 1992. Dual initiation of protein synthesis on foot-and-mouth disease virus RNA are selected following internal entry and scanning of ribosomes in vivo. EMBO J. 11:1105–1110.
12. Belsham, J. G., and N. Sonenberg. 1996. RNA-protein interactions in reg-ulation of picornavirus RNA translation. Microbiol. Rev. 60:499–511. 13. Bishop, N. E., and D. A. Anderson. 1993. RNA-dependent cleavage of VP0
capsid protein in provirions of hepatitis A virus. Virology 197:616–623. 14. Brown, E. A., A. J. Zajac, and S. M. Lemon. 1994. In vitro characterization
of an internal ribosomal entry site (IRES) present within the 59 nontrans-lated region of hepatitis A virus RNA: comparison with the IRES of en-cephalomyocarditis virus. J. Virol. 68:1066–1074.
15. Burkot, T. R., Z. W. Da, H. M. Geysen, R. A. Wirtz, and A. Sau. 1991. Fine specificities of monoclonal antibodies against the Plasmodium falciparum circumsporozoite protein: recognition of both repetitive and non-repetitive regions. Parasite Immunol. 13:161–170.
16. Chow, M., J. F. Newman, D. J. Filman, J. M. Hogle, D. J. Rowlands, and F.
Brown.1987. Myristylation of picornaviruses capsid protein VP4 and its structural significance. Nature 327:482–486.
17. Cohen, J. I., J. R. Ticehurst, S. M. Feinstone, B. Rosenblum, and R. H.
Purcell.1987. Hepatitis A virus cDNA and its RNA transcripts are infectious in cell culture. J. Virol. 61:3035–3039.
18. Dedieu, J. F., J. Ronco, S. van der Werf, J. M. Hogle, Y. Henin, and M.
Girard.1992. Poliovirus chimeras expressing sequences from the principal neutralization domain of human immunodeficiency virus type 1. J. Virol.
66:3161–3167.
19. Dotzauer, A., S. M. Feinstone, and G. Kaplan. 1994. Susceptibility of non-primate cell lines to hepatitis A virus infection. J. Virol. 68:6064–6068. 20. Evans, D. J., and J. W. Almond. 1991. Design, construction, and
character-ization of poliovirus antigen chimeras. Methods Enzymol. 203:386–400. 21. Francki, R. I. B., C. M. Fauquet, D. L. Knudson, and F. Brown. 1991.
Classification and nomenclature of viruses. Arch. Virol. 2:320–326. 22. Gauss-Muller, V., F. Lottspeich, and F. Deinhardt. 1986. Characterization of
hepatitis A virus structural proteins. Virology 155:732–736.
23. Gust, I. D., and S. M. Feinstone. 1988. Hepatitis A. CRC Press, Inc., Boca Raton, Fla.
24. Innis, B. L., R. Snitbhan, P. Kunasol, T. Laorakpongse, W. Poopatanakool,
C. A. Kozik, S. Suntayakorn, T. Suknuntapong, A. Safary, D. B. Tang, and et al.1994. Protection against hepatitis A by an inactivated vaccine. JAMA
271:1328–1334.
25. Jackson, R. J., S. L. Hunt, C. L. Gibbs, and A. Kaminski. 1994. Internal
initiation of translation of picornavirus RNAs. Mol. Biol. Rep. 19:147–159. 26. Jia, X. Y., M. Tesar, D. F. Summers, and E. Ehrenfeld. 1996. Replication of hepatitis A viruses with chimeric 59nontranslated regions. J. Virol. 70:2861– 2868.
27. Kaplan, G., M. S. Freistadt, and V. R. Racaniello. 1990. Neutralization of poliovirus by cell receptors expressed in insect cells. J. Virol. 64:4697–4702. 28. Kaplan, G., A. Totsuka, P. Thompson, T. Akatsuka, Y. Moritsugu, and S. M.
Feinstone.1996. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J. 15:4282– 4296.
29. Kusov, Y., Y. A. Kazachkov, G. K. Dzagurov, G. A. Khozinskaya, M. S.
Balayan, and V. Gauss-Muller.1992. Identification of precursors of struc-tural proteins VP1 and VP2 of hepatitis A virus. J. Med. Virol. 37:220–227. 30. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of
the head of bacteriophage T4. Nature 227:680–685.
31. Le, S. Y., J. H. Chen, N. Sonenberg, and J. Maizel, Jr. 1993. Conserved tertiary structural elements in the 59nontranslated region of cardiovirus, aphthovirus and hepatitis A virus RNAs. Nucleic Acids Res. 21:2445–2451. 32. MacGregor, A., M. Kornitschuk, J. G. Hurrell, N. I. Lehmann, A. G.
Coul-epis, S. A. Locarnini, and I. D. Gust.1983. Monoclonal antibodies against hepatitis A virus. J. Clin. Microbiol. 18:1237–1243.
33. Malcolm, B. A., S. M. Chin, D. A. Jewell, J. R. Stratton-Thomas, K. B.
Thudium, R. Ralston, and S. Rosenberg.1992. Expression and characteriza-tion of recombinant hepatitis A virus 3C proteinase. Biochemistry 31:3358– 3363.
34. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
35. Mattion, N. M., E. C. Harnish, J. C. Crowley, and P. A. Reilly. 1996. Foot-and-mouth disease virus 2A protease mediates cleavage in attenuated Sabin 3 poliovirus vectors engineered for delivery of foreign antigens. J. Vi-rol. 70:8124–8127.
36. Mattion, N. M., P. A. Reilly, E. Camposano, S. L. Wu, S. J. DiMichele, S. T.
Ishizaka, S. E. Fantini, J. C. Crowley, and C. Weeks-Levy.1995. Character-ization of recombinant polioviruses expressing regions of rotavirus VP4, hepatitis B surface antigen, and herpes simplex virus type 2 glycoprotein D. J. Virol. 69:5132–5137.
37. Mattion, N. M., P. A. Reilly, S. J. DiMichele, J. C. Crowley, and C.
Weeks-Levy.1994. Attenuated poliovirus strain as a live vector: expression of re-gions of rotavirus outer capsid protein VP7 by using recombinant Sabin 3 viruses. J. Virol. 68:3925–3933.
38. Miceli, R. M., M. E. DeGraaf, and H. D. Fischer. 1994. Two-stage selection of sequences from a random phage display library delineates both core residues and permitted structural range within an epitope. J. Immunol. Methods 167:279–287.
39. Porter, D. C., D. C. Ansardi, W. S. Choi, and C. D. Morrow. 1993. Encap-sidation of genetically engineered poliovirus minireplicons which express human immunodeficiency virus type 1 Gag and Pol proteins upon infection. J. Virol. 67:3712–3719.
40. Sangar, D. V., S. E. Newton, D. J. Rowlands, and B. E. Clarke. 1987. All foot-and-mouth disease virus serotypes initiate protein synthesis at two sep-arate AUGs. Nucleic Acids Res. 15:3305–3315.
41. Schultheiss, T., W. Sommergruber, Y. Kusov, and V. Gauss-Muller. 1995. Cleavage specificity of purified recombinant hepatitis A virus 3C proteinase on natural substrates. J. Virol. 69:1727–1733.
42. Sinnis, P., and B. L. Sim. 1997. Cell invasion by the vertebrate stages of Plasmodium. Trends Microbiol. 5:52–58.
43. Tesar, M., S. A. Harmon, D. F. Summers, and E. Ehrenfeld. 1992. Hepatitis A virus polyprotein synthesis initiates from two alternative AUG codons. Virology 186:609–618.
44. Tesar, M., X. Y. Jia, D. F. Summers, and E. Ehrenfeld. 1993. Analysis of a potential myristoylation site in hepatitis A virus capsid protein VP4. Virology
194:616–626.
45. Vaheri, A., and J. S. Pagano. 1965. Infectious poliovirus RNA: a sensitive method of assay. Virology 27:435–436.
46. Weitz, M., B. M. Baroudy, W. L. Maloy, J. R. Ticehurst, and R. H. Purcell. 1986. Detection of a genome-linked protein (VPg) of hepatitis A virus and its comparison with other picornaviral VPgs. J. Virol. 60:124–130. 47. Werzberger, A., B. Mensch, B. Kuter, L. Brown, J. Lewis, R. Sitrin, W.
Miller, D. Shouval, B. Wiens, G. Calandra, et al.1992. A controlled trial of a formalin-inactivated hepatitis A vaccine in healthy children. N. Engl. J. Med. 327:453–457.
48. Zhang, L., S. Sato, J. I. Kim, and R. P. Roos. 1995. Theiler’s virus as a vector for foreign gene delivery. J. Virol. 69:3171–3175.