0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
Multiple Effects of Mutations in Human Immunodeficiency
Virus Type 1 Integrase on Viral Replication
ALAN ENGELMAN,
1GEORGE ENGLUND,
2JAN M. ORENSTEIN,
3MALCOLM A. MARTIN,
2AND
ROBERT CRAIGIE
1*
Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases,
1and Laboratory of
Molecular Microbiology, National Institute of Allergy and Infectious Diseases,
2Bethesda, Maryland 20892, and
Department of Pathology, George Washington University, Washington, D.C. 20037
3Received 5 December 1994/Accepted 26 January 1995
The integration of a DNA copy of the human immunodeficiency virus type 1 (HIV-1) genome into a
chromosome of an infected cell is a pivotal step in virus replication. Integration requires the activity of the
virus-encoded integrase, which enters the cell as a component of the virion. Results of numerous mutagenesis
studies have identified amino acid residues and protein domains of HIV-1 integrase critical for in vitro activity,
but only a few of these mutants have been studied for their effects on HIV replication. We have introduced
site-directed changes into an infectious DNA clone of HIV-1 and show that integrase mutations can affect virus
replication at a variety of steps. We identified mutations that altered virion morphology, levels of
particle-associated integrase and reverse transcriptase, and viral DNA synthesis. One replication-defective mutant
virus which had normal morphology and protein composition displayed increased levels of circular viral DNA
following infection of a T-cell line. This virus also had a significant titer in a CD4-positive indicator cell assay,
which requires the viral Tat protein. Although unintegrated viral DNA can serve as a template for Tat
expression in infected indicator cells, this level of expression is insufficient to support a spreading viral
infection in CD4-positive lymphocytes.
Three pol gene products, protease (PR), reverse
tran-scriptase (RT), and integrase (IN), perform functions essential
for human immunodeficiency virus type 1 (HIV-1) replication
at different steps in the virus life cycle. PR processes viral
polyprotein precursors into the mature virion. Soon after
in-fection, RT, and its associated RNase H activity, copy the viral
RNA genome into double-stranded blunt-ended DNA. In an
initial processing reaction, IN cleaves this DNA near each 3
9
end, adjacent to the conserved dinucleotide CA. Following
nuclear localization, IN joins the recessed CA
OHends to two
59-phosphates in the two strands of a chromosomal target site
by a pair of DNA strand transfer reactions. The 5
9
ends of the
viral DNA and the 3
9
ends of the host DNA remain unjoined
in the resulting integration intermediate. Repair of the
inter-mediate, which is thought to be carried out by cellular
en-zymes, generates the integrated provirus. See reference 20 for
a recent review of the roles of PR, RT, and IN in virus
repli-cation.
Purified HIV-1 IN exhibits both 3
9
processing and DNA
strand transfer activities in vitro (3, 44, 53). Reactions require
substrates which mimic the ends of linear viral DNA, IN, and
a divalent metal ion cofactor. HIV-1 IN also catalyzes an
ap-parent reversal of the strand transfer reaction in vitro, a
pro-cess termed disintegration (6). In this reaction, branched
oli-gonucleotide DNA substrates, composed of both viral and
target DNA sequences, are resolved into their separate viral
and target DNA components.
Mutational analyses of HIV-1 IN indicate that the protein
consists of three functional domains: the N-terminal, core, and
C-terminal domains. Deletion mutagenesis has shown that all
three domains are required for 3
9
processing and DNA strand
transfer in vitro (7, 43, 52), whereas the core domain is
suffi-cient for disintegration activity (4, 32). The core domain is
composed of two subdomains; one part, originally identified as
a relatively protease resistant fragment of HIV-1 IN (9),
con-tains the D,D-35-E amino acid sequence motif (9, 24). This
motif is conserved in the integrases of retroviruses and
retro-transposons, as well as in the transposases of some bacterial
transposons (2, 14, 24, 36, 40). Certain single amino acid
sub-stitutions of the Asp and Glu residues which compose the
motif abolish the 3
9
processing, DNA strand transfer, and
disintegration activities of HIV-1 IN in vitro (9, 26, 48),
dem-onstrating the importance of each residue in the catalysis of
polynucleotidyl transfer. The other part of the core domain lies
C terminal to the protease-resistant fragment and is necessary
for both the DNA binding and disintegration activities of the
domain (11). In addition to its central role in catalysis, the core
domain contributes to interactions between integrase
pro-tomers (8, 19, 32, 49).
The hypothesis that the conserved residues of the D,D-35-E
sequence motif form part of a common IN active site is
sup-ported by the result that both 3
9
processing and DNA strand
transfer are one-step transesterification reactions (12). The
functions of the N- and C-terminal domains in the 3
9
process-ing and DNA strand transfer reactions in vitro are less well
understood than those of the core domain. The C-terminal
domain is important for nonspecific binding of intact HIV-1 IN
to DNA in vitro (11, 52, 57) and therefore may interact with
target DNA during DNA strand transfer. Although the precise
roles of the terminal domains in the 3
9
processing and DNA
strand transfer reactions are unknown, the N- or C-terminal
functions, and the core functions, can be provided by separate
polypeptides in reactions that contain a pair of mutant IN
proteins (8, 49).
In this study, the effects of mutations on IN function were
investigated by introducing changes into an infectious
molec-ular clone of HIV-1
NL4-3and assaying virus replication. Our
* Corresponding author. Mailing address: NIDDK, LMB, Building 5, Room 301, 5 Center Dr. MSC 0560, Bethesda, MD 20892-0560. Phone: (301) 496-4081. Fax: (301) 496-0201. Electronic mail address: [email protected].
2729
on November 9, 2019 by guest
http://jvi.asm.org/
Construction of mutant HIV-1 proviral DNAs.A PCR-based mutagenesis strategy was used to introduce site-directed mutations in the pol gene of pNL4-3, an infectious plasmid DNA clone of HIV-1NL4-3(1). Each mutation was incor-porated into a convenient restriction site fragment by using two successive PCRs essentially as previously described (17). The first set of reactions used pNL4-3 as a template to generate the two halves of the fragment, each half containing a restriction site near one end and the mutation near the other end. The mutagenic primers were chosen so that the ends of the halves which contained the mutation overlapped. The full-length fragment was generated by using the overlapping halves as the template and the two primers that contained the restriction sites. PCR mixtures contained 4 ng of DNA template and 5 ng of each primer per ml and 2 U of Vent DNA polymerase (New England Biolabs, Beverly, Mass.), using buffer conditions specified by the manufacturer. Reaction mixtures were kept at 968C for 2 min and then subjected to 20 cycles of denaturation at 968C (15 s), annealing at 588C (1 min), and extension at 748C (45 s). Amplified fragments were purified following polyacrylamide gel electrophoresis (PAGE).
IN mutations were incorporated into the AgeI-PflMI fragment between bases 3485 and 5297 (33). The 1-234 and 1-4 deletion mutations were made by intro-ducing termination codons after the codons encoding Val-234 and Gly-4, respec-tively. For the 1-234 mutation, 59-GTTTGGAAAGGA was changed to 59-GTT TGATAATGA, where 59-GTT encodes Val-234. For 1-4, 59-GGAATAGATA AGGCC was changed to 59-GGATAGTAATAGCC, where 59-GGA encodes Gly-4 and the underlined base was deleted.
The single base substitution G3A at position 3105, encoding the amino acid substitution Asp-1863Asn in the active site of RT, was incorporated into the
ApaI-AgeI fragment between bases 2006 and 3485. Following digestion with
restriction endonucleases, the PCR-amplified fragments were ligated with the appropriately digested, gel-purified pNL4-3 DNA backbone by using standard techniques (41). The regions of the resulting plasmids synthesized by PCR were sequenced by the chain termination method (42).
Cells and viruses.HeLa and 293 cells were grown in Dulbecco modified Eagle
medium (DMEM) supplemented to contain 5 and 10% fetal calf serum, respec-tively, 100 U of penicillin G sodium, and 0.1 mg of streptomycin sulfate per ml. Cells were plated at 3.63104
cells per cm2
16 h prior to transfection. Virus stocks were prepared by transfecting cells with 10mg of plasmid DNA in the presence of calcium phosphate, using the Stratagene (La Jolla, Calif.) mamma-lian transfection kit and 106
293 cells or the Pharmacia (Piscataway, N.J.) Cell-Phect kit and 3.53105HeLa cells. Culture supernatants were tested for Mg21
-dependent 32P-RT activity 48 h posttransfection essentially as previously described (55). Supernatants were filtered through 0.45-mm-pore-size nitrocel-lulose, and equal32P-RT counts per minute of wild-type HIV-1
NL4-3and mutant viruses were used in subsequent infections. Phenotypically mixed virus particles were made by mixing plasmid DNAs prior to transfection.
CEM-12D7 cells (37), a subclone of the CEM-A3.01 human T-cell line (15), were grown in RPMI 1640 supplemented to contain 10% fetal calf serum, 100 U of penicillin G sodium, and 0.1 mg of streptomycin sulfate per ml. Cells (2.53
106) were infected with 2.53105 32P-RT cpm in 0.5 ml of RPMI 1640 for 1 h at 378C prior to addition of 5 ml of RPMI 1640 supplemented with 10% fetal calf serum. Cultures were fed at 2-day intervals, and aliquots of the medium were assayed for32P-RT activity. Each mutant virus was tested in at least two inde-pendent experiments.
The CD4-LTR/b-gal indicator cell line, obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Al-lergy and Infectious Diseases, from Michael Emerman (22), was maintained in DMEM supplemented to contain 10% fetal calf serum, 100 U of penicillin G sodium, 0.1 mg of streptomycin sulfate, 0.2 mg of G418 sulfate, and 0.1 mg of hygromycin B per ml. Cells were seeded at 0.43105cells per well of a 24-well plate 24 h prior to infection. Virus (150ml) was adsorbed in the presence of 3mg of DEAE-dextran for 2 h at 378C prior to addition of 1 ml of medium. The cells were fixed and stained with 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal) 48 h postinfection as previously described (22).
Analysis of HIV-1 proteins.Both cell- and particle-associated HIV-1 proteins were analyzed by radiolabeling transfected HeLa cells and immunoprecipitation with AIDS patient antisera (obtained through the AIDS Research and Refer-ence Reagent Program from Alfred Prince) (34, 35) essentially as previously described (16, 54). Lysates of pelleted virions were also immunoprecipitated with 40ml of rabbit polyclonal antisera (Assay Research, Inc., College Park, Md.) generated against purified recombinant HIV-1 IN.
Quantitation of immunoprecipitated proteins was done with a PhosphorIm-ager using ImageQuant 3.3 (Molecular Dynamics, Sunnyvale, Calif.) and on a Macintosh Quadra 650 using the public domain NIH Image program (written by Wayne Rasband at the National Institutes of Health and available from the
CEM-12D7 cells (3.0310) were infected with 3.0310 P-RT cpm of each virus as described above. The cells were washed with 1 ml of DMEM 18 h postinfection and resuspended in 0.25 ml of DMEM. RQ1 RNase-free DNase (20 U; Promega Corporation, Madison, Wis.) was added for 20 min at 378C to digest residual plasmid DNA. Trypsin (Gibco BRL Life Technologies) was added to the final concentration 0.005% (wt/vol), and the mixture was incubated at 378C for 10 min to degrade the DNase. The cells were washed twice with 1 ml of phosphate-buffered saline (PBS) and lysed in 0.5 ml of 10 mM Tris-HCl (pH 8.1)–1 mM EDTA–0.001% (vol/vol) Triton X-100–0.0001% (wt/vol) sodium do-decyl sulfate (SDS). Proteinase K (50mg) was added, and the lysates were heated at 568C for 1 h and then at 958C for 10 min.
PCR mixtures contained 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.75 mM MgCl2, 0.02% (wt/vol) gelatin, 0.2 mM each deoxynucleoside triphosphate, 1mM each primer, 25ml of infected cell lysate (corresponding to approximately 1mg of total DNA), and 1.25 U of AmpliTaq DNA polymerase (Perkin-Elmer, Roche Molecular Systems, Inc., Branchburg, N.J.) in a volume of 50ml. The reaction components were mixed at room temperature, overlaid with 70 ml of light mineral oil, and heated to 948C prior to addition of the DNA polymerase. Thirty-four cycles of denaturation, annealing, and polymerization were per-formed at 948C (45 s), 608C (30 s), and 728C (2 min), respectively. Portions (10
ml) of each reaction mixture were electrophoresed in 6% polyacrylamide gels (Novex, San Diego, Calif.). Gels were stained with 0.5mg of ethidium bromide per ml to visualize the DNA.
Electron microscopy.HeLa cells were transfected with 10mg of plasmid DNA
as described above. Two days posttransfection, cells were washed once with cold PBS, and 2.5% (vol/vol) glutaraldehyde (Sigma no. G5882) in PBS was added. Cells were fixed at 48C for at least 24 h and processed for thin-section microscopy as previously described (16).
RESULTS
Mutagenesis strategy.
HIV-1 IN contains three distinct
functional domains: the N-terminal, core, and C-terminal
do-mains. Each of the domains is required for IN activity in assays
for 3
9
processing and DNA strand transfer in vitro (4, 7, 9, 24,
26, 43, 48, 51, 52). Mixtures of certain pairs of mutant IN
proteins, however, are able to complement these in vitro
ac-tivities (8, 49). The original goal of this study was to test the
effects of mutations in each of the IN protein domains on
replication of HIV-1 in cell culture. A separate goal was to
explore whether virions containing two different mutant IN
proteins within the same virus particle could integrate HIV
DNA in a single round of infection.
Mutations were introduced into pNL4-3, an infectious
pro-viral DNA clone of HIV-1 (1), and virus stocks were produced
by transfecting HeLa or 293 cells. Transient transfection of
CD4-negative cells bypasses the early steps in replication, such
as receptor-mediated entry, reverse transcription, and
integra-tion. The infectivity of virus released into cell supernatants was
assayed in the human T-cell leukemia line CEM-12D7 (37).
Infectivity was also evaluated in a single cycle by using the
HeLa-derived indicator cell line CD4-LTR/
b
-gal (22). This cell
line contains a copy of the
b
-galactosidase gene under the
control of the HIV-1 LTR. Following infection and presumed
integration of viral DNA, newly synthesized Tat protein
trans-activates the expression of
b
-galactosidase, which in turn
causes the nuclei of infected cells to appear blue after staining
with X-Gal. This assay has been termed MAGI, for
multinu-clear activation of galactosidase indicator (22).
HIV-1
IN/H12Nwas selected as an N-terminal domain IN
point mutant, and HIV-1
IN/1-234(1-234 refers to the amino
acid residues retained) was selected as a C-terminal domain
deletion mutant (Fig. 1); an N-terminal deletion mutant was
not included since it would disrupt the protease cleavage site
on November 9, 2019 by guest
http://jvi.asm.org/
between the RT and IN domains in the Gag-Pol polyprotein
precursor. HIV-1
IN/D116Nwas chosen as a core domain mutant
on the basis of the putative role of Asp-116 in the catalysis of
3
9
processing, DNA strand transfer, and disintegration (9, 26,
48). A single amino acid residue in the C-terminal domain,
Trp-235, was targeted because it is conserved among retroviral
IN proteins (Fig. 1). Finally, a single base deletion and three
stop codons were engineered after the fourth codon in the IN
reading frame, to produce the integrase-minus mutant
HIV-1
IN/1-4.
It has recently been reported that phenotypically mixed
Moloney murine leukemia virus (Mo-MLV) particles,
pro-duced by transfecting two viral genomes carrying separate
mu-tations in pol, are capable of complementing for virus growth
(47). In that study, infectivity was demonstrated with virus
inocula containing defective forms of RT and IN within
indi-vidual particles. Might similar phenotypically mixed HIV-1
particles function in a single round of infection? We
engi-neered the amino acid substitution Asp-186
3
Asn in one of the
active-site residues (23, 27, 31) of HIV-1 RT to test the
infec-tivity of phenotypically mixed particles containing this mutant
RT and the separate mutant INs.
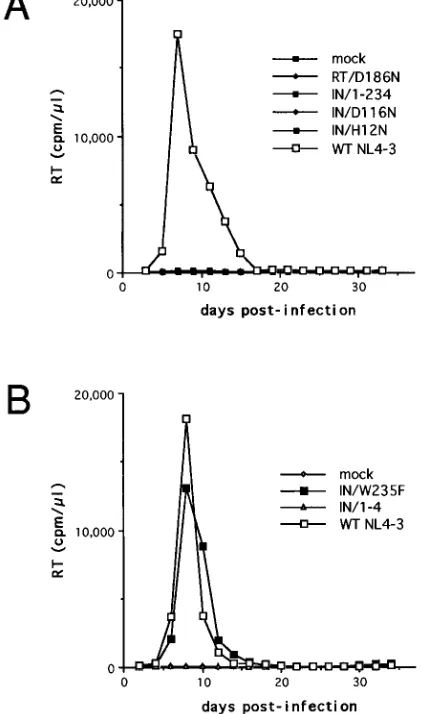
Most of the IN mutations render HIV-1 replication defective
in CEM-12D7 cells.
HeLa or 293 cells were transfected with
plasmid DNA to generate virus stocks. Each of the IN mutant
plasmids yielded detectable Mg
21-dependent
32P-RT activity
in cell supernatants 48 h posttransfection. In three
indepen-dent experiments, the RT activity in HIV-1
IN/D116Nsuperna-tants was approximately 85% of wild-type HIV-1
NL4-3activity.
HIV-1
IN/H12N, HIV-1
IN/1-234, and HIV-1
IN/1-4each exhibited
about 25% of wild-type activity. HIV-1
IN/W235Fyielded
approx-imately 50% of wild-type activity in two independent
experi-ments. As expected, transfected cell supernatants of
HIV-1
RT/D186Ndid not display detectable
32P-RT activity.
After normalization for
32P-RT activity, the infectivity of
each virus stock was measured in CEM-12D7 cells by
32P-RT
production. Cells infected with HIV-1
NL4-3reached peak levels
of
32P-RT activity about 8 days after infection, as did cells
infected with HIV-1
IN/W235F(Fig. 2). None of the other IN
mutant viruses showed any sign of replication over a 2-month
observation period.
The H12N, 1-234, and 1-4 mutations reduce the levels of
virus-associated RT and IN.
Four of the five mutant IN viruses
failed to infect CEM-12D7 cells. This was expected for
HIV-1
IN/D116N, HIV-1
IN/1-234, and HIV-1
IN/1-4, since these viruses
do not encode an IN protein capable of 3
9
processing or DNA
strand transfer activity in vitro. The replication-defective
phe-notypes may be due solely to the inability of the mutant INs to
promote these reactions after infection. However, it is also
possible these mutations affect other steps in the virus life
cycle. For example, proper processing of the Gag-Pol
precur-sor during virus assembly, or the reverse transcription reaction,
may be impaired. Although we knew that each mutant IN
plasmid produced detectable
32P-RT activity in culture
super-natants following transfection, the integrity of the resulting
virions, as well as the capacity of RT to function in the next
infectious cycle, was unknown.
Transfected cells were radiolabeled, and cell- and
particle-associated lysates were analyzed by immunoprecipitation to
visualize HIV-1 proteins. Analysis of HeLa cells expressing
HIV-1
NL4-3revealed the presence of the expected precursor
and mature forms of the envelope and Gag proteins (Fig. 3A,
lane 1). Each of the mutant plasmid DNAs also directed the
expression of these proteins in proportions roughly equal to
wild type, although the overall level of expression was reduced
two- to threefold in the cases of HIV-1
IN/H12N, HIV-1
IN/1-234,
and HIV-1
IN/1-4(Fig. 3A). Analysis of pelleted culture
super-FIG. 1. IN protein domains and mutations. In the diagram of the 288-residue wild-type protein, the three domains defined by complementation in vitro (8, 49) are shown. The N-terminal domain contains two His and two Cys residues which are conserved in retroviral and retrotransposon IN proteins (9, 18, 21, 51). The core domain contains three putative active-site residues, Asp-64, Asp-116, and Glu-152, which are conserved in certain bacterial transposases in addition to being conserved in retroviral and retrotransposon IN (2, 14, 24, 36, 40). The C-terminal domain contains a single amino acid residue, Trp-235, which is conserved only among retroviral IN (10, 30). The residues altered by point mutation are marked (*). The lines alongside the two deletion mutations depict the sizes of the proteins relative to wild-type IN.
FIG. 2. Replication kinetics of wild-type (WT) and mutant HIV-1 in CEM-12D7 cells. Cells were infected with equal32P-RT counts per minute of the indicated virus stocks, and culture supernatants were monitored for 32P-RT activity at the indicated time points. Since HIV-1RT/D186Ndid not display de-tectable32P-RT activity following transfection, the volume of virus inoculum was arbitrarily chosen as that for HIV-1IN/1-234.
on November 9, 2019 by guest
http://jvi.asm.org/
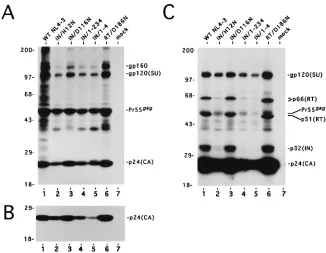
[image:3.612.328.539.77.434.2]natants revealed each contained near normal levels of the
capsid (CA) protein, except for HIV-1
IN/1-4, which had
approx-imately threefold less (Fig. 3B).
A longer exposure of the gel in Fig. 3B revealed additional
virion proteins, including two products of pol, the p66 subunit
of RT and IN (Fig. 3C, lane 1). HIV-1
IN/D116Nand
HIV-1
RT/D186Neach contained wild-type levels of protein (lanes
3 and 6). In contrast, HIV-1
IN/H12N, HIV-1
IN/1-234, and
HIV-1
IN/1-4particles contained reduced amounts of certain
pro-teins. When normalized to the levels of CA and surface (SU)
envelope glycoprotein, it was evident these viruses contained
between three- and fivefold reductions of p66(RT) (lanes 2, 4,
and 5). The level of IN in HIV-1
IN/H12Nwas also reduced to at
least this level. Detection of the 1-234 truncated mutant IN
protein, which is close in size to CA, was difficult using AIDS
patient antisera. Radiolabeled particles were therefore also
analyzed by immunoprecipitation using a polyclonal antisera
raised against purified recombinant HIV-1 IN.
This experiment again showed that HIV-1
NL4-3,
HIV-1
IN/D116N, and HIV-1
RT/D186Ncontained similar levels of IN
(Fig. 4, lanes 1, 3, and 6). The C-terminal truncated mutant
protein was associated with HIV-1
IN/1-234particles at a level
similar to that of p66(RT) (compare Fig. 4, lane 4, with Fig. 3C,
lane 4), indicating RT and IN were reduced to similar levels in
this virus. The level of IN in HIV-1
IN/H12Nwas at least
three-fold less than that observed in Fig. 3C. We conclude that
HIV-1
IN/H12N, HIV-1
IN/1-234, and HIV-1
IN/1-4contained
ap-proximately 3- to 10-fold reductions in the levels of RT and IN
relative to CA and SU.
Effects of IN mutations on virion morphology.
To obtain a
more detailed view of the effects that some of the IN mutations
had on particle formation, thin sections of transfected HeLa
cells were analyzed by electron microscopy. Analysis of cells
expressing wild-type HIV-1
NL4-3revealed mature particles
[image:4.612.143.469.69.322.2]containing typical conical nucleoids (Fig. 5A). HIV-1
IN/D116NFIG. 3. Analysis of cell- and particle-associated HIV-1 proteins by radiolabeling and immunoprecipitation with AIDS patient antisera. (A) Cell-associated proteins. The proteins in lane 1 were from HeLa cells transfected with wild-type (WT) pNL4-3. Lanes 2 to 6 contain proteins after transfection with plasmids encoding the indicated site-directed mutations. Lane 7 is from cells which were mock transfected. The migration positions of precursor and mature forms of envelope, gp160 and gp120(SU), respectively, and capsid, Pr55gagand p24(CA), respectively, are indicated on the right. The gel (12% polyacrylamide) was exposed to Kodak XAR 2 film at2708C for approximately 19 h. The migration positions of molecular mass standards are indicated in kilodaltons on the left. (B) Particle-associated proteins. The proteins in lanes 1 to 6 were from particle lysates following transfection with the plasmids described for lanes 1 to 6 in panel A. Lane 7 was from a mock transfection. The 12% polyacrylamide gel was exposed to film for approximately 19 h. Other labeling is the same as in panel A. (C) Longer exposure of the gel in panel B. The migration positions of the p66(RT) and p32(IN) are indicated on the right. The p51 subunit of RT is most likely also present and comigrates with Pr55gagunder our electrophoresis conditions. These two proteins were separated in cells expressing HIV-1RT/D186N, probably because the D186N amino acid substitution caused both the p66 and p51 subunits to display slightly faster electrophoretic mobilities. Similar changes in electrophoretic mobility have been observed for recombinant RT/D186N expressed in Escherichia coli (27, 31). The exposure was for approximately 5 weeks at2708C. Other labeling is the same as in panel A.
FIG. 4. Analysis of particle-associated proteins, using polyclonal antisera against wild-type IN. Lanes 1 to 7 contain particle lysates as described for lanes 1 to 7 in Fig. 3B. The immunoprecipitants were electrophoresed in a 8 to 16% gradient SDS-polyacrylamide gel (Novex). The gel was exposed to film for ap-proximately 10 weeks at2708C. The migration positions of wild-type (WT) and the C-terminal truncation mutant IN proteins are indicated p32(IN) and p26(IN/ 1-234), respectively, on the right. The migration positions of molecular mass standards are indicated in kilodaltons on the left.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.330.537.536.659.2]and HIV-1
RT/D186Nparticles for the most part had a similar
morphology (Fig. 5B and data not shown).
In contrast to the relatively benign effects of the IN and RT
active-site mutations on morphology, the H12N, 1-234, and 1-4
IN mutations affected the appearance of virus particles.
Al-though there were some late-budding forms, most were either
immature rings or aberrant mature particles (Fig. 5C). Either
the latter were totally devoid of electron-dense nucleoid
ma-terial or the mama-terial was situated between the empty nucleoid
and the membrane (Fig. 5C to E). The empty nucleoids were
pleomorphic instead of the typical conical or bar shape.
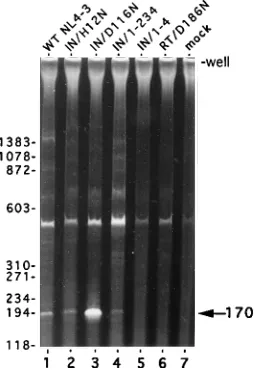
IN mutations and viral DNA synthesis.
Viral DNA synthesis
was analyzed by PCR 18 h after infection, using primers which
amplify sequences unique to the two-LTR-containing circle
formed in the nuclei of infected cells by end-to-end ligation of
linear viral DNA (50). This analysis revealed amplified
frag-ments of the appropriate sizes in cells infected with
HIV-1
NL4-3, HIV-1
IN/H12N, HIV-1
IN/D116N, and HIV-1
IN/1-234but
not in cells infected with either HIV-1
IN/1-4or HIV-1
RT/D186N(Fig. 6). Interestingly, the signal from cells infected with
HIV-1
IN/D116Nwas consistently more intense than the signal
follow-ing infection with the other viruses, includfollow-ing the wild type
(compare lane 3 with lane 1). PCR using primers which amplify
sequences between R and gag (58) showed that this region of
viral DNA was present in equal amounts in cells infected with
HIV-1
NL4-3and HIV-1
IN/D116N(10). Cells infected with
HIV-1
IN/D116Napparently contained relatively more of the two-LTR
circular form of viral DNA.
HIV-1
IN/D116Ndisplays a significant titer in the MAGI
as-say.
The infectivity of HIV-1
NL4-3and each mutant virus was
measured in a single round by using the MAGI assay. As
FIG. 5. Ultrastructural analysis of wild-type (A) and mutant (B to E) virions. (A and B) Thin sections of HeLa cells expressing wild type HIV-1NL4-3(A) and HIV-1RT/D186N(B). Typical mature particles contain a conical nucleoid with a dense broad end (arrow). (C and D) HIV-1IN/H12Nvirus particles. (E) HIV-1IN/1-4virus particles. Note the almost completed budding particle (C, large arrowhead), completed immature ring forms (C, smaller arrowheads), and ab-errant mature particles with empty nucleoids, with or without ectopic dense material (C, D, and E, arrows). Magnifications: A, 354,000; B, C, and D,372,000; E,378,000.
on November 9, 2019 by guest
http://jvi.asm.org/
expected, infection of CD4-LTR/
b
-gal cells with HIV-1
NL4-3yielded large numbers of blue cells after staining with X-Gal.
The titer of HIV-1
NL4-3was approximately 2
3
10
4
blue cells
per 10
6 32P-RT cpm of input virus (Table 1). HIV-1
IN/1-234
and
HIV-1
IN/1-4each yielded titers of approximately 30 blue cells
per 10
6 32P-RT cpm, whereas the titer of HIV-1
IN/H12N
was
about 170 blue cells per 10
6 32P-RT cpm. Infection with
HIV-1
RT/D186Nyielded the same number of stained cells as a mock
infection (on average one blue cell per 6 wells of a 24-well
plate). In contrast to the relatively low titers displayed by these
replication-defective viruses, HIV-1
IN/D116Ndisplayed an
un-expectedly high titer, approximately 2
3
10
3blue cells per 10
6 32P-RT cpm (Table 1). Similar titers were observed when virus
stocks derived from three independent IN/D116N mutant
plas-mid DNA constructs were used.
Although we originally planned to test complementation of
individual virus particles containing a pair of mutant IN
pro-teins for the ability to integrate viral DNA, our protein,
ultra-structural, and DNA analyses suggested that the majority of
monitored with the MAGI assay. We also assayed the
infec-tivity of viruses containing a mixture of the separate IN
mu-tants and RT/D186N, as it has recently been reported that RT
and IN mutants of Mo-MLV are able to complement virus
growth (47).
Infection with phenotypically mixed particles did not yield
any increase in the number of blue-stained cells from that
observed with the individual mutant viruses (10). This was the
case for viruses produced by transfecting two IN mutant
plas-mids, as well as mixtures of the RT/D186N and different
mu-tant IN plasmids. The ratios of the two plasmids were varied
prior to transfection to reflect the relative differences of CA in
the individual mutant viruses (Fig. 3B). Particles produced
from these transfections also did not show any sign of
comple-mentation.
DISCUSSION
Our results emphasize the importance of exercising caution
in interpreting the effects of IN mutations on virus replication.
Although most of the mutant virions that we constructed were
expected to be replication defective because the mutation
di-minishes IN activity in vitro, many were found to exhibit
de-fects at other stages of the viral replication cycle. One
muta-tion, IN/D116N, that maintained normal virion morphology
and protein composition exhibited two interesting properties.
The two-LTR circular form of viral DNA accumulated to
higher levels than with the wild-type virus following infection
of CEM-12D7 cells, and although the mutant virus was unable
to spread throughout the culture and generate progeny virions
as detected by RT assays (Fig. 2A), a significant titer of blue
cells was observed with the MAGI assay (Table 1). We
con-clude that IN-mediated DNA integration is essential for
infec-tivity in CD4-positive lymphocytes but not in HeLa-CD4 cells
as monitored by the MAGI assay.
Replication of HIV-1 containing IN mutations.
A single
amino acid residue in the C-terminal domain of HIV-1 IN,
Trp-235, is conserved among retroviral IN proteins (10, 30).
Since the amino acid substitution Trp-235
3
Glu has no
detect-able effects on the activities of IN in vitro (26), it was of interest
to determine whether this residue might be functionally
im-portant for replication of HIV-1 in cell culture. We found that
HIV-1
IN/W235Fexhibited wild-type replication kinetics in
CEM-12D7 cells. It is possible that the high degree of
conser-vation of this residue reflects a selective advantage under some
growth conditions which are not detected in this assay. It is
interesting that HIV-1 carrying the substitution Trp-235
3
Ala
is unable to replicate in cell culture (5), suggesting the
require-ment for an aromatic side chain at this position for replication
in T-cell lines.
HIV-1
IN/D116N, HIV-1
IN/1-234, and HIV-1
IN/1-4were unable
[image:6.612.114.241.73.258.2]to replicate in CEM-12D7 cells. This finding was expected in
view of the fact these mutations abolish the 3
9
processing and
DNA strand transfer activities of recombinant IN in vitro and
agrees with previous reports that IN function is essential for
HIV-1 replication in T-cell lines (5, 25, 45). The substitution of
Asn for His-12 decreases in vitro 3
9
processing and DNA
strand transfer activities of purified IN only about threefold
FIG. 6. PCR analysis of the two-LTR-containing circle in CEM-12D7 cells.Reaction mixtures in lanes 1 to 6 contained lysates of cells infected with the indicated viruses. The HIV-1RT/D186Ninoculum was normalized to the wild-type (WT) level on the basis of the levels of particle-associated CA (Fig. 3B). The PCR in lane 7 contained a lysate from cells which were mock infected. The migration positions of DNA standards are indicated in base pairs on the left. The position of the polyacrylamide gel well and the 170-bp U5/U3 circle junction PCR product are indicated on the right. The origin of the amplified band with an electrophoretic mobility of approximately 500 bp is unknown. However, because this band is present in the reaction mixture containing the lysate from cells which were mock infected, it most likely is not due to the amplification of DNA formed by reverse transcription.
TABLE 1. MAGI assay with wild-type and mutant HIV-1
Virus Titer
a(blue cells/ 106 32P-RT cpm)
Wild-type NL4-3 ... 19,736 IN/H12N ... 168 IN/D116N ... 2,276 IN/1-234 ... 27 IN/1-4 ... 35 RT/D186Nb... ,5
a
Cells which contained dark blue nuclei were counted. Each value is the average number from at least three independent infections.
b
HIV-1RT/D186Ndid not display detectable 32
P-RT activity. The virus inocu-lum was based on the level of particle-associated CA relative to HIV-1NL4-3(Fig. 3B).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.56.298.604.686.2](9). However, replication of HIV-1
IN/H12Nwas not detected in
CEM-12D7 cells.
IN mutations that give rise to defective virions.
Three of the
IN mutations reported in this study, H12N, 1-234, and 1-4,
reduced the levels of virion-associated RT relative to CA. The
H12N and 1-234 mutations caused similar reductions in each
of the mutant IN proteins (Fig. 3 and 4). These mutations
probably perturbed the levels at which the Gag-Pol precursors
were incorporated into virus particles and/or the proper
pro-cessing of the precursors after incorporation. Electron
micros-copy of cells expressing HIV-1
IN/H12N, HIV-1
IN/1-234, and
HIV-1
IN/1-4revealed alterations in virion morphogenesis and
maturation. Typical conical nucleoids with electron-dense
broad ends were rarely, if ever, formed. The electron-dense
material was either extranucleoid or apparently absent, and the
core was often misshapen (Fig. 5). These results demonstrate
that relatively small changes in IN, for example, the single
amino acid substitution His12
3
Asn, can markedly affect virus
assembly and maturation.
Certain Mo-MLV and HIV-1 IN mutations have previously
been shown to alter the protein composition of virus particles.
For Mo-MLV, some N-terminal, core, and C-terminal domain
mutations have led to lower levels of RT and IN (38, 39).
Similar affects have been described for some core domain
mutants of HIV-1 (5, 45, 46). An integrase-minus deletion
mutant similar to HIV-1
IN/1-4displayed a comparable
pheno-type (13).
HIV-1 gene expression from unintegrated viral DNA.
The
phenotype of HIV-1
IN/D116Nwas particularly interesting. PCR
with a pair of primers that amplify across the two-LTR circle
junction revealed that CEM-12D7 cells infected with
HIV-1
IN/D116Ncontained more circular viral DNA than did cells
infected with wild-type HIV-1
NL4-3. Although virus replication
was not detected by RT assays of culture supernatants after
infection of CEM-12D7 cells, an unexpectedly high infectivity
was obtained when the MAGI assay was used. Unlike the other
IN mutations reported here which rendered HIV-1 replication
defective, mutation of this putative catalytic site residue did
not affect the incorporation and/or processing of Gag-Pol
dur-ing particle assembly and maturation. Therefore, the early
phase of the virus life cycle most likely proceeded normally up
to the 3
9
processing step of integration. Two factors may
ac-count for the increased abundance of two-LTR-circle DNA in
CEM-12D7 cells infected with HIV-1
IN/D116N. First, the
ab-sence of 3
9
processing activity by the mutant IN would result in
maintenance of the blunt viral DNA ends, which would be a
better substrate for host DNA ligase than a pair of processed
ends. Second, because HIV-1
IN/D116NDNA is unable to
inte-grate, potentially more linear molecules would be available for
ligation.
The significant titer in the MAGI assay indicates the
pres-ence of Tat in the nuclei of CD4-LTR/
b
-gal cells infected with
HIV-1
IN/D116N. Expression of
b
-galactosidase is not induced
when these cells are infected with wild-type HIV-1 in the
presence of azidothymidine (28), apparently demonstrating a
requirement for reverse transcription and excluding the
possi-bility that expression can be turned on by Tat which may enter
the cell with the virus. Consistent with this finding, HIV-1
RT/ D186Ndid not transactivate
b
-galactosidase expression
follow-ing infection (Table 1). A simple interpretation of our results
is CD4-LTR/b-gal cells infected with HIV-1
IN/D116Naccumu-lated unintegrated DNA, and this DNA served as a template
for Tat expression. This level of expression, even if it occurs in
CEM-12D7 cells infected with HIV-1
IN/D116N, is insufficient to
support a spreading infection.
ACKNOWLEDGMENTS
We thank A. Buckler-White for sequencing plasmid DNA and E. Freed for technical advice. We also thank E. Freed, D. Dimitrov, and K. Mizuuchi for careful reading of the manuscript and I. Sunila and C. Dye III for assistance with the electron microscopy studies.
This work was supported in part by the National Institutes of Health Intramural AIDS Targeted Antiviral Program.
ADDENDUM
A recent paper by Wiskerchen and Muesing (56) also
re-ports that HIV-1 variants carrying mutations in active-site
res-idues of IN accumulate unintegrated DNA after infection and
display a significant titer in the MAGI assay.
REFERENCES
1. Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and
M. A. Martin.1986. Production of acquired immunodeficiency
syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291.
2. Baker, T. A., and L. Luo. 1994. Identification of residues in the Mu trans-posase essential for catalysis. Proc. Natl. Acad. Sci. USA 91:6654–6658. 3. Bushman, F. D., and R. Craigie. 1991. Activities of human immunodeficiency
virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. USA 88:1339–1343.
4. Bushman, F. D., A. Engelman, I. Palmer, P. Wingfield, and R. Craigie. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding. Proc. Natl. Acad. Sci. USA 90:3428–3432.
5. Cannon, P. M., W. Wilson, E. Byles, S. M. Kingsman, and A. J. Kingsman. 1994. Human immunodeficiency virus type 1 integrase: effect on viral repli-cation of mutations at highly conserved residues. J. Virol. 68:4768–4775. 6. Chow, S. A., K. A. Vincent, V. Ellison, and P. O. Brown. 1992. Reversal of
integration and DNA splicing mediated by integrase of human immunode-ficiency virus. Science 255:723–726.
7. Drelich, M., R. Wilhelm, and J. Mous. 1992. Identification of amino acid residues critical for endonuclease and integration activities of HIV-1 IN protein in vitro. Virology 188:459–468.
8. Engelman, A., F. D. Bushman, and R. Craigie. 1993. Identification of dis-crete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 12:3269–3275.
9. Engelman, A., and R. Craigie. 1992. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66:6361–6369.
10. Engelman, A., G. Englund, M. A. Martin, and R. Craigie. Unpublished data. 11. Engelman, A., A. B. Hickman, and R. Craigie. 1994. The core and carboxyl-terminal domains of the integrase protein of human immunodeficiency virus type 1 each contribute to nonspecific DNA binding. J. Virol. 68:5911–5917. 12. Engelman, A., K. Mizuuchi, and R. Craigie. 1991. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211– 1221.
13. Englund, G., and R. L. Willey. Unpublished data.
14. Fayet, O., P. Ramond, P. Polard, M. F. Pre`re, and M. Chandler. 1990. Functional similarities between the IS3 family of bacterial insertion ele-ments? Mol. Microbiol. 4:1771–1777.
15. Folks, T., S. Benn, A. Rabson, T. Theodore, M. D. Hoggan, M. A. Martin, M. Lightfoote, and K. Sell.1985. Characterization of a continuous T-cell line susceptible to the cytopathic effects of the acquired immunodeficiency syn-drome (AIDS)-associated retrovirus. Proc. Natl. Acad. Sci. USA 82:4539– 4543.
16. Freed, E. O., J. M. Orenstein, A. J. Buckler-White, and M. A. Martin. 1994. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle formation. J. Virol. 68:5311–5320. 17. Higuchi, R., B. Krummel, and R. K. Saiki. 1988. A general method of in vitro
preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 16:7351–7367.
18. Johnson, M. S., M. A. McClure, D.-F. Feng, J. Gray, and R. F. Doolittle. 1986. Computer analysis of retroviral pol genes: assignment of enzymatic functions to specific sequences and homologies with nonviral enzymes. Proc. Natl. Acad. Sci. USA 83:7648–7652.
19. Kalpana, G. V., and S. P. Goff. 1993. Genetic analysis of homomeric inter-actions of human immunodeficiency virus type 1 integrase using the yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 90:10593–10597.
20. Katz, R. A., and A. M. Skalka. 1994. The retroviral enzymes. Annu. Rev. Biochem. 63:133–173.
21. Khan, E., J. P. G. Mack, R. A. Katz, J. Kulkosky, and A. M. Skalka. 1991. Retroviral integrase domains: DNA binding and the recognition of LTR sequences. Nucleic Acids Res. 19:851–860.
22. Kimpton, J., and M. Emerman. 1992. Detection of replication-competent
on November 9, 2019 by guest
http://jvi.asm.org/
highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 12:2331–2338.
25. Lafemina, R. L., C. L. Schneider, H. L. Robbins, P. L. Callahan, K. LeGrow, E. Roth, W. A. Schleif, and E. A. Emini.1992. Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cells. J. Virol. 66:7414–7419.
26. Leavitt, A. D., L. Shiue, and H. E. Varmus. 1993. Site-directed mutagenesis of HIV-1 integrase demonstrates differential effects on integrase function in vitro. J. Biol. Chem. 268:2113–2119.
27. Le Grice, S. F. J., T. Naas, B. Wohlgensinger, and O. Schatz. 1991. Subunit-selective mutagenesis indicates minimal polymerase activity in heterodimer-associated p51 HIV-1 reverse transcriptase. EMBO J. 10:3905–3911. 28. Lewis, P., M. Hensel, and M. Emerman. 1992. Human immunodeficiency
virus infection of cells arrested in the cell cycle. EMBO J. 11:3053–3058. 29. Lightfoote, M. M., J. E. Coligan, T. M. Folks, A. S. Fauci, M. A. Martin, and
S. Venkatesan.1986. Structural characterization of reverse transcriptase and endonuclease polypeptides of the acquired immunodeficiency syndrome vi-rus. J. Virol. 60:771–775.
30. Lin, T.-H., and D. P. Grandgenett. 1991. Retrovirus integrase: identification of a potential leucine zipper motif. Protein Eng. 4:435–441.
31. Lowe, D. M., V. Parmar, S. D. Kemp, and B. A. Larder. 1991. Mutational analysis of the two conserved motifs in HIV-1 reverse transcriptase. FEBS Lett. 282:231–234.
32. Mazumder, A., A. Engelman, R. Craigie, M. Fresen, and Y. Pommier. 1994. Intermolecular disintegration and intramolecular strand transfer activities of wild-type and mutant HIV-1 integrase. Nucleic Acids Res. 22:1037–1043. 33. Meyers, G., S. Wain-Hobson, B. Korber, R. F. Smith, and G. N. Pavlakis.
1993. Human retroviruses and AIDS. A compilation and analysis of nucleic acid and amino acid sequences. Theoretical biology and biophysics group T-10, Los Alamos National Laboratory, Los Alamos, N.Mex.
34. Prince, A. M., B. Horowitz, L. Baker, R. W. Shulman, H. Ralph, J. Valinsky, A. Cundell, B. Brotman, W. Boehle, F. Rey, M. Piet, H. Reesink, N. Lelie, M. Tersmette, F. Miedema, L. Barbosa, G. Nemo, C. L. Nastala, J. S. Allan,
D. R. Lee, and J. W. Eichberg.1988. Failure of a human immunodeficiency
virus (HIV) immune globin to protect chimpanzees against experimental challenge with HIV. Proc. Natl. Acad. Sci. USA 85:6944–6948.
35. Prince, A. M., H. Reesink, D. Pascual, B. Horowitz, I. Hewlett, K. K. Murthy, K. E. Cobb, and J. W. Eichberg.1991. Prevention of HIV infection by passive immunization with HIV immunoglobulin. AIDS Res. Hum. Retroviruses 7:971–973.
36. Radstro¨m, P., O. Sko¨ld, G. Swedberg, J. Flensburg, P. H. Roy, and L.
Sundstro¨m.1994. Transposon Tn5090 of plasmid R571, which carries an
integron, is related to Tn7, mu, and the retroelements. J. Bacteriol. 176: 3257–3268.
37. Ross, E. K., A. J. Buckler-White, A. Rabson, G. Englund, and M. A. Martin. 1991. Contribution of NF-kB and Sp1 binding motifs to the replicative capacity of human immunodeficiency virus type 1: distinct patterns of viral growth are determined by T-cell types. J. Virol. 65:4350–4358.
38. Roth, M. J. 1991. Mutational analysis of the carboxyl terminus of the Molo-ney murine leukemia virus integration protein. J. Virol. 65:2141–2145. 39. Roth, M. J., P. Schwartzberg, N. Tanese, and S. P. Goff. 1990. Analysis of
mutations in the integration function of Moloney murine leukemia virus:
is required for integration activity, but not for DNA binding. Biochem. Biophys. Res. Commun. 185:874–880.
44. Sherman, P. A., and J. A. Fyfe. 1990. Human immunodeficiency virus inte-gration protein expressed in Escherichia coli possesses selective DNA cleav-ing activity. Proc. Natl. Acad. Sci. USA 87:5119–5123.
45. Shin, C., B. Taddeo, W. A. Haseltine, and C. M. Farnet. 1994. Genetic analysis of the human immunodeficiency virus type 1 integrase protein. J. Virol. 68:1633–1642.
46. Taddeo, B., W. A. Haseltine, and C. M. Farnet. 1994. Integrase mutants of human immunodeficiency virus type 1 with a specific defect in integration. J. Virol. 68:8401–8405.
47. Telesnitsky, A., and S. P. Goff. 1993. Two defective forms of reverse tran-scriptase can complement to restore retroviral infectivity. EMBO J. 12:4433– 4438.
48. van Gent, D. C., A. A. M. Oude Groeneger, and R. H. A. Plasterk. 1992. Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc. Natl. Acad. Sci. USA 89:9598–9602.
49. van Gent, D. C., C. Vink, A. A. M. Oude Groeneger, and R. H. A. Plasterk. 1993. Complementation between HIV integrase proteins mutated in differ-ent domains. EMBO J. 12:3261–3267.
50. Varmus, H., and P. Brown. 1989. Retroviruses, p. 53–108. In D. E. Berg and M. M. Howe (ed.), Mobile DNA. American Society for Microbiology, Wash-ington, D.C.
51. Vincent, K. A., V. Ellison, S. A. Chow, and P. O. Brown. 1993. Character-ization of human immunodeficiency virus type 1 integrase expressed in
Escherichia coli and analysis of variants with amino-terminal mutations. J.
Virol. 67:425–437.
52. Vink, C., A. A. M. Oude Groeneger, and R. H. A. Plasterk. 1993. Identifica-tion of the catalytic and DNA-binding region of human immunodeficiency virus type 1 integrase protein. Nucleic Acids Res. 21:1419–1425. 53. Vink, C., D. C. van Gent, Y. Elgersma, and R. H. A. Plasterk. 1991. Human
immunodeficiency virus integrase protein requires a subterminal position of its viral DNA recognition sequence for efficient cleavage. J. Virol. 65:4636– 4644.
54. Willey, R. L., J. S. Bonifacino, B. J. Potts, M. A. Martin, and R. D. Klausner. 1988. Biosynthesis, cleavage and degradation of the human immunodefi-ciency virus type 1 envelope glycoprotein gp160. Proc. Natl. Acad. Sci. USA 85:9580–9584.
55. Willey, R. L., D. H. Smith, L. A. Lasky, T. S. Theodore, P. L. Earl, B. Moss, D. J. Capon, and M. A. Martin.1988. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 62:139–147.
56. Wiskerchen, M., and M. A. Muesing. 1995. Human immunodeficiency virus type 1 integrase: effects of mutations on viral ability to integrate, direct gene expression from unintegrated viral DNA templates, and sustain propagation in primary cells. J. Virol. 69:376–386.
57. Woerner, A. M., and C. J. Marcus-Sekura. 1993. Characterization of a DNA binding domain in the C-terminus of HIV-1 integrase by deletion mutagen-esis. Nucleic Acids Res. 21:3507–3511.
58. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y.
Chen.1990. HIV-1 entry into quiescent primary lymphocytes: molecular
analysis reveals a labile, latent viral structure. Cell 61:213–222.