0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
Human Immunodeficiency Virus Type 1 Integrase: Effects of
Mutations on Viral Ability To Integrate, Direct Viral Gene
Expression from Unintegrated Viral DNA Templates,
and Sustain Viral Propagation in Primary Cells†
MARYANN WISKERCHEN‡ANDMARK A. MUESING*
Infectious Disease Research, Lilly Research Laboratories, Indianapolis, Indiana 46285-0438
Received 6 June 1994/Accepted 5 October 1994
Integrase is the only viral protein necessary for integration of retroviral DNA into chromosomal DNA of the host cell. Biochemical analysis of human immunodeficiency virus type 1 (HIV-1) integrase with purified protein and synthetic DNA substrates has revealed extensive information regarding the mechanism of action of the enzyme, as well as identification of critical residues and functional domains. Since in vitro reactions are carried out in the absence of other viral proteins and they analyze strand transfer of only one end of the donor substrate, they do not define completely the process of integration as it occurs during the course of viral infection. In an effort to further understand the role of integrase during viral infection, we initially constructed a panel of 24 HIV-1 mutants with specific alanine substitutions throughout the integrase coding region and analyzed them in a human T-cell line infection. Of these mutant viruses, 12 were capable of sustained viral replication, 11 were replication defective, and 1 was temperature sensitive for viral growth. The replication defective viruses express and correctly process the integrase and Gag proteins. Using this panel of mutants and an additional set of 18 mutant viruses, we identified nine amino acids which, when replaced with alanine, destroy integrase activity. Although none of the replication-defective mutants are able to integrate into the host genome, a subset of them with alterations in the catalytic triad are capable of Tat-mediated transactivation of an indicator gene linked to the viral long terminal repeat promoter. We present evidence that integration of the HIV-1 provirus is essential not only for productive infection of T cells but also for virus passage in both cultured peripheral blood lymphocytes and macrophage cells.
Efficient expression and stable maintenance of retroviral genomes are dependent on an obligatory integration step in the viral life cycle in which viral DNA is permanently joined to infected host chromosomal DNA (for recent reviews, see references 23 and 56). Reconstruction of the integration reaction in vitro has demonstrated that integration can be mediated by the viral integrase protein in the absence of any added eucaryotic host proteins and has revealed mechanistic aspects of the integration reaction (7, 9, 13, 19, 21, 26, 27, 46, 51, 54). Retroviral integrase cleaves the blunt-ended termini of linear viral DNA in an early endonucleolytic reaction that
removes two nucleotides from the 39 ends of viral DNA
duplexes (4, 7, 13, 16, 27, 43, 46, 55). This event activates the
39-recessed termini for a subsequent strand transfer reaction in
which the 39end of the viral DNA is covalently linked to the 59
end of breaks produced by integrase in the target DNA molecule (4, 7, 9, 13, 22, 26, 31, 34). In vivo, both ends of the viral DNA are inserted into the host chromosome in a con-certed fashion and the gapped intermediate, unjoined at the
viral DNA 59 ends, is then repaired (presumably by host
enzymes) to complete the integration process. In vitro, purified integrase can also use the gapped intermediate substrate to perform a disintegration reaction, in which the intermediate
structure is resolved into the initial substrates of the forward strand transfer reaction (12, 18).
Retroviral integrase can be subdivided into three domains on the basis of both domain behavior in vitro and phylogenetic comparisons between other retroviral integrases and related proteins which can perform polynucleotidyl transfer reactions (15, 20, 24, 26). The first domain contains a zinc finger-like motif of two histidine residues, an intervening region of variable length followed by two cysteine residues [the HHCC
domain; H-X(3–7)-H-X(23–32)-C-X(2)-C] (24). Truncations in
this region and single-amino-acid substitutions at the con-served histidine or cysteine residues in human immunodefi-ciency virus type 1 (HIV-1) integrase result in the production of mutant proteins unable to bind zinc efficiently in vitro (6, 8). Alterations at the conserved histidine and cysteine residues
abolish both integration and 39processing activities in vitro but
retain disintegration activity, suggesting a role for this region in mediating the interaction of HIV-1 integrase with viral DNA (8, 18, 52). The second domain is the catalytic center of the protein and is located approximately between residues 50 and 212 (18). It is a relatively protease-resistant domain (18) and contains the active site for all polynucleotidyl transfer activities (16, 18, 30, 49, 53). Essential to the active site is the highly
conserved D,D-35-E region [D-X(39–58)-D-X(35)-E] with
invari-ant residues at D-64, D-116, and E-152 (18, 28, 30). The region of integrase necessary for homodimer formation has been mapped to this core domain by both genetic and biochemical analyses (17, 25, 50). The third domain is a relatively noncon-served region varying in both length and primary sequence within individual members of the retrovirus subfamily groups. This domain is, however, required to be present in cis to an
* Corresponding author. Mailing address: Infectious Disease Re-search, Lilly Research Laboratories, Lilly Corporate Center, Indianap-olis, IN 46285-0438. Phone: (317) 276-6714. Fax: (317) 276-1743.
† Dedicated to the memory of Joel Dalrymple.
‡ Present address: Aaron Diamond AIDS Research Center, New York, NY 10016.
376
on November 9, 2019 by guest
http://jvi.asm.org/
active catalytic core for efficient 39processing and DNA strand transfer reactions (17, 50).
Although much information has been gathered regarding the relative contribution of each of the conserved regions of integrase to its activities in vitro, little is known about the consequence of specific mutations in vivo. To complement existing in vitro data, we have produced a panel of specific mutations in the HIV-1 integrase gene and reconstructed each mutant integrase gene into a complete infectious clone of HIV-1 (HXB2d). We analyzed the ability of the altered integrase protein to function during viral infection of cultured T cells. Charge-cluster-to-alanine scanning mutagenesis (2) was used to replace groups of charged amino acids in the primary sequence. The charged amino acids were targeted because they are likely to occupy exposed positions in tertiary structure and their substitution is less likely to disrupt the overall conformation and inherent stability of the molecule (2, 15). Using this approach, we have identified a set of nine amino acids and four charge clusters of the HIV-1 integrase protein necessary for function. The replication-defective mutants do not induce syncytium formation by T cells; however, they contain stable Gag-Pol proteins of the correct size and an active reverse transcriptase. We also compare the integration frequency of the replication-competent viruses with that of wild-type virus.
Integrated proviral DNA maintained within host chromo-somal DNA is an active template for the expression of retroviral gene products. Circular forms of viral DNA can be found in the nucleus after retroviral infection of host cells (23, 56). These viral DNAs are generally believed to be transcrip-tionally silent remnants of a failed attempt at retroviral inte-gration. There is a report in the literature, however, suggesting that certain mutants defective in integrase function are capable of limited viral gene expression (48). The replication-defective mutants described here fall into two distinct classes based on their ability to direct the expression of the tat gene from an unintegrated viral circular template DNA. Mutants with alter-ations in the catalytic triad residues, D-64, D-116, and E-152, are capable of tat gene expression in the absence of integra-tion, whereas mutants with alterations elsewhere are not. The Tat expression generated from unintegrated mutant template DNA is correlated with the recovery of single- and double-long terminal repeat (LTR) viral circles from cells nonproductively infected with mutants containing an alteration in the catalytic triad. One representative member from each class of replica-tion-defective integrase mutants was used to demonstrate that integration is required not only for viral replication of cultured human T-lymphoid cells but also for a productive infection of cultured peripheral blood lymphocytes and macrophages of primary cell origin.
MATERIALS AND METHODS
Cells and reagents.CEM cells, a cell line derived from human T cells, were grown in RPMI 1640 medium (GIBCO) supplemented with 10% fetal bovine serum (JRH Biosciences). COS-7 and 293 cells were grown in Dulbecco modified Eagle medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum. HeLa-CD4-LTR-b-gal cells were received from M. Emerman (29) through the AIDS Research and Reference Reagent Program and grown in DMEM supple-mented with 20% newborn calf serum (JRH Biosciences), 0.2 mg of G418 per ml, and 0.1 mg of hygromycin B per ml (Sigma). Fresh human adult monocytes from elutriated source leukocytes, which were 96.4% pure as determined by differen-tial count with Wright’s stain, were purchased from Advanced Biotechnologies, Inc. These cells were seeded in a 24-well plate (106
cells per well) in DMEM– high-glucose medium supplemented with 20% fetal bovine serum and 10% human AB serum and incubated at 378C in a 5% CO2atmosphere. Every 4 days
the cells were refed with fresh medium. After infection, the human serum was omitted from the medium and the cells were incubated in sealed bags at 378C. Adult human lymphocytes from elutriated source leukocytes, which were 95%
pure and phytohemagglutinin stimulated, were purchased from Advanced Bio-technologies, Inc. These cells were grown in RPMI 1640 medium supplemented with 20% fetal bovine serum and 10% conditioned medium containing interleu-kin-2.
Viral p24 levels were measured by enzyme-linked immunosorbent assay with the HIV-1 p24 antigen assay kit and kinetic standard purchased from Coulter Immunology. The following reagents were received from the AIDS Research and Reference Reagent Program: HIV-gpt from K. Page and D. Littman (41), SV-A-MLV-env plasmid from N. Landau and D. Littman (33), HIV-1 HXB2 integrase antiserum raised against integrase amino acids 23 to 34 from D. Grandgenett (5), and pYU-2 plasmid from B. Hahn and G. Shaw (36, 37). Rabbit polyclonal antibody raised against HIV-1 p24 (CA) was purchased from Amer-ican Bio-Technologies, Inc., and murine monoclonal antibody to HIV-1 p17 (MA) was purchased from Dupont.
Construction of integrase mutants.Recombinant plasmids were constructed and used to transform bacteria by standard procedures (45). The wild-type virus used in these experiments was generated from plasmid R7-3, which is a derivative of the HIV-1 HXB2d isolate (20). R7-3 encodes a truncated Vpr protein and has no ATG initiation codon for vpu. It also encodes a functional nef gene (39). Plasmid PSKSII1was constructed by inserting the PstI-SalI (HXB2d nucleotides [nt] 1414 to 5785) fragment of R7-3 into the same sites of Bluescript IIKS1
phagemid (Stratagene). Mutations were introduced into PSKSII1with the oligonucleotide-directed mutagenesis system as specified by the manufacturer (Amersham). For the two mutations introduced into the YU-2 genome (36), a Bst1107I-EcoRI fragment of pYU-2 (nt 2925 to 5741) was subcloned into the EcoRI-SmaI-digested Bluescript phagemid KSII1. Single-stranded DNA gener-ated from this phagemid was mutagenized with the Mutagene kit as specified by the manufacturer (Bio-Rad), and the altered fragment was then reinserted into the pYU-2 plasmid with a SnaBI-NcoI fragment. Mutagenic oligonucleotides used to generate mutants are as follows (the sequences which introduced alanine codons are underlined, and sequences which differ from HXB2d are in lowercase
letters): GCCAGCTGTGcTgcATGTCAGCTAAAAGGAG (D41A/K42A);
GCCATGgcTGGACAAGTAGcCTGTAGTCCAGG (H51A/D55A); GTAC ACATTTAGcAGGAgcAGTTATCCTGG (E69A/K71A); GCCAGTGGATA TATAGcAGCAGcAGTTATTCCAGC (E85A/E87A); CAATACATACAGc CAATGGCAGC (D116A); AATTCCAAATgCCTGCgcGATTCCCGC (K136 A/E138A); GGGCGGGAATCgcGCAGGAATTTGG (K136A); GGAATC AAGCAGGcATTTGGAATTCCC(E138A); CCTATAATTTTCTTTAATTC TgcATTCATAGATgCTACTAC (E152A/K156A); CCTATAATTTTCTTTAA TgCTgcATTCATAGATTCTACTAC (K156A/E157A); CCTATAATTTT CgcTAATgCTTTATTCATAGATTCTACTAC (E157A/K159A); CCTATAAT TgcCgcTAATTCTTTATTCATAGATTCTACTAC (K159A/K160A); CAGCC TGAgCTgcTACCTGTCC (R166A/D167A); CTGCTGTCTTAAGAgcTgC AGCCTG (E170A/H171A); CTGCTGTCgcAAGATGTgCAGCCTG (E170A/ K173A); CTGCTGTCgcAAGAgcTTCAGCCTG (H171A/K173A); GTCTACT ATTgcTgCCCCTGCAC (E198A/R199A); GCTATTATGgCTACTATTgcTT CCCC (R199A/D202A); TTGTAATgCTgcAGTTTGTATGTCTGTTGC (K211A/E212A); CCAGAGGAGCgcTGCTGGTCCTgcCCAAAGTGG (K236A/K240A); CTACTGCCCCgCACCggcCCAGAGGAG (K244A/E246A); GGCACTACTTTTATGgCACTATTAgCTTGTATTACTACTGCCCC (D253A/D256A); GCCATCTGTTTTCCATAAgCCgcAATGATCTTTGC (R269A/D270A); GCCCAAGATGAAgcTGAGAAATATC (H12A); GAG AAATATgcCAGTAATTGGAG(H16A); GGAGAAGCCgcGCATGGACA AG (M50A); GGACAAGTAGcCTGTAGTCCAGG (D55A); TGGCAAC TAgcTTGTACACATTTAG (D64A); GGAAGATGGgCAGTAAAAA CAATAC (P109A); GCTACGGTTgcGGCCGCCTGT (K127A); GGAATTC CCTttAATCCCCAAAG (Y143F); GGAGTAGTAGcATCTATGAATAAAG (E152A); GAATCTATGAATgcAGAATTAAAG (K156A); GGACAGGTAg cAGATCAGGCTGAAC (R166A); GTGCAGGGGAAgcAATAGTAGAC (R199A); CAATACATACAGcCAATGGCAGC (D116A, YU-2); and GGG GAAgcAATAGTAGcCATAATAGC (R199A/D202A,YU-2). The presence of the correct alteration for each mutant infectious proviral clone was determined by sequencing with the double-stranded DNA cycle sequencing system (Bethesda Research Laboratories). The mutation was reintroduced into R7-3 with either an EcoRI fragment (HXB2d nt 4648 to 5743) or a SpeI-SalI fragment (HXB2d nt 1506 to 5785) and sequenced again to confirm the presence of the correct substitutions.
Construction of the HXB2d/YU-2 chimeric infectious clone.The infectious clone containing the HXB2d/YU-2 chimeric viral genome has nt 6223 to 8473 of HXB2d substituted with nt 6224 to 8435 from YU-2, a primary HIV-1 macro-phage-tropic isolate with full-length reading frames for all viral proteins. It was constructed as follows. An EcoRI fragment containing the 59half of the YU-2 genome contained on pYU-2 was deleted, creating pDRI/YU-2. pDRI/YU-2 was used as template for single-stranded DNA preparation and mutagenized to place a BamHI site into YU-2 DNA at the analogous position found in HXB2d, creating pDRI/YU-2(BamHI). The BbsI-BamHI fragment from pD RI/YU-2(BamHI), containing 5 nt upstream of the initiating ATG of the YU-2 env reading frame to the BamHI site in gp41, was cloned into an HXB2d-derived plasmid by replacing the HXB2d BbsI-BamHI fragment with the BbsI-BamHI fragment from pDRI/YU-2(BamHI), creating pMM10. The SalI-BamHI frag-ment from pMM10 was cloned into the unique SalI and BamHI sites of R7-3, creating pHXB2d/YU-2.
on November 9, 2019 by guest
http://jvi.asm.org/
Transient-expression T-cell infection.Viral stocks were prepared by two methods. COS-7 cells (23106
) were electroporated with 20mg of DNA of the wild-type and mutant proviral clones in 1 ml of DMEM supplemented with 20% fetal bovine serum, incubated on ice for 10 min, transferred to 10 ml of medium, split into two 25-cm2
flasks, and incubated at 358C in a 5% CO2 atmosphere. Cell-free medium was harvested 36 h after electroporation and assayed for p24 content (Coulter). Virus stocks generated by COS-7 electroporation were used for the data reported in Table 1. These stocks yielded a low p24 content but were highly infectious; they required small amounts of p24 to obtain syncytium formation during infection. Virus stocks generated by calcium phosphate-mediated transfection of 293 cells were used in all other experiments. 293 cells (one 75-cm2
flask) at 40% confluency were transfected with 20mg of DNA from the infectious proviral wild-type or mutant clones with the Cellphect kit (Pharmacia). At 4 h after addition of DNA, the cells were washed with DMEM and shocked for 3 min with 15% glycerol in 10 mM N-2-hydroxyethylpiperazine-N9-2-ethanesulfonic acid (HEPES; pH 8.0)–150 mM NaCl. The glycerol was removed, and the cells were washed once with DMEM and incubated in DMEM–10% fetal bovine serum at the designated temperature for 48 h. The supernatants were then harvested, and the cells were removed by low-speed centrifugation. All virus stocks were generated at 358C, normalized for p24 content, filtered through a 0.45-mm filter, and stored in aliquots at2708C.
CEM cells (103) were infected with each of the wild-type or mutant virus
stocks, normalized for equal p24 content, in 200ml of medium in a 96-well format or mock infected with medium alone. All infections were performed in duplicate. The cells were observed for syncytium formation and split 1:5 7 days postinfec-tion. For infections which proceeded for longer than 11 days, the cells were split 1:5 every 4 or 5 days as needed to prevent cell death. Virus production was evaluated by measuring p24 content or reverse transcriptase (RT) activity in the culture supernatant as indicated.
Macrophage and PBL infections.Confluent macrophage cells in a 24-well plate were infected with 100 ng of p24 from the mutant and wild-type virus stocks 8 days after the initial seeding of the monocytes. The infection proceeded for 3 h at 378C. The virus was removed, and the cells were washed once with medium, overlaid with 2 ml of fresh media, and incubated at 378C in sealed bags. Every 4 days the culture supernatant was removed and assayed for p24 content, and cells were refed with new medium. Phytohemagglutinin-stimulated peripheral blood lymphocytes (PBLs) (23105
) were infected for 2 h at 378C with 100 ng of p24 of the mutant and wild-type virus stocks. The cells were centrifuged for 5 min at 3,000 rpm in a Sorvall H1000B rotor, and the medium was removed. The cells were resuspended in 2 ml of fresh medium, transferred to a 24-well plate, and incubated at 378C. PBLs were split 1:4 every 4 days, and the p24 content in the culture supernatant was monitored.
Western blot analysis.293 cells (one 75-cm2
flask) at 40% confluency were transfected with calcium phosphate and 20mg of DNA from the infectious proviral wild-type or mutant clones by using the Cellphect kit (Pharmacia). After 4 h of transfection, cells were glycerol shocked, refed with 10 ml of medium, and incubated at the appropriate temperature for 3 days. Culture supernatants were harvested, and cells were removed by low-speed centrifugation. Virions were pelleted from cell-free medium by centrifuging through a 10% sucrose cushion at
25,0003g for 2 h in an SW41 Ti rotor at 48C. Medium alone was concentrated as a negative control. The virion pellet was lysed in 200ml of RIPA buffer (10 mM Tris [pH 7.5], 150 mM NaCl, 1% deoxycholate, 1% Triton X-100, 0.1% sodium dodecyl sulfate [SDS]), and p24 levels were determined. The lysed virions were incubated on ice for 10 min and boiled with 40ml of 63sample buffer for 3 min. Each virion sample (20 ng of p24) was subjected to SDS-polyacrylamide gel electrophoresis followed by electrophoretic transfer to nitrocellulose mem-brane (Hybond C plus; Amersham). An SDS–12.5% polyacrylamide gel was used for analysis of integrase content, and a 15% gel was used for analysis of p17 and p24 content. A standard ECL-Western immunoblot analysis was performed as specified by the manufacturer (Amersham). Antibody specific for p17 was diluted to 5mg/ml, p24 antibody was used at a 1:1,000 dilution, and integrase antibody was used at a 1:100 dilution.
RT activity. HIV-1 RT activity in cell-free culture supernatant and in concentrated virus stock preparations was determined as previously described (1). This determination involved incubating 10ml of sample with 50ml of RT cocktail for 1.5 h at 378C and spotting 5ml onto NA45 paper (Schleicher & Schuell). RT cocktail consists of 50 mM Tris (pH 7.5), 75 mM KCl, 2 mM dithiothreitol, 5 mM MgCl2, 5mg of polyadenylic acid (Pharmacia) per ml, 1.6mg
of poly(dT)12–18(Pharmacia) per ml, 10mCi of [ 32
P]TTP per ml, and 0.05% Nonidet P-40. Filters were washed four times for 5 min each with 23SSC (13
SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and once with 95% ethanol, dried, and autoradiographed. Individual spots were cut and Cerenkov counted to determine the counts per minute. Samples were assayed in duplicate.
Pseudotyped mutant construction and infection.The SalI-BamHI fragment (HXB2d nt 5785 to 8475) from R7-3 and each of the integrase mutant DNAs was replaced with the SalI-BamHI fragment from HIV-gpt that includes the simian virus 40 origin of replication and promoter and codes for the gpt resistance marker. To prepare pseudotyped amphotropic stocks, 293 cells (one 75-cm2
flask) were cotransfected with 10mg of SV-A-MLV-env and 10mg of each of the mutant integrase proviral DNA-gpt clones. As a negative control, R7-3–gpt was cotransfected with a plasmid unable to provide a retrovirus envelope in trans. After 60 h of transfection the supernatant was collected, filtered through a 0.45-mm-pore-size filter, and normalized to 20 ng of p24 core antigen per ml. Target HeLa-CD4-LTR–b-gal cells at 30% confluency in six-well format were infected with 20 ng of p24 from each cotransfection supernatant in the presence of 8mg of Polybrene per ml. After a 4-h infection period at the indicated temperature, the virus was removed and the cells were washed once with serum-free medium and fed with nonselective medium. All subsequent incuba-tions were continued at the original infection temperature. The cells were grown for an additional 40 h and then split 1:2, 1:20, and 1:200 into gpt selection medium (33). The cells were refed with gpt selection medium every 3 days, and gpt-resistant colonies were stained with 0.2% crystal violet–25% isopropanol–5% acetic acid and counted 12 days postinfection. The values reported were adjusted for the dilutions but were not adjusted for expansion in the initial 40-h incubation in nonselective medium. The integration frequency values have not been modified to reflect the different generation times for the cells at each assay temperature.
MAGI cell assay.HeLa-CD4-LTR–b-gal cells (0.43105) (29) in a 24-well
format were infected in duplicate with 5 ng and 50 ng of p24 for both the mutant and wild-type virus stocks in the presence of 20mg of DEAE-dextran per ml. At 2 h after infection at 378C, virus was removed, the cells were washed once with medium, refed with fresh medium that was not supplemented with G418 or hygromycin B, and incubated at 378C for an additional 40 h. The cells were fixed and stained with 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal) as previously described (29), and blue cells were counted.
Southern blot analysis.HeLa-CD4-LTR–b-gal cells at 70% confluency in one 75-cm2flask were infected with 1.5mg of p24 (200 ng/ml) from the virus stock in
the presence of 20mg of DEAE-dextran per ml. The virus was allowed to adsorb for 4 h at 378C. The virus was then removed, and the cells were washed once with serum-free medium, refed with complete medium, and incubated at 378C. At 36 h after the initiation of virus infection, the medium was removed and the cells were washed with serum-free medium and trypsinized. Pelleted cells were washed twice with serum-free medium and transferred to microcentrifuge tubes. A standard plasmid alkaline lysis miniprep procedure (3) was performed to isolate covalently closed circular DNA molecules from the infected cells. Approximately 25% of each preparation was cleaved with ClaI and NheI, electrophoresed through a 0.8% agarose gel, blotted to nylon (Duralon-UV; Stratagene), and probed with a32
P-labeled restriction fragment containing one complete copy of the LTR. To control for potential bacterial plasmid DNA contamination originating from the DNA used to generate the viral stocks, a control transfection stock was generated with R7-3–gpt in the absence of a cotransfected plasmid providing envelope function. Also included on the gel was a DNA preparation from CEM cells infected with R7-3 for 3 days. Only DNA synthesized by reverse transcription will be a substrate for ClaI cleavage because of dam-mediated methylation in the bacterial plasmids used to make virus stocks. For an R7-3 plasmid, the LTR-containing ClaI-NheI fragments are expected to be 12.2 and 4.2 kb. The LTR-containing fragment from a similar digest of a viral single-LTR circle is 2.6 kb, and the fragment from a viral double-LTR circle is 3.3 kb. To control for equal amounts of DNA loading, the blot was stripped of the LTR probe and rehybridized with a32
[image:3.612.60.297.82.261.2]P-labeled restriction fragment containing
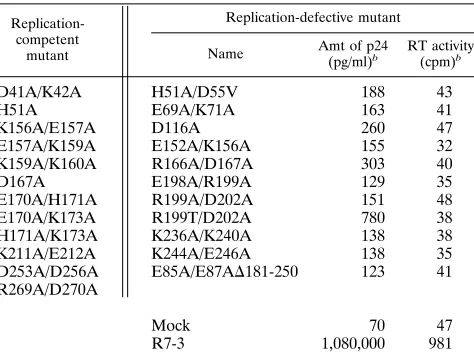
TABLE 1. Summary of HIV-1 integrase mutationsa
Replication-competent
mutant
Replication-defective mutant
Name Amt of p24
(pg/ml)b
RT activity (cpm)b
D41A/K42A H51A/D55V 188 43
H51A E69A/K71A 163 41
K156A/E157A D116A 260 47
E157A/K159A E152A/K156A 155 32
K159A/K160A R166A/D167A 303 40
D167A E198A/R199A 129 35
E170A/H171A R199A/D202A 151 48
E170A/K173A R199T/D202A 780 38
H171A/K173A K236A/K240A 138 38
K211A/E212A K244A/E246A 138 35
D253A/D256A E85A/E87AD181-250 123 41 R269A/D270A
Mock 70 47
R7-3 1,080,000 981
aMutants are named such that the first letter is the amino acid changed, the
number is its location in the integrase protein, and the last letter is the amino acid to which it was changed.
bp24 and RT values were measured 11 days after infection of CEM cells with
1.5 ng of p24 from the mutant and wild-type viruses generated by electroporation of COS-7 cells.
on November 9, 2019 by guest
http://jvi.asm.org/
nt 6941 to 7445 of the human mitochondrial DNA genome. The mitochondrial band migrates as a 13-kb linear fragment.
RESULTS
General strategy of mutant construction.
Charge-cluster-to-alanine scanning mutagenesis was used to generate mutants with mutations of the HIV-1 integrase function. This technique substitutes alanines for any two charged amino acids within a 5-amino-acid scan window. Initially, 24 specific alterations in the coding region of the HIV-1 integrase gene were con-structed (Table 1). One of these exhibited a temperature-sensitive phenotype and is described in the accompanying paper (57). The mutations were designed so as not to interrupt the following regions of integrase: the DNA sequence of the central polypurine tract (11), the resident splice acceptor and splice donor sites (40), and the region coding for the amino terminus of the Vif protein (40). In some cases aberrant mutagenesis occurred, resulting in single alanine substitutions (H51A and D167A), substitutions with amino acids other than alanine (R199T/D202A, K51A/D55V), and an in-frame dele-tion of 69 amino acids near the carboxy terminus (E85A/
E87AD181–250). All mutations were inserted into R7-3, a
full-length infectious HIV-1 proviral clone. To prepare mutant virus stocks regardless of the integrity of the integrase function, the proviral constructs were transfected into a COS-7 cell line
and incubated at 358C for 36 h. CEM cells, a cell line derived
from human T cells, were infected with cell-free virus stocks normalized for 1.5 ng of p24 virion core protein per infection
and incubated at 35, 37, and 39.58C for up to 20 days. The
mutants were scored as either replication competent or repli-cation defective on the basis of formation of syncytia in the infected cultures (Table 1). Twelve of the mutant viruses are replication competent in T cells at all three temperatures. Three of these 12 viruses, D41A/K42A, H51A, and K156A/ E157A develop syncytia as rapidly as the wild type does. The remaining nine form syncytia but with delayed onset of 2 to 5 days compared with that for wild-type virus.
Identification and characterization of replication-defective
integrase mutants.Eleven of the mutant viruses were
replica-tion defective at the three temperatures analyzed. The levels of p24 virion core protein expression and RT activity in cell-free culture supernatant at the end of an 11-day growth period at
378C were measured (Table 1). Less than 0.1% of the wild-type
level of p24 and background RT levels were obtained from the replication-defective mutant viruses, indicating that these vi-ruses are incapable of progeny virion formation.
To investigate if changes in integrase of the replication-defective viruses alter aspects of viral replication other than integration, mutant viruses generated from transient transfec-tion of 293 cells with proviral plasmid DNA were examined for their ability to express and process the Gag-Pol precursor protein correctly. The virus stocks were concentrated by cen-trifugation and normalized for p24 content. Western blot analysis was performed with antibody raised against the amino terminus of HIV-1 integrase (Fig. 1A) or a pool of antibodies to MA (p17) and CA (p24) (Fig. 1B). No difference in the level of 34-kDa integrase protein, relative to the wild type, was detected in the mutant virion preparations. The virus with a
69-amino-acid deletion, E85A/E87AD181–250, produces a
27-kDa integrase protein as expected, but in diminished amounts, perhaps indicating instability of this truncated form of the protein. Charge-cluster-to-alanine scanning mutagenesis did not introduce mutations that rendered the integrase protein unstable.
Similar results were obtained with virus preparations
ana-lyzed by Western blotting for MA and CA proteins (Fig. 1B). Wild-type levels of both proteins were found for all of the replication-defective viruses, indicating that the mutations had no detectable effect on Gag processing or incorporation of MA and CA into the integrase mutant virions. Notably, E85A/
E87AD181–250, a virus that contains a deletion of 25% of the
integrase protein, had normal expression and processing of the Gag proteins.
Since integrase and Gag proteins, which have cleavage sites flanking RT in the Gag-Pol precursor polyprotein, appear to be processed correctly, it is likely that authentic RT also exists in the mutant virions. The same virion preparations used for Western blotting analysis were tested for RT activity. Replica-tion-competent virions were prepared identically. The RT activity of any of the mutant viruses did not vary from that of wild-type virus by more than twofold (Fig. 2). These results suggest that the primary defect associated with the replication-defective phenotype is not due to loss of Gag-Pol processing or of RT activity or to alteration of levels of MA, CA, or integrase
FIG. 1. Presence of correct Gag-Pol proteins in replication-defective inte-grase mutant virions. 293 cells were transfected with 20 mg of DNA from wild-type R7-3 or mutant infectious proviral clones. Virions produced after 72 h were concentrated, normalized for p24 content, and screened for integrase (IN) protein (A) or Gag proteins (B) by Western blot analysis. The positions of viral protein are indicated at the left margin, and molecular size standards are shown at the right.
on November 9, 2019 by guest
http://jvi.asm.org/
proteins packaged into virions but, rather, may be specific to integrase function.
Residues of integrase necessary for replication-competent
phenotype. The charged residues at positions D-55, D-116,
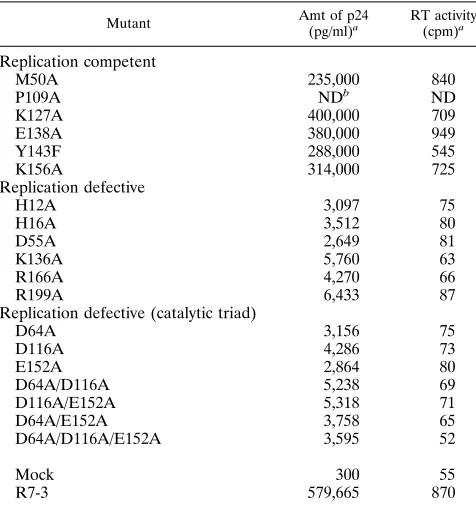
E-152, R-166, R-199, and K-236 are implicated as being essential for integrase function. The other residue forming the pair in the defective double mutant either is present in a replication-competent mutant or is not conservatively substi-tuted in other HIV-1 strains. To determine conclusively whether these residues are essential for viral replication, a panel of 14 mutants with single alanine substitution were generated (Table 2). Certain other residues of interest were also targeted for mutation because they are absolutely con-served in all retroviruses and have been previously defined as essential for integrase function by biochemical analyses with purified mutant integrase proteins. H-12 and H-16 are two residues of a zinc finger-like domain near the amino terminus of the integrase protein that have been shown to be required for zinc binding (8). D-64, D-116, and E-152 are residues which define the catalytic triad of the enzyme (18, 30). Purified enzymes with mutations in any of these residues are defective for catalytic functions of integrase (8, 18, 30, 35). This second set of viral mutants was analyzed for their ability to produce syncytia during CEM cell infection (Table 2). Also, p24 content and RT activity in the culture supernatant 7 days postinfection were determined. As expected, E152A, R166A, and R199A alterations result in a replication-defective pheno-type. All mutant viruses constructed with mutations in the conserved residues of the zinc-binding domain and the cata-lytic triad residues are unable to produce progeny virion. Although H12A and H16A viruses do not form syncytia upon CEM cell infection, the Gag-Pol polyprotein precursor and integrase proteins are expressed and processed correctly (Fig. 1). All of the mutant virus stocks of this set have RT activity at approximately wild-type levels (data not shown).
Unintegrated viral DNA can be a template for viral gene
expression.To directly investigate the frequency of integration
of the original panel of mutant viruses, the proviral clones were reconstructed to include a dominant selectable marker cas-sette, SV40-gpt, in place of most of the coding region of the envelope gene. These viruses were pseudotyped with an
am-photropic envelope and used to infect HeLa-CD4-LTR–b-gal
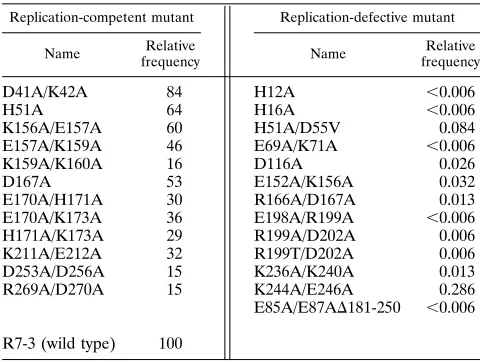
cells in a single-step infection. The number of gpt-resistant colonies obtained from an infection with pseudotyped R7-3 is used as a reference value and set to equal 100 (Table 3). All
other values in Table 3 are relative to this determination. The gpt-resistant colonies, resulting from an integration event, were quantified 12 days postinfection. Culture supernatant from a mock transfection with R7-3–gpt DNA was used as a negative-control virus stock. None of the replication-compe-tent mutants was as efficient as the virus with wild-type integrase in its ability to integrate into host cell DNA as determined by this assay. Mutant viruses D41A/K42A and H51A form syncytia as rapidly as the wild-type does, whereas the other replication-competent mutants exhibit delayed syn-cytium formation relative to the wild type. These visual obser-vations correlate with their integration frequency relative to
wild-type integration in HeLa-CD4-LTR–b-gal cells.
Replica-tion-competent viruses with integration frequencies as low as
15% of that of the wild type in HeLa-CD4-LTR–b-gal cells
(K159A/K160A, D253A/D256A, and R269A/D270A) are ca-pable of sustaining a productive infection of CEM cells. The replication-defective mutants were unable to integrate their DNA into the host genome. However, very low levels of gpt-resistant integrants were detected for some of the replica-tion-defective mutants. These are unlikely to represent wild-type revertants, since integrase in the incoming virion was generated in the transfected cell.
To determine if the integration-defective mutants are capa-ble of expressing Tat, a MAGI cell assay was performed. The
HeLa-CD4-LTR–b-gal cell line was infected with each of the
replication-defective mutant viruses normalized for equal amounts of p24 (Table 4). The replication-defective integrase mutants fall into two distinct phenotypes. Although none of them is able to integrate the viral genome into that of the host (Table 3), a subgroup of these viruses with alterations within
[image:5.612.316.554.83.337.2]FIG. 2. RT activity in mutant virions. Wild-type R7-3 and charge-cluster-to-alanine mutant virions were produced by transfection of 293 cells with 20mg of DNA from infectious proviral clones. Virions produced after 72 h were concen-trated, normalized for p24 content, and screened for RT activity at 378C. Replication-competent and replication-defective virus samples are in the same order from top to bottom as listed in Table 1.
TABLE 2. Single-amino-acid and catalytic triad integrase mutants
Mutant Amt of p24
(pg/ml)a RT activity (cpm)a
Replication competent
M50A 235,000 840
P109A NDb ND
K127A 400,000 709
E138A 380,000 949
Y143F 288,000 545
K156A 314,000 725
Replication defective
H12A 3,097 75
H16A 3,512 80
D55A 2,649 81
K136A 5,760 63
R166A 4,270 66
R199A 6,433 87
Replication defective (catalytic triad)
D64A 3,156 75
D116A 4,286 73
E152A 2,864 80
D64A/D116A 5,238 69
D116A/E152A 5,318 71
D64A/E152A 3,758 65
D64A/D116A/E152A 3,595 52
Mock 300 55
R7-3 579,665 870
a
p24 content and RT activity of progeny virion were measured 7 days after infection of 103
CEM cells with 5 ng of p24 from the mutant and wild-type viruses generated by calcium phosphate transfection of 293 cells. p24 levels less than 10,000 pg/ml were considered to be background, resulting from virus introduced in the initial infection.
b
ND, not determined.
on November 9, 2019 by guest
http://jvi.asm.org/
the catalytic center of integrase are able to express Tat protein
during infection of HeLa-CD4-LTR–b-gal cells whereas the
other viruses are not (Table 4). Those with a change in the catalytic triad residues D-64, D-116, and E-152 or within K-136, which is part of the temperature-sensitive mutant K136A/E138A, yielded the largest number of blue cells in this assay. While these viruses express Tat in the MAGI cell assay, no p24 or RT activity can be detec ted when these viral stocks are used to infect CEM cells (Table 2). The Tat levels expressed may be too low to transactivate further viral gene expression.
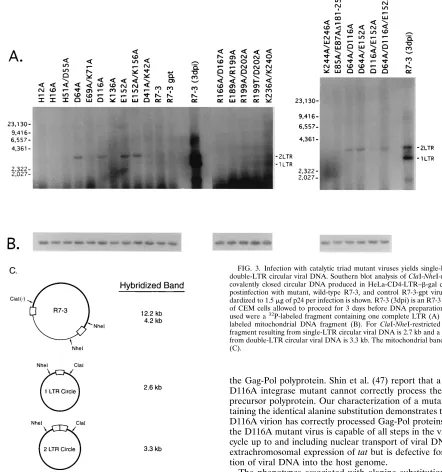
To investigate the possibility that the form of viral DNA in the nucleus differs between the two classes of replication-defective mutants distinguished by Tat expression in
HeLa-CD4-LTR–b-gal cells, Southern blot analysis was performed
(Fig. 3). HeLa-CD4-LTR–b-gal cells were infected with equal
amounts of p24 virus stocks. At 36 h postinfection, a plasmid alkaline lysis miniprep procedure was used to isolate covalently closed circular DNA from infected cells. The DNA was restricted with NheI and ClaI, electrophoresed through a 0.8%
agarose gel, blotted to nylon, and probed with a32P-labeled
fragment containing one complete copy of the LTR. The sizes of fragments expected to hybridize are depicted in Fig. 3C. The single-LTR and double-LTR circular forms of viral DNA in lane R7-3 (3dpi) of Fig. 3A originate from an R7-3 infection of CEM cells harvested 3 days postinfection. The DNA prepara-tions did not differ significantly in the amount of mitochondrial DNA that was also recovered (Fig. 3B). Only plasmids with an alanine substitution at the conserved residues D-64, D-116, and E-152 contain significant amounts of the circular forms of viral DNA detectable by this type of analysis. The results presented in Fig. 3 further explain the results in Table 4. Mutants with mutations at D-64, D-116 and E-152 are capable of providing a circular template for the expression of Tat, even though infection with these mutants is abortive with respect to integration. Contrary to this correlation, the K136A mutant expresses Tat, but circular forms of the viral DNA were not detected in this assay. K136A integrates at levels 7% of that of the wild type but does not result in a productive infection (57). The replication-competent viruses D41A/K42A and R7-3 do
not contain significant levels of single-LTR or double-LTR circular DNA after 36 h of infection (Fig. 3A), suggesting that integration is quite efficient during initial infection. However, these forms are quite pronounced if an infection is allowed to proceed for longer periods in CEM cells (Fig. 3A).
HIV-1 integration is required for viral growth in
macro-phages and PBLs in primary cell culture.While it has been
established both here and elsewhere (32, 47) that integration is required for viral propagation in cultured human T-lymphoid cells, little is known about the requirement for integration in primary cultures of macrophage cells and PBLs. These exper-iments have been hampered because an HIV-1 molecular clone specifying macrophage tropism has been unavailable until the recent cloning of YU-2, a macrophage-tropic isolate (36, 37). Proviral clones were constructed to include the YU-2 envelope in the replication-defective D116A or R199A/D202A proviral clones originating from HXB2d. Identical mutations were also engineered directly into the integrase coding region of YU-2 (Fig. 4A). This set of macrophage-tropic viruses was used to infect primary human macrophages and PBLs with standardized amounts of p24. The infections were monitored for up to 20 days and assayed for the presence of p24 in the culture supernatant (Fig. 4). These results show that neither class of integrase-defective mutants can replicate productively in macrophages or PBLs. The same results were obtained if the integrase mutations were constructed into the YU-2 genome, which encodes a complete copy of the vpr and vpu genes, or if a YU-2 envelope fragment containing the macrophage tropism domain was transferred to the vpr- and vpu-defective HXB2d genome also containing the integrase mutations.
DISCUSSION
[image:6.612.57.297.91.272.2]Charge-cluster-to-alanine mutagenesis of the HIV-1 inte-grase gene was performed to identify residues of inteinte-grase necessary for function during infection. Since the integrase polypeptide is synthesized as a component of a large precursor protein (Gag-Pol), it is possible that minor mutational alter-ations in the integrase portion of the polyprotein will result in aberrant processing or instability of the products of the Gag-Pol polyprotein and/or will influence their incorporation into mutant virions. To this end, mutant virions were examined
TABLE 3. Relative integration frequencyaof pseudotyped HIV-gpt
derivatives in HeLa-CD4-LTR–b-gal cells
Replication-competent mutant Replication-defective mutant
Name Relative
frequency Name
Relative frequency
D41A/K42A 84 H12A ,0.006
H51A 64 H16A ,0.006
K156A/E157A 60 H51A/D55V 0.084
E157A/K159A 46 E69A/K71A ,0.006
K159A/K160A 16 D116A 0.026
D167A 53 E152A/K156A 0.032
E170A/H171A 30 R166A/D167A 0.013
E170A/K173A 36 E198A/R199A ,0.006
H171A/K173A 29 R199A/D202A 0.006
K211A/E212A 32 R199T/D202A 0.006
D253A/D256A 15 K236A/K240A 0.013
R269A/D270A 15 K244A/E246A 0.286
E85A/E87AD181-250 ,0.006 R7-3 (wild type) 100
a
The value obtained for R7-3–gpt is arbitrarily set to 100. All other values are relative to this determination. The value for R7-3–gpt1SV-A-MLV-env is 100, and the value for R7-3–gpt1pUC is,0.006. The absolute value of gpt-resistant colonies for R7-3–gpt is 1.53104
[image:6.612.316.555.92.258.2]. A total of 77 colonies resulted from a 1:200 dilution of the virus. No gpt-resistant colonies were obtained from the negative control infection when the infected cells were split 1:2 into gpt selection medium.
TABLE 4. Tat expression during infection of replication-defective HIV-1 integrase mutants in HeLa-CD4-LTR–b-gal cellsa
Viral mutant No. of blue cells
Viral mutant (catalytic triad)
No. of blue cells
H12A 9 D64A 1,230
H16A 5 D116A 1,354
H51A/D55V 6 E152A 1,389
D55A 12 D64A/D116A 1,293
E69A/K71A 1 D116A/E152A 740
K136A 1,650 D64A/E152A 800
E152A/K156A 1,073 D64A/D116A/E152A 625
R166A 28
R166A/D167A 1 R7-3 7,540
E198A/R199A 0 Mock 0
R199A/D202A 0
R199T/D202A 1
R199A 3
K236A/K240A 0
K244A/E246A 4
a
HeLa-CD4-LTR–b-gal cells were infected with 5 and 50 ng of p24 for each virus. The MAGI cell assay was performed 40 h after infection. The number of blue cells reported represents duplicate measurements resulting from infection with 50 ng of p24.
on November 9, 2019 by guest
http://jvi.asm.org/
for major products of the Gag-Pol precursor protein (Fig. 1). Virion proteins from all replication-defective mutant viruses examined contain wild-type levels of both MA and CA protein. Identical results were obtained for the integrase protein in all of the alanine substitution mutants. Although the mutant virus containing a 70-amino-acid deletion in integrase (E85A/
E87AD181–251) contained diminished amounts of a truncated
integrase protein, the amount of virion-specific MA and CA protein remain unaffected relative to the wild-type virus, R7-3. All mutant viruses examined were capable of providing nearly wild-type levels of polymerase activity when compared with R7-3 (Fig. 2). These results indicate that paired alanine substitutions in HIV-1 integrase do not disrupt normal pro-cessing or impose detectable packaging constraints on the incorporation of wild-type levels of the protein derivatives of
the Gag-Pol polyprotein. Shin et al. (47) report that a HIV-1
D116A integrase mutant cannot correctly process the p55gag
precursor polyprotein. Our characterization of a mutant con-taining the identical alanine substitution demonstrates that the D116A virion has correctly processed Gag-Pol proteins. Here the D116A mutant virus is capable of all steps in the viral life cycle up to and including nuclear transport of viral DNA and extrachromosomal expression of tat but is defective for inser-tion of viral DNA into the host genome.
[image:7.612.86.528.79.551.2]The phenotypes associated with alanine substitution at 31 amino acid positions in the HIV-1 integrase protein are identified from the combined analysis of the replicative poten-tial of 42 integrase mutant viruses and are summarized in Table 5. We are able to identify nine specific amino acids and four charged regions essential for integrase function during the infection of cultured T cells. It is noteworthy that all but one (K-136) of the nine amino acids identified as necessary for viral propagation are absolutely conserved in a comparative study of HIV-1 isolates whereas half of the nonessential residues described here are substituted both conservatively and noncon-servatively among these isolates (38). We are unable to deter-mine which residue of the charged-cluster pairs E-69/K-71, E-85/E-87, K-244/E-246, and K-236/K-240 may be more im-portant to the function of integrase in these replication defec-tive viral mutants. The E85A/E87A mutant was defecdefec-tive when
the integrase deletion in the E85A/E87AD181–250 mutant was
repaired (data not shown).
FIG. 3. Infection with catalytic triad mutant viruses yields single-LTR and double-LTR circular viral DNA. Southern blot analysis of ClaI-NheI-restricted covalently closed circular DNA produced in HeLa-CD4-LTR–b-gal cells 36 h postinfection with mutant, wild-type R7-3, and control R7-3-gpt viruses stan-dardized to 1.5mg of p24 per infection is shown. R7-3 (3dpi) is an R7-3 infection of CEM cells allowed to proceed for 3 days before DNA preparation. Probes used were a32
P-labeled fragment containing one complete LTR (A) or a32
P-labeled mitochondrial DNA fragment (B). For ClaI-NheI-restricted DNA, a fragment resulting from single-LTR circular viral DNA is 2.7 kb and a fragment from double-LTR circular viral DNA is 3.3 kb. The mitochondrial band is 13 kb (C).
on November 9, 2019 by guest
http://jvi.asm.org/
A survey of essential and nonessential amino acids required during infection might be expected to reveal a slightly different set of residues from those determined in vitro. Although five of the residues (H-12, H-16, D-64, D-116, and E-152) identified here were expected to be essential during viral infection, two of the mutant viruses described here exhibit behavior not pre-dicted from previous in vitro studies. Mutant viruses E157A/ K159A and K159A/K160A are replication competent. How-ever, in vitro studies with purified integrase protein containing the K159A mutation show that the mutant protein retained only about 10% of the DNA-processing activity of the wild-type protein (30). Conversely, mutants containing alterations at R-199 are replication defective during infection of cultured T cells (Table 1). This was unexpected since recombinant mutant integrase protein containing substitutions at R-199
demonstrate in vitro 39processing, strand transfer, and
disin-tegration activities indistinguishable from those of the wild
type (35). These results serve to highlight the importance of integrase mutant analysis during infection for interpretation of the contribution of particular integrase residues to the virus life cycle.
Cells infected with three of the replication-competent mu-tants (D41A/K42A, H51A, and K156A/E157A) formed syncy-tia as rapidly as did the wild-type virus. The remaining replication-competent viruses exhibited a delay of onset of syncytium formation relative to that of wild-type virus, but all were capable of sustained viral spread upon reinfection. Visual evidence of viral passage in CEM cells can be correlated with the integration frequency determined for each HIV-gpt
mu-tant in the genetic integration assay in HeLa-CD4-LTR–b-gal
cells (Table 3). Efficient replication of mutant virus in CEM cells for D41A/K42A, H51A, and K156A/E157A was reflected in integration frequencies between 60 and 84% of those obtained for R7-3–gpt. Slower viral growth in this T-cell line is
FIG. 4. Functional integrase required for HIV-1 infection of macrophages and PBLs of primary-cell origin. (A) The macrophage-tropic HXB2d/YU-2 chimeric infectious clone is schematically represented. (B to E) HXB2d/YU-2 has nt 6223 to 8473 of HXB2d substituted with nt 6224 to 8435 of YU-2 with a BbsI-BamHI fragment. Integrase mutations D116A and R199A/D202A were introduced into YU-2 and into HXB2d/YU-2. Mutant virus from these infectious clones was produced by calcium phosphate transfection of 293 cells, standardized for p24 content, and used to infect macrophage cells (B and C) or PBLs (D and E). The progression of the infections was monitored by measuring p24 of progeny virion produced up to 20 days postinfection. p24 values represent the average of duplicate measurements, with the value at day zero subtracted.
on November 9, 2019 by guest
http://jvi.asm.org/
reflected in lower integration values obtained on
HeLa-CD4-LTR–b-gal cells. If the relative integration frequencies
deter-mined in HeLa-CD4-LTR–b-gal cells model integration
fre-quencies during viral infection of CEM cells, then frefre-quencies as low as 15% relative to those for wild-type virus are capable of maintaining viral replication in this cultured T-cell line. However, when the HIV-gpt derivative of the K136A mutant was tested by using this system, an integration frequency of 6% was correlated with a replication-defective phenotype in CEM cells (57).
The characterization of the replication-defective integrase mutants included an assay for detection of single- and double-LTR circles to indicate viral DNA translocation to the nucleus (Fig. 3). Significant amounts of circular viral DNA forms were detected only in cells infected with viruses containing alanine substitutions at residues of the conserved catalytic triad, D-64, D-116, and E-152. Southern blot analysis showed that little, if any, circular viral DNA was obtained from cells infected with the remaining mutants of this set. There is no in vitro precedence for these results with purified integrase. Three possible explanations for this result are offered. First, the mutant viruses not producing circular viral DNAs are unable to synthesize full-length viral DNA upon entry into the host cell. However, since all the mutant virions contain RT activity that is not significantly different from that of wild type (Fig. 2), it seems unlikely that this possibility is correct. Second, the mutants which do not produce viral circular forms may catalyze the synthesis of viral DNA but might be unable to transport the DNA into the cell nucleus. This possibility also seems unlikely since it has been reported that HIV-1 mutants containing significant deletions within the integrase coding region still retain their ability to translocate their reverse-transcribed DNA to the nucleus (48). Lastly, mutations at the catalytic triad residues might render the mutant preintegration complex more likely to release, or never initially bind, the linear viral DNA ends and thereby allow access of host nuclease and ligase to circularize the molecule. The fact that viral circular DNAs are not readily detected in other replication-defective mutants by Southern analysis may not reflect an inherent inability for the viral DNA to be a substrate for circularization but, rather, might indicate an association of these mutant integrase pro-teins with the linear viral DNA termini, thereby preventing release of the viral DNA. Other explanations for these data have not been ruled out.
Unintegrated linear or circular viral DNAs are not generally believed to be templates for retroviral gene expression. For HIV-1, the expression of viral protein originating from
unin-tegrated viral templates has been measured (44). Viral gene expression could not be detected when a sensitive reporter gene was present in place of nef in an HIV-1 genome which also contained a frameshift mutation after amino acid 171 of integrase (44). However, the data presented in Table 4 argue that under certain circumstances, unintegrated viral DNA expresses Tat. All viruses with mutations in residues of the catalytic triad express Tat, although they do not integrate. There is a correlation between the ability of an integration-defective mutant virus to produce single- and double-LTR circles (Fig. 3) and the ability to express Tat from unintegrated viral template DNA (Table 4) for mutants containing alter-ations in the catalytic triad. Although the replication-defective mutant, K136A, produces blue plaques in a MAGI assay, we were unable to detect significant amounts of unintegrated circular viral DNA from a K136A infection. However, the number of the blue cells formed after a K136A infection must result in part from the expression of integrated copies of the mutant viral genome, since K136A-gpt exhibits an integration frequency of 7% that of the wild type when assayed under identical experimental conditions (57). The number of blue cells, resulting from extrachromosomal expression of Tat
ob-tained from an infection of HeLa-CD4-LTR–b-gal cells with
any of the integrase catalytic triad mutants, is five- to sevenfold smaller than that resulting from an infection with the wild-type virus, R7-3. This discrepancy may reflect a lower efficiency of infection per unit of p24 with the replication-defective mutants or alternatively may result from a relatively low level of Tat production from unintegrated viral DNA templates. It remains to be determined if Tat expression from unintegrated circular viral templates has pathogenic relevance.
The amount of Tat protein produced from an infection with the catalytic triad integrase mutants is unknown. However, it must be smaller than that synthesized from a replication-competent viral infection, since this protein is rate limiting for the production of all subsequent viral structural gene products (14). We have been unable to detect de novo synthesis of any viral structural gene products during infection with any of the replication-defective integrase mutants (Tables 1 and 2). In contrast, Stevenson et al. (48) have produced integrase mu-tants containing EcoRI frameshift deletion mutations at inte-grase amino acids 140 to 141 which are unable to sustain an active infection of cultured T cells but express low levels of capsid protein in the absence of integration. We have recon-structed two of the Stevenson et al. (48) mutants containing
EcoRI frameshift deletion mutations in R7-3 (Dint2 andDint5
[48]). Neither R7-3Dint2 nor R7-3Dint5 integrates in a gpt
integration assay, and they do not produce blue plaques in a MAGI cell assay (data not shown).
[image:9.612.57.297.88.228.2]Although replication-competent integration-defective mu-tants have been reported for spleen necrosis virus (42), HIV-1 must retain integrase activity for propagation in cultured human T cells (32, 47). Whether or not HIV-1 integration is required for efficient viral production in primary cells has previously been unknown. One representative member from each class of tat-expressing or tat-nonexpressing replication-defective integrase mutants (D116A or R199A/D202A, respec-tively) was used to determine whether HIV-1 derivatives specifying these mutations could replicate in cultured primary cells. In the first set of experiments, HXB2d-derived integrase mutant clones were altered to include determinants for mac-rophage tropism and tested for growth in cultured PBLs (Fig. 4E) and macrophages (Fig. 4C). A second set of experiments was undertaken to preclude any contribution that the HXB2d genetic background might have on the interpretation of results. To this end, the identical integrase mutations were engineered
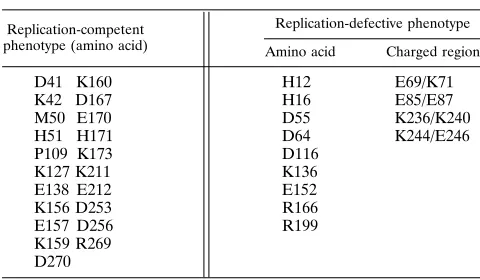
TABLE 5. Phenotype associated with alanine-substituted amino acids
Replication-competent phenotype (amino acid)
Replication-defective phenotype
Amino acid Charged region
D41 K160 H12 E69/K71
K42 D167 H16 E85/E87
M50 E170 D55 K236/K240
H51 H171 D64 K244/E246
P109 K173 D116
K127 K211 K136
E138 E212 E152
K156 D253 R166
E157 D256 R199
K159 R269 D270
on November 9, 2019 by guest
http://jvi.asm.org/
into infectious proviral clones of primary origin (YU-2) (Fig. 4B and D). For both approaches, functional integrase is required for propagation of HIV-1 in primary cell culture of the major cell types associated with HIV-1 infection in hu-mans. This suggests that chemotherapy targeted to retrovirus integrase may have clinical relevance for HIV-1 infection in vivo.
ADDENDUM
An additional study characterizing HIV-1 integrase mutant viruses has recently been reported (10).
ACKNOWLEDGMENTS
We thank Steve Hatch and Chris Mundy for technical contributions and assistance with graphics. We thank Syd Johnson for helpful suggestions. We are grateful to Joe Colacino and Richard Jaskunas for critical review of the manuscript and to Carlos Lopez for support of this work.
REFERENCES
1. Aldovini, A., and B. D. Walker (ed.). 1990. Techniques in HIV research. Stockton Press, New York.
2. Bass, S. H., M. G. Mulkerrin, and J. A. Wells. 1991. A systematic mutational analysis of hormone-binding determinants in the human growth hormone receptor. Proc. Natl. Acad. Sci. USA 88:4498–4502.
3. Birnboim, H. C., and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7:1513–1519. 4. Brown, P. O., B. Bowerman, H. E. Varmus, and J. M. Bishop. 1989.
Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc. Natl. Acad. Sci. USA
86:2525–2529.
5. Bukrinsky, M. I., N. Sharova, T. L. McDonald, T. Pushkarskaya, W. G.
Tarpley, and M. Stevenson. 1993. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc. Natl. Acad. Sci. USA
90:6125–6129.
6. Burke, C. J., G. Sanyal, M. W. Bruner, J. A. Ryan, R. L. LaFemina, H. L.
Robbins, A. S. Zeft, C. R. Middaugh, and M. G. Cordingley.1992. Structural implications of spectroscopic characterization of a putative zinc finger peptide from HIV-1 integrase. J. Biol. Chem. 267:9639–9644.
7. Bushman, F. D., and R. Craigie. 1991. Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. USA 88:1339–1343.
8. Bushman, F. D., A. Engelman, I. Palmer, P. Wingfield, and R. Craigie. 1993. Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotide transfer and zinc binding. Proc. Natl. Acad. Sci. USA 90:3428–3432.
9. Bushman, F. D., T. Fujiwara, and R. Craigie. 1990. Retroviral DNA integration directed by HIV integration protein in vitro. Science 249:1555– 1558.
10. Cannon, P. M., W. Wilson, E. Byles, S. M. Kingsman, and A. J. Kingsman. 1994. Human immunodeficiency virus type 1 integrase: effect on viral replication of mutations at highly conserved residues. J. Virol. 68:4768–4775. 11. Charneau, P., and F. Clavel. 1991. A single-stranded gap in human immu-nodeficiency virus unintegrated linear DNA defined by a central copy of the polypurine tract. J. Virol. 65:2415–2421.
12. Chow, S. A., K. A. Vincent, V. Ellison, and P. O. Brown. 1992. Reversal of integration and DNA splicing mediated by integrase of human immunode-ficiency virus. Science 255:723–726.
13. Craigie, R., T. Fujiwara, and F. D. Bushman. 1990. The IN protein of moloney murine leukemia virus processes the viral DNA ends and accom-plishes their integration in vitro. Cell 62:829–837.
14. Cullen, B. R. 1989. Regulatory pathways governing HIV-1 replication. Cell
58:423–426.
15. Cunningham, B. C., and J. A. Wells. 1989. High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science
244:1081–1085.
16. Drelich, M., R. Wilhelm, and J. Mous. 1992. Identification of amino acid residues critical for endonuclease and integration activities of HIV-1 IN protein in vitro. Virology 188:459–468.
17. Engelman, A., F. D. Bushman, and R. Craigie. 1993. Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex. EMBO J. 12:3269–3275.
18. Engelman, A., and R. Craigie. 1992. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66:6361–6369.
19. Engelman, A., K. Mizuuchi, and R. Craigie. 1991. HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67:1211– 1221.
20. Feinberg, M. B., D. Baltimore, and A. D. Frankel. 1991. The role of Tat in the HIV life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. USA 88:4045–4050.
21. Fujiwara, T., and R. Craigie. 1989. Integration of mini-retroviral DNA: a cell-free reaction for biochemical analysis of retroviral integration. Proc. Natl. Acad. Sci. USA 86:3065–3069.
22. Fujiwara, T., and K. Mizuuchi. 1988. Retroviral DNA integration: structure of an integration intermediate. Cell 54:497–504.
23. Goff, S. P. 1992. Genetics of retroviral integration. Annu. Rev. Genet. 26: 527–544.
24. Johnson, M. S., M. A. McClure, D. F. Fend, J. Gray, and R. F. Doolittle. 1986. Computer analysis of retroviral pol genes: assignment of enzymatic functions to specific sequences and homologies with nonviral enzymes. Proc. Natl. Acad. Sci. USA 83:7648–7652.
25. Kalpana, G. V., and S. P. Goff. 1993. Genetic analysis of homomeric interactions of human immunodeficiency virus type 1 integrase using the yeast two-hybrid system. Proc. Natl. Acad. Sci. USA 90:10593–10597. 26. Katz, R. A., G. Merkel, J. Kulkosky, and A. M. Skalka. 1990. The avian
retroviral IN protein is both necessary and sufficient for integrative recom-bination in vitro. Cell 63:87–95.
27. Katzman, M., R. A. Katz, A. M. Skalka, and J. Leis. 1989. The avian retroviral integration protein cleaves the terminal sequences of linear viral DNA at the in vivo sites of integration. J. Virol. 63:5319–5327.
28. Khan, E., J. P. G. Mack, R. A. Katz, J. Kulkosky, and A. M. Skalka. 1991. Retroviral integrase domains: DNA binding and the recognition of the LTR sequences. Nucleic Acids Res. 19:851–860.
29. Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integratedb-galactosidase gene. J. Virol. 66: 2232–2237.
30. Kulkosky, J., K. S. Jones, R. A. Katz, J. P. G. Mack, and A. M. Skalka. 1992. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 12:2331–2338.
31. LaFemina, R. L., P. L. Callahan, and M. G. Cordingley. 1991. Substrate specificity of recombinant human immunodeficiency virus integrase protein. J. Virol. 76:5624–5630.
32. LaFemina, R. L., C. L. Schneider, H. L. Robbins, P. L. Callahan, K. LeGrow,
E. Roth, W. A. Schleif, and E. Emini.1992. Requirement of active human immunodeficiency virus type 1 integrase enzyme for productive infection of human T-lymphoid cells. J. Virol. 66:7414–7419.
33. Landau, N. R., K. A. Page, and D. R. Littman. 1991. Pseudotyping with human T-cell leukemia virus type 1 broadens the human immunodeficiency virus host range. J. Virol. 65:162–169.
34. Leavitt, A. D., R. B. Rose, and H. Varmus. 1992. Both substrate specificity and target oligonucleotide sequences affect in vitro integration mediated by human immunodeficiency virus type 1 integrase protein produced in Sac-charomyces cerevisiae. J. Virol. 66:2359–2368.
35. Leavitt, A. D., L. Shiue, and H. E. Varmus. 1993. Site-directed mutagenesis of HIV-1 integrase demonstrated differential effects on integrase functions in vitro. J. Biol. Chem. 268:2113–2119.
36. Li, Y., H. Hui, C. J. Burgess, R. W. Price, P. M. Sharp, B. H. Hahn, and
G. M. Shaw.1992. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. J. Virol. 66:6587– 6600.
37. Li, Y., J. C. Kappes, J. A. Conway, R. W. Price, G. M. Shaw, and B. H. Hahn. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973–3980. 38. Meyers, G., B. Korber, S. Wain-Hobson, R. F. Smith, and G. N. Pavlakis
(ed.).1993. Human retroviruses and AIDS. Los Alamos National Labora-tory, Los Alamos, N.M.
39. Miller, M. D., M. T. Warmerdam, I. Gaston, W. C. Greene, and M. B.
Feinberg.1994. The human immunodeficiency virus-1 nef gene product: a positive factor for viral infection and replication in primary lymphocytes and macrophages. J. Exp. Med. 179:101–113.
40. Muesing, M. A., D. H. Smith, C. D. Cabriadilla, C. V. Benton, L. A. Lasky,
and D. J. Capon.1985. Nucleic acid structure and expression of the human AIDS/lymphadenophathy retrovirus. Nature (London) 313:450–458. 41. Page, K. A., N. R. Landau, and D. R. Littman. 1990. Construction and use of
a human immunodeficiency virus vector for analysis of virus infectivity. J. Virol. 64:5270–5276.
42. Panganiban, A. T., and H. M. Temin. 1983. The terminal nucleotides of retrovirus DNA are required for integration but not virus production. Nature (London) 306:155–160.
43. Roth, M. J., P. L. Schwartzberg, and S. P. Goff. 1989. Structure of the termini of DNA intermediates in the integration of retroviral DNA: dependence on IN function and terminal DNA sequence. Cell 58:47–54.
on November 9, 2019 by guest
http://jvi.asm.org/
44. Sakai, H., M. Kawamura, J.-I. Sakuragi, S. Sakuragi, R. Shibata, A.
Ishimoto, N. Ono, S. Ueda, and A. Adachi.1993. Integration is essential for efficient gene expression of human immunodeficiency virus type 1. J. Virol.
67:1169–1174.
45. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
46. Sherman, P. A., and J. A. Fyfe. 1990. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. USA 87:5119–5123.
47. Shin, C.-G., B. Taddeo, W. A. Haseltine, and C. M. Farnet. 1994. Genetic analysis of the human immunodeficiency virus type 1 integrase protein. J. Virol. 68:1633–1642.
48. Stevenson, M., S. Haggerty, C. A. Lamonica, C. M. Meier, S. K. Welch, and
A. J. Wasiak.1990. Integration is not necessary for expression of human immunodeficiency virus type 1 protein products. J. Virol. 64:2421–2425. 49. van Gent, D. C., A. Oude Groeneger, and R. H. A. Plasterk. 1992. Mutational
analysis of the integrase protein of human immunodeficiency virus type 2. Proc. Natl. Acad. Sci. USA 89:9598–9602.
50. van Gent, D. C., C. Vink, A. A. M. Oude Groeneger, and R. H. A. Plasterk. 1993. Complementation between HIV integrase proteins mutated in differ-ent domains. EMBO J. 12:3261–3267.
51. Varmus, H., and P. Brown. 1989. Retroviruses, p. 53–108. In D. E. Berg and
M. M. Howe (ed.), Mobile DNA. American Society for Microbiology, Washington, D.C.
52. Vincent, K. A., V. Ellison, S. A. Chow, and P. O. Brown. 1993. Character-ization of human immunodeficiency virus type 1 integrase expressed in Escherichia coli and analysis of variants with amino-terminal mutations. J. Virol. 67:425–437.
53. Vink, C., A. A. M. Oude Groeneger, and R. H. A. Plasterk. 1993. Identifica-tion of the catalytic and DNA-binding region of the human immunodefi-ciency virus type 1 integrase protein. Nucleic Acids Res. 21:1419–1425. 54. Vink, C., E. Yeheskiely, G. A. van der Marel, J. H. van Boom, and R. H. A.
Plasterk.1991. Site-specific hydrolysis and alcoholysis of human immunode-ficiency virus DNA termini mediated by the viral integrase protein. Nucleic Acids Res. 19:6691–6698.
55. Vora, A. C., M. L. Fitzgerald, and D. P. Grandgenett. 1990. Removal of 39-OH terminal nucleotides from blunt-ended long terminal repeat termini by the avian retrovirus integration protein. J. Virol. 64:5656–5659. 56. Whitcomb, J. M., and S. H. Hughes. 1992. Retroviral reverse transcription
and integration: progress and problems. Annu. Rev. Cell Biol. 8:275–306. 57. Wiskerchen, M., and M. A. Muesing. 1995. Identification and
characteriza-tion of a temperature-sensitive mutant of human immunodeficiency virus type 1 by alanine scanning mutagenesis of the integrase gene. J. Virol.
69:597–601.