ABSTRACT
PÉREZ, DAMARIS ESTHER. Synthesis, Characterization and Reactivity of Rhenium(III) Complexes for Catalytic, Stoichiometric and Insertion Reactions. (Under the direction of Elon A. Ison).
This document focuses on the synthesis and characterization of new rhenium(III) complexes bearing tridentate pincer ligands. These organometallic complexes were subject to comprehensive studies of their reactivity towards catalytic, stoichiometric and insertion reactions. Mechanistic studies of these transformations will lead to the development of more efficient catalytic systems.
In Chapter 2, cationic bisacetonitrile diamidoamine (DAAm) rhenium(III) and cationic diamineamido (DAmA) rhenium(III) complexes were synthetized, and characterized. Their reactivity towards the hydrosilylation reaction of aldehydes was studied. It was proposed through experimental and kinetic data that the most likely pathway for this transformation follows a non-hydride ionic hydrosilylation mechanism. Various aliphatic and aromatic aldehydes were tested. Excellent yields were achieved at ambient temperature in neat conditions using dimethylphenylsilane. The reaction affords TONs of up to 9,200 and a TOF of up to 126 h-1.
In Chapter 3, a computational mechanistic study of the hydrosilylation reaction of aldehydes by novel cationic rhenium(III) bisacetronitrile complex is presented. DFT analysis supports a non-hydride ionic hydrosilylation mechanism which was previously proposed based on experimental and kinetic data.
In Chapter 4, the reactivity of novel DAAm-C6F5 rhenium(III) acetate complex towards Lewis acidic boron reagents was investigated. The role of water as an oxygen source was studied. Novel rhenium(III) and rhenium(V) complexes bearing a C6F5 ligand were synthesized, characterized, and proposed as intermediates. Mechanistic transformations were proposed base on experimental data and previous reports in the literature.
Synthesis, Characterization and Reactivity of Rhenium(III) Complexes for Catalytic, Stoichiometric and Insertion Reactions
by
Damaris Esther Pérez
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
Chemistry
Raleigh, North Carolina 2018
APPROVED BY:
______________________________ _____________________________
Dr. Elon A. Ison Dr. Reza Ghiladi
Committee Chair
DEDICATION
To my husband, Hernán
To my baby girl, Sofía Esther
To my parents, Zory and Fidel
To my sisters, Sandra and Vivian
To my grandparents, Mama Candy (1928 - 2012) and Papa Tato
BIOGRAPHY
Damaris Esther Pérez (formerly Rivera-Santos) was born on September 13th, 1986 in
Aibonito, Puerto Rico. She and her sisters, Sandra and Vivian were raised by their parents, Fidel
and Zoraida. From an early age, she had a passion for math and science. Damaris graduated from
Bonifacio Sánchez Jiménez high school in May 2004 and moved to San Juan, Puerto Rico to
complete her undergraduate degree at University of Puerto Rico at Río Piedras (UPR-RP). She
graduated with a Bachelor of Science degree in Chemistry in May 2009. Upon graduation, she
stayed at UPR-RP took graduate courses and worked as a teaching assistant. Then, in August 2010,
she moved to Oswego, New York to pursue a Master’s Degree in Chemistry at the State University
of New York (SUNY) at Oswego. She conducted graduated research under the supervision of Dr.
Fehmi Damkaci in organic chemistry and worked as a teaching assistant. She graduated with a
M.S. in chemistry in May 2012 and then moved to Lincoln, Nebraska. In August 2012, she started
graduated research under the supervision of Dr. James Takacs in organic chemistry and worked as
a teaching assistant at University of Nebraska Lincoln (UNL). In May 2013, she moved to Raleigh,
North Carolina to pursue her Ph.D. in Chemistry at North Carolina State University. Since arriving
to NC State University, she has been conducting her doctoral research in organometallic chemistry
under the direction of Dr. Elon A. Ison. Upon graduation in 2018, Damaris plans to pursue a
TABLE OF CONTENTS
LIST OF TABLES ... vi
LIST OF FIGURES ... vii
LIST OF SCHEMES ... ix
Chapter 1: General Introduction ... 1
1.1 Organometallic Chemistry ... 2
1.2 Scope of this dissertation ... 3
1.3 References ... 5
Chapter 2: Cationic Rhenium(III) Complexes: Synthesis, Characterization, and Reactivity for Catalytic Hydrosilylation Reaction of Aldehydes ... 7
2.1 Abstract ... 8
2.2 Introduction ... 8
2.3 Results and Discussion ... 10
2.4 Conclusions ... 34
2.5 Experimental Section ... 35
2.6 References ... 42
Chapter 3: Mechanistic Investigation of the Hydrosilylation Reaction of Aldehydes by a Bisacetonitrile Cationic Rhenium(III) Complex: A Computational Study ... 44
3.1 Abstract ... 45
3.2 Introduction ... 45
3.3 Results and Discussion ... 49
3.4 Conclusions ... 59
3.5 Experimental Section ... 60
3.6 References ... 61
Chapter 4: Reactivity of Novel DAAm Rhenium(III) Acetate Complex Towards Lewis Acidic Boron Reagents ... 64
4.1 Abstract ... 65
4.2 Introduction ... 65
4.4 Conclusions ... 83
4.5 Experimental Section ... 84
4.6 References ... 91
5.1 Abstract ... 94
5.2 Introduction ... 94
5.3 Results and Discussion ... 97
5.5 Conclusions ... 115
5.6 Experimental Section ... 116
LIST OF TABLES
Table 2.1. Catalytic hydrosilylation of benzaldehyde according to equation 1 ... 18
Table 2.2. Catalytic hydrosilylation of benzaldehyde with various silane substrates ... 19
Table 2.3. Catalytic hydrosilylation of benzaldehyde according to equation 3 ... 20
Table 2.4. Catalytic hydrosilylation of benzaldehyde with various silane substrates under neat conditions ... 21
Table 2.5. Selected Crystallographic Data and Collection Parameters for 5a, 6b, and 8. ... 41
Table 3.1 Comparison of the calculated DFT (B3PW91-D3) bond lengths (Å) for 4a with the X-ray crystallography ... 50
Table 3.2 Comparison of the calculated DFT (B3PW91-D3) bond angles (°) for 4a with the X-ray crystallography.………...………. 51
Table 4.1 Selected Crystallographic Data and Collection Parameters for 17 and 18. ... 90
Table 4.2 Selected Crystallographic Data and Collection Parameters for 19 and 21. ... 90
Table 5.1 Results for the CO Insertion Reaction of (DAAm)ReIII(CO)(R) complexes ... 104
Table 5.2 Results for the CO Insertion Reaction with 26 ... 112
Table 5.3 Selected Crystallographic Data and Collection Parameters for 23a and 23b. ... 121
LIST OF FIGURES
Figure 2.1. 1H NMR spectrum of cationic rhenium(III) bisacetronitrile complex
6a at room temperature in acetonitrile-d3 ... 12
Figure 2.2. FTIR spectrum (KBr Pellets cm-1) of [(DAAm)Re(CO)(NCCH3)2][BF4] (aryl = C6F5) (6a)…………..……… 13
Figure 2.3.X-ray structure of 5a………...………... 14
Figure 2.4. X-ray structure of 6b ... 14
Figure 2.5. Thermal ellipsoid plot of 8 ... 16
Figure 2.6. 1H NMR of DAmA rhenium(III) dichloride complex (8) in acetonitrile-d3 ... 17
Figure 2.7. Time profile for the catalytic hydrosilylation of benzaldehyde with dimethylphenylsilane and 2-bromomesitylene using 6a as the catalyst. ... 23
Figure 2.8. Rate law dependence on rhenium concentration. Plot kobs (s-1) versus Rhenium concentration (mM) ... 25
Figure 2.9. Kinetic plot of the concentration of benzaldehyde vs reaction time (s). ... 26
Figure 2.10. Hammett plot for the competition experiments between para-substituted benzaldehyde ... 30
Figure 2.11. Eyring plot for the hydrosilylation of benzaldehyde with 6a ... 31
Figure 2.12. Calibration curve to calculate the concentration of benzaldehyde by Gas Chromatography (GC) ... 39
Figure 3.1 X-ray crystallography determined structure of 4a ... 49
Figure 3.2 B3PW91-D3 calculated structure of intermediate 13 ... 52
Figure 3.3 B3PW91-D3 calculated structure of intermediate 14 ... 53
Figure 3.4 B3PW91-D3 calculated structure of TS4 ... 55
Figure 3.5 B3PW91-D3 calculated structure of ion pair 15 ... 56
Figure 3.6 B3PW91-D3 calculated structure of TS5 ... 57
Figure 3.7 B3PW91-D3 calculated structure of 16 ... 58
Figure 3.8 DFT (B3PW91-D3) calculated reaction progress for the hydrosilylation reaction of benzaldehyde by cationic rhenium(III) complex. ... 59
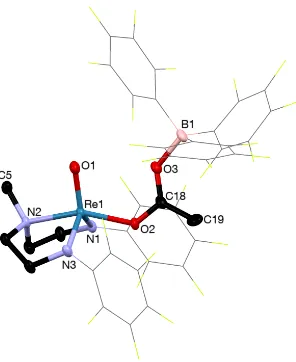
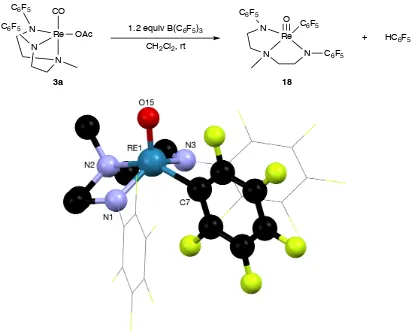
Figure 4.1 Thermal ellipsoid plot of 17 ... 68
Figure 4.2 1H NMR (400 MHz, CD2Cl2) spectrum of [(DAAm)Re(O)(OC(CH3)OB(C6F5)3] (17)……….. 69
Figure 4.4 Decomposition of complex 17 monitored by 1H NMR spectroscopy overtime. ... 72
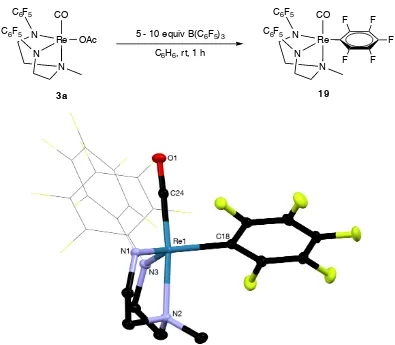
Figure 4.5 X-ray structure of 19 ... 73
Figure 4.6 1H NMR (300 MHz, C6D6) spectrum of [(DAAm-C6F5)Re(CO)(C6F5)] (19). ... 74
Figure 4.7 19F NMR (376 MHz, C6D6) spectrum of tris(pentafluorophenyl)borane monohydrate (20) ... 76
Figure 4.8 1H NMR (300 MHz, C6D6) spectrum of [DAAmRe(CO)(OB(C6F5)2OC(CH3)O] (21) ... 78
Figure 4.9 X-ray structure of complex 21 ... 79
Figure 4.10 19F NMR (376 MHz, C6D6) spectrum of bispentafluoroborinic acid (22). ... 81
Figure 5.1 1H NMR spectrum of complex 23a in methylene chloride-d2 ... 98
Figure 5.2 Thermal ellipsoid plot of 23a ... 99
Figure 5.3 1H NMR spectrum of complex 23b in methylene chloride-d2 ... 100
Figure 5.4 Thermal ellipsoid plot of 23b ... 101
Figure 5.5 1H NMR spectrum of complex 24 in methylene chloride-d2 ... 102
Figure 5.6 1H NMR spectrum of complex 25 in methylene chloride-d2 ... 102
Figure 5.7 Thermal ellipsoid plot of 25 ... 103
Figure 5.8 Spectral comparison: (a) 1H NMR spectrum of the crude reaction of 23b with 60 psi CO at 80 °C after 30 min in CD2Cl2. (b) 1H NMR spectrum of complex 23b in CD2Cl2 ... 106
Figure 5.9 Integrated 1H NMR spectrum of the reaction of 23b with 60 psi CO at 80 °C after 30 minutes ... 107
Figure 5.10 FTIR spectrum of the acyl-product ... 108
Figure 5.11 FTIR spectrum of the complex 23b ... 109
Figure 5.12 1H NMR spectrum of complex 26 in methylene chloride-d2 ... 110
Figure 5.13 Thermal ellipsoid plot of 26 ... 111
LIST OF SCHEMES
Scheme 1.1 Variety of rhenium complexes synthesized in the Ison group. ... 3
Scheme 2.1. Catalytic comparison between neutral rhenium(V) complex and cationic rhenium(V) complexes for hydrosilylation reaction by Abu Omar group. ... 9
Scheme 2.2. Catalytic competency of DAAm rhenium(III) complex versus rhenium(V) complexes in hydrosilylation reactions by Ison group. ... 10
Scheme 2.3. Synthesis of novel cationic rhenium(III) bisacetronitrile derivatives ... 11
Scheme 2.4. Synthesis of DAmA rhenium(III) dichloride complex 8. ... 15
Scheme 2.5. Substrate scope using the optimized conditions according to equation 5. ... 22
Scheme 2.6. Stoichiometric reaction of 6a and dimethylphenylsilane. ... 27
Scheme 2.7. Proposed mechanism for the formation of 9 from 3a by Ison group. ... 28
Scheme 2.8. Proposed mechanism for the formation of 9 from 6a. ... 28
Scheme 2.9. Stoichiometric reaction of 6a and benzaldehyde. ... 29
Scheme 2.10. Competition experiments between para-substituted benzaldehyde. ... 29
Scheme 2.11. Variable temperature kinetic experiments. ... 31
Scheme 2.12. Proposed mechanism for the hydrosilylation reaction of aldehydes catalyzed by 6a. ... 32
Scheme 3.1 Hydride mechanisms for the hydrosilylation reaction of carbonyl compounds. ... 46
Scheme 3.2 Proposed mechanism for catalytic ionic hydrosilylation by Bullock et al. ... 47
Scheme 3.3 Catalytic Hydrosilylation Reaction of Aldehydes by 6a. ... 47
Scheme 3.4 Proposed mechanism for the hydrosilylation reaction of aldehydes catalyzed by 6a. ... 48
Scheme 4.1 Synthesis of DAAm rhenium(III) acetate complexes 3a and 3b previously reported by our group. ... 65
Scheme 4.2 Synthesis of Lewis acid/base adducts previously reported by the Ison group. ... 66
Scheme 4.3 Preliminary investigation of the reactivity of 3a towards Lewis acidic boron reagents. ... 67
Scheme 4.4 Synthesis of [(DAAm)Re(O)(OC(CH3)OB(C6F5)3)], 17. ... 69
Scheme 4.5 Attempts to reproduce previous results by our group. ... 70
Scheme 4.6 Proposed reaction pathway for the formation of 18. ... 71
Scheme 4.7 Synthesis of [(DAAm-C6F5)Re(CO)(C6F5)] (aryl = C6F5), 19. ... 73
Scheme 4.9 Proposed decarbonylation for the formation of 18 from 19. ... 75
Scheme 4.10 Synthesis of tris(pentafluorophenyl)borane monohydrate, 20. ... 75
Scheme 4.11 Reactivity of 19 with [H2O·B(C6F5)3] ... 76
Scheme 4.12 Reactivity of 19 in the presence of oxygen ... 77
Scheme 4.13 Probing 20 as the active species in the reaction of 3a towards Lewis acidic boric reagents. ... 77
Scheme 4.14 Proposed mechanism pathway for the transformation of 21 to 19. ... 80
Scheme 4.15 Synthesis of bis(pentafluorophenyl)borinic acid, 22. ... 81
Scheme 4.16 Probing 22 as the active species in the reaction of 3a towards Lewis acidic boron reagents. ... 82
Scheme 4.17 Equilibrium between the κ1 and κ2 isomers of 3a. ... 82
Scheme 4.18 Proposed transformation of 3a’ in the presence of the conjugated base [HOB(C6F5)3]- to lead 21. ... 83
Scheme 4.19 Intermediates identified in this investigation and proposed mechanism for each transformation. ... 83
Scheme 5.1 Synthesis of DAAm rhenium(III) acetate complexes 3a and 3b. ... 95
Scheme 5.2 Carbonylation Reaction of DAAm Oxorhenium(V) Complexes by the Ison Group ... 95
Scheme 5.3 Carbonylation Reaction of DAP Oxorhenium(V) Complex by the Ison Group ... 96
Scheme 5.4 Carbonylation Reaction of SSS Oxorhenium(V) Complex by the Ison Group ... 96
Scheme 5.5 Carbonylation Reaction of PNP Nitridorhenium(V) Complex by the Ison Group ... 96
Scheme 5.6 Synthesis of DAAm rhenium(III) derivatives ... 97
Scheme 5.7 General CO insertion reaction with (DAAm-aryl)ReIII(CO)(R) complexes. ... 104
Scheme 5.8 Synthesis of complex (DAAm-Mes)Re(O)(CH2Ph), 26. ... 109
Scheme 5.9 General CO insertion reaction with (DAAm-Mes)ReV(O)(CH2Ph)(26) complex. ... 111
Scheme 5.10 General Mechanism for Direct CO Insertion. ... 113
Scheme 5.11 General Mechanism for CO Adduct formation. ... 114
Chapter 1: General Introduction
1.1 Organometallic Chemistry
Organometallic chemistry is the field that combines inorganic and organic chemistry. It involves the interaction of a metal center surrounded by an organic ligand to generate an organometallic complex. Transition metal complexes have been shown to be of great importance in the catalytic conversion of various chemical transformations, increasing reactivity, selectivity, and creating new pathways for the formation of organic molecules.1,2 Unlike typical metal complexes, organometallic complexes are often easily prepared and have a good solubility in a variety of solvents, which makes them attractive species for homogenous catalysis. Also, their contribution to the field of green chemistry includes their capacity to: increase reaction rates, efficiency, selectivity of reactions, maximize atom economy and minimize waste of synthetic processes.3
Rhenium catalysts are attractive due to their unique properties. One of the benefits is that they possess properties of both early and late transition metals. The chemical properties of rhenium resemble the metals in the manganese group (Group 7) of the Periodic Table. However, the physical properties of rhenium are much more similar to those of the refractory metals of Groups 5 and 6, particularly molybdenum and tungsten. Also, it has access to a variety of oxidation states from -1 to +7. Rhenium easily change from one valence to another, a property which makes it ideal for use as a catalyst.4
Recently, our group reported the synthesis of a series of rhenium complexes that exhibit catalytic activity and the mechanisms of these transformations were described.5-18 Examples of rhenium(V) and rhenium(III) complexes synthesized in the Ison group are shown in Scheme 1.1. The stability and reactivity of organometallic compounds are associated with the nature of the
Scheme 1.1 Variety of rhenium complexes synthesized in the Ison group.
The use of rhenium complexes as catalysts has numerous advantages. Rhenium complexes have a large functional group tolerance,1,2 and the ability to tolerate a variety of ligands. Rhenium can generally form various stable complexes, which is beneficial for the study of reaction mechanisms. The synthesis of these complexes serves as an inexpensive and air and moisture stable alternative to low-valent late transition metals. These characteristics make the development of new rhenium complexes a promising option for current organometallic systems. Rhenium catalysts have been the subject of many studies, and subsequently, a variety of rhenium complexes bearing diverse ligand frameworks have been synthesized by many research groups.19
1.2 Scope of this dissertation
The work in this thesis explores the reactivity of rhenium(III) complexes. As shown in Scheme 1.1 (right) a variety of rhenium(III) complexes can be synthesized. The goal of this dissertation is to study new rhenium(III) complexes and discover potential catalytic applications based on experimental observations and data analysis. These reactivity studies will help to tune, improve and understand the behavior of these organometallic complexes. The majority of the investigations in this dissertation are based on the reactivity of [(DAAm-aryl)Re(CO)(OAc)] (aryl = C6F5, Mes) complexes and their analogues.
Previous studies on rhenium(III) acetate complexes helped us to improve and design a better and more efficient cationic DAAm rhenium(III) bisacetonitrile catalyst for the hydrosilylation reaction of aldehydes (Chapter 2). The mechanism for this reaction is explored computationally utilizing Density Functional Theory (DFT) and is presented in Chapter 3. The reactivity of DAAm-C6F5 rhenium(III) acetate complex with Lewis acids is investigated in Chapter
Re N L N R CO R R Re L L L X R
Oxo (O2-) Nitrido (N3-)
R
R
Alkyl (-R) Aryl (-Ph) Hydride (-H)
-Mes, -C6F5
-Dipp, -Dbhp, -iPr
Tridentate Ligand mono or dianionic chelate
(DAAm, DAP, SSS, PNP)
Alkyl (-R) Aryl (-Ph) Halogens (-X) Re(V)
Re(III)
-Mes, -C6F5
Tridentate Ligand mono or dianionic chelate
1.3 References
(1) Hartwig, J. F. Organotransition Metal Chemistry; University Science Books: Mill Valley, CA, 2010.
(2) Crabtree, R. H. The Organometallic Chemistry of Transition Metals; John Wiley & Sons, Inc.: Hoboken, NJ, 2009.
(3) Lancaster, M., Green Chemistry: An Introductory Text. The Royal Society of Chemistry: Cambridge, UK, 2002.
(4) Millensifer, T.; Sinclair, D.; Jonasson, I.; Lipmann, A. Rhenium. In Critical Metals Handbook; Gunn, G., Ed.; Wiley: New York, 2014; p 340.
(5) Feng, Y.; Aponte, J.; Houseworth, P. J.; Boyle, P. D.; Ison, E. A. Inorg. Chem., 2009, 48
(23), 11058-11066.
(6) Smeltz, J. L.; Boyle, P. D.; Ison, E. A. J. Am. Chem. Soc., 2011, 133, 13288-13291. (7) Lilly, C. P; Boyle, P. D.; Ison, E. A. Dalton Trans., 2011, 40, 11815-11821.
(8) Lilly, C. P; Boyle, P. D.; Ison, E. A. Organometallics, 2012, 31, 4295-4301. (9) Smeltz, J. L.; Boyle, P. D.; Ison, E. A. Organometallics. 2012, 31, 5994–5997.
(10)Smeltz, J. L.; Lilly, C. P.; Boyle, P. D.; Ison, E. A. J. Am. Chem. Soc. 2013, 135, 9433-9441.
(11)Robbins, L. K.; Lilly, C. P.; Smeltz, J. L.; Boyle, P. D.; Ison, E. A. Organometallics
2015, 34, 3152-3158.
(12)Lambic, N. S.; Sommer, R. D.; Ison, E. A. J. Am. Chem. Soc., 2016, 138, 4832-42 (13)Lambic, N. S.; Lilly, C. P.; Robbins, L. K.; Sommer, R. D.; Ison, E. A.
Organometallics 2016, 35, 2822-2829.
(14)Lambic, N. S.; Lilly, C. P.; Sommer, R. D.; Ison, E. A. Organometallics 2016, 35(17), 3060-3068.
(15)Pérez, D. E.; Smeltz, J. L.; Sommer, R. D.; Boyle, P. D.; Ison, E. A. Dalton Trans. 2017, 46(14), 4609-4616.
(16)Lambic, N. S.; Sommer, R. D.; Ison, E. A. ACS Catalysis 2017, 7, 1170-1180. (17)Lambic, N. S.; Brown, C. A.; Sommer, R. D.; Ison, E. A. Organometallics 2017, 36,
2042-2051.
(18)Lambic, N. S.; Sommer, R. D.; Ison, E.A. Dalton Trans. 2018, 47, 758-768.
Gangopadhyay, J.; Sengupta, S.; Bhattacharyya, S.; Chakraborty, I.; Chakravorty, A.
Inorg. Chem. 2002, 41, 2616-2622; (d) Koshino, N.; Espenson, J. H. Inorg. Chem. 2003,
42, 5735-5742; (e) Du, G.; Abu-Omar, M. M. Curr. Org. Chem. 2008, 12, 1185-1198; (f) Ison, E. A.; Cessarich, J. E.; Du, G.; Fanwick, P. E.; Abu-Omar, M. M. Inorg. Chem. 2006,
45, 2385-2387; (g) Du, G.; Fanwick, P. E.; Abu-Omar, M. M. Inorg. Chim. Acta 2008,
361, 3184-3192; (h) Sousa, S. C.; Cabrita, I.; Fernandes, A. C. Chem. Soc. Rev. 2012, 41, 5641-5653; (i) Ahmad, I.; Chapman, G.; Nicholas, K. M. Organometallics 2011, 30, 2810-2818; (j) Wang, Y.; Espenson, J. H. Org. Lett. 2000, 2, 3525-3526; (k) Schröckeneder, A.; Traar, P.; Raber, G.; Baumgartner, J.; Belaj, F.; Mösch-Zanetti, N. C. Inorg. Chem. 2009,
48, 11608-11614; (l) Kirillov, A. M.; Haukka, M.; Kirillova, M. V.; Pombeiro, A. J. L.
Adv. Synth. Catal. 2005, 347, 1435-1446; (m) Abu-Omar, M. M.; Appelman, E. H.; Espenson, J. H. Inorg. Chem. 1996, 35, 7751-7757; (n) Abu-Omar, M. M.; Espenson, J. H.
Organometallics 1996, 15, 3543-3549; (o) Abu-Omar, M. M.; Khan, S. I. Inorg. Chem.
Chapter 2: Cationic Rhenium(III) Complexes: Synthesis, Characterization,
and Reactivity for Catalytic Hydrosilylation Reaction of Aldehydes
Portions of this chapter were published in:
Pérez, D. E.; Smeltz, J. L.; Sommer, R. D.; Boyle, P. D.; Ison, E. A.
Dalton Trans.
,
2017
,
46
, 4609-4616
2.1 Abstract
The catalytic efficiency for the hydrosilylation of benzaldehyde with organosilanes with the cationic rhenium(III) complex [(DAAm-aryl)Re(CO)(NCCH3)2][BF4] (DAAm = N,N-bis(2-arylaminoethyl)methylamine; aryl = C6F5) (6a) has been demonstrated. In addition, the catalytic competency of the complexes [(DAAm-aryl)Re(CO)(NCCH3)2][BF4] (DAAm = bis(2-arylaminoethyl)methylamine; aryl = Mes) (6b) and (DAmA-aryl)Re(CO)(Cl)2 (DAmA = N,N-bis(2-arylamineethyl)methylamino; aryl = C6F5) (8) were compared with 6a. Data suggest that electron-withdrawing substituents at the diamidoamine ligand increases the catalytic reactivity of the complexes. Various aliphatic and aromatic aldehydes were tested. Excellent yields were achieved at ambient temperature in neat conditions using dimethylphenylsilane. The reaction affords TONs of up to 9,200 and a TOF of up to 126 h-1. Kinetic and mechanistic studies were performed and possible reaction mechanisms for the hydrosilylation of benzaldehyde are proposed.
2.2 Introduction
Scheme 2.1. Catalytic comparison between neutral rhenium(V) complex and cationic rhenium(V) complexes for hydrosilylation reaction by Abu Omar group.
However, recently our group reported a rhenium(III) complex that is significantly more active that its rhenium mono-oxo precursors. The results show that the rhenium(III) complex 3 is more reactive towards the hydrosilylation of benzaldehydes than rhenium(V) complexes 1 and 2 (Scheme 2.2) under similar reaction conditions.5 We hypothesized that when oxorhenium complexes are employed as catalysts, the active species are not high-valent rhenium intermediates, but species that are generated upon deoxygenation with the organosilane. Just a few examples of low-valent rhenium complexes not bearing an oxo ligand have been shown to be efficient catalysts for hydrosilylation reaction of carbonyl compounds.6,7
5 mol% [Re] 1.5 equiv HSiEt3 ,CD2Cl2, rt O H Re N O N O L O B(C6F5)4
L = H2O or CH3CN Re O L N N O O Re O O N N O O Re O N N O O
n = 0 or 1 n n
B(C6F5)4
NO REACTION 100% conversion
Scheme 2.2. Catalytic competency of DAAm rhenium(III) complex versus rhenium(V) complexes in hydrosilylation reactions by Ison group.
In this chapter we report the synthesis and catalytic competency of low-valent cationic DAAm rhenium(III) complexes for the hydrosilylation reaction of aldehydes. We demonstrate that novel cationic DAAm rhenium(III) complexes are more efficient hydrosilylation catalysts than the neutral DAAm rhenium(III) and DAAm rhenium(V) complexes previously reported in the literature by our group. We examine how changing the electronics and sterics of the aryl substituents on the diamidoamine nitrogens affects the catalytic reactivity of the complex. Also, in this chapter we reported the synthesis of a novel diamineamido (DAmA) ligand and investigated how the hybridization in one of the nitrido in the ligand framework affect the reactivity of the complex towards hydrosilylation.
2.3 Results and Discussion
2.3.1 Synthesis and Characterization of Cationic DAAm Rhenium(III) Complexes and Cationic DAmA Rhenium(III) Complex
The synthesis of cationic DAAm rhenium(III) complexes by the reaction of 3 with acids bearing non-coordinating anions in acetonitrile was investigated. A series of cationic [(DAAm-aryl)Re(CO)(NCCH3)2][X] (aryl = C6F5, Mes; X = OTf, BArF, BF4, PF6) were synthetized from the novel DAAm rhenium(III) complexes 3a and 3b (Scheme 2.3). The corresponding DAAm rhenium(III) acetate complex was treated with 1 equiv of the non-coordinating acid at ambient
H O
1.2 equiv HSiMe2Ph, neat, 80 °C 0.1 mol% [Re]
H OSiMe2Ph
H
Re O N
N N C6F5
C6F5
CH3
Re O N
N N C6F5
C6F5
O N Re
N N C6F5 CO
C6F5
OAc
1 3 d, 94 %
2 2 d, 98 %
temperature. A color change from brown (3a) to yellow, or dark red (3b) to light orange, was observed immediately after the addition of acetonitrile to the reaction mixture, which was indicative of the cationic rhenium(III) bisacetonitrile complex formation.
Scheme 2.3. Synthesis of novel cationic rhenium(III) bisacetronitrile derivatives
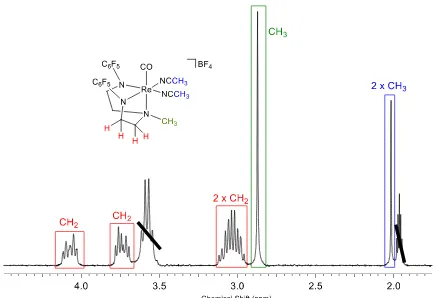
The representative 1H NMR spectrum of 6a is shown in Figure 2.1. The methylene protons from the ligand backbone resonate as 3 distinct multiplets at δ 4.08 (2H), δ 3.73 (2H) and δ 3.04 (4H) ppm. A singlet at δ 2.89 ppm (3H) is representative of the methyl group on the amine from the ligand backbone. The singlet corresponding to the methyl groups from the acetonitrile ligands resonate at δ 2.00 ppm (6H).
1 equiv HX Re
N N
N
R CO
R
OAc
CH3CN, rt, 5 min
Re N
N N
R CO
R NCCH3
NCCH3 X
(3a) R = C6F5 (3b) R = Mes
(4a) R = C6F5, X = OTf; (4b) R = Mes, X = OTf (5a) R = C6F5, X = BArF4; (5b) R = Mes, X = BArF4
(6a) R = C6F5, X = BF4; (6b) R = Mes, X = BF4 (7a) R = C6F5, X = PF6; (7b) R = Mes, X = PF6
Figure 2.1. 1H NMR spectrum of cationic rhenium(III) bisacetronitrile complex 6a at room temperature in acetonitrile-d3.
Figure 2.2. FTIR spectrum (KBr Pellets cm-1) of [(DAAm)Re(CO)(NCCH3)2][BF4] (aryl = C6F5) (6a).
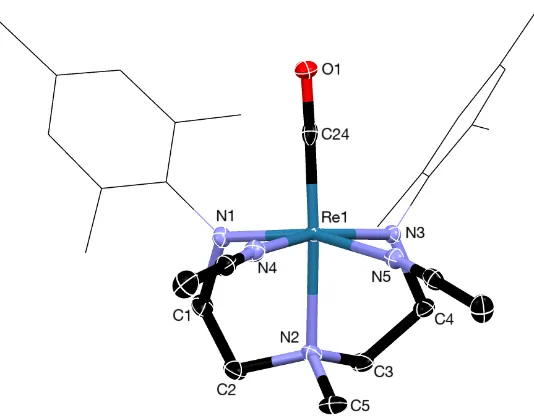
Figure 2.3. X-ray structure of 5a. Thermal ellipsoids are at 50%. H atoms and counter ion have been omitted and the pentafluorophenyl groups on the ligand are shown in wireframe format for clarity. Selected bond lengths (Å) and angles (deg): C1, 1.867(4); N1, 1.932(3); Re1-N2, 2.220(3); Re1-N3, 1.928(1); Re1-N4, 2.167(3); Re1-N5, 2.120(2); N1-Re1-N3, 105.04(15); N1-Re1-N2, 80.79(13); N1-Re1-C1, 94.26(16); N2-Re1-C1, 172.91(16); N1-Re1-N4, 85.09(13); N1-Re1-N5, 160.96(15); Re1-N1-C8, 119.2(2); Re1-N3-C12, 116.9(3).
The synthesis of 4a-b, 5a-b, 6a-b, and 7a-b provided a diverse set of cationic rhenium(III) complexes bearing either electron withdrawing (R = C6F5) or electron donating (R = Mes) analogues of the DAAm ligand framework. Some of the benefits of this series of cationic rhenium(III) complexes include: (1) the rhenium(III) precursors are air and moisture stable; (2) the synthesis is facile; (3) different non-coordinating acids can be used; and (4) a direct comparison of the reactivity could be made based on the electronics of the ligand framework.
From previous reports, we hypothesized that in order to design a highly active rhenium catalyst for hydrosilylation reactions, the metal center must have an oxidation state number of three combined with a cationic charge. The main two characteristics necessary for a more efficient catalyst are present in these novel cationic DAAm rhenium(III) complexes. We proposed that cationic DAAm rhenium(III) complexes will be more efficient hydrosilylation catalysts than its precursors and the cationic rhenium(V) complex reported by Abu-Omar.2-4
The DAAm analogue, diamineamido (DAmA) ligand was also synthetized. Complex 3a was treated with an excess of hydrochloric acid (HCl) in methylene chloride at room temperature (Scheme 2.4). X-ray crystallography of the green crystals confirmed the formation of 8 in solution (Figure 2.5). The geometry about rhenium in 8 is best described as a distorted octahedral. The acetate ligand is protonated and replaced by two chloride ligands in the equatorial plane of the complex. X-ray studies also provided evidence for the protonation of one amide nitrogen and a change in hybridization at nitrogen from sp2 to sp3. Evidence for this protonation and the change in hybridization from sp2 to sp3 is provided by the analysis of the angles around N1 and N3 in 8. A comparison between 3a and 8 reveals a decrease in the Re-N1-C8 angle from 118° to 108°. Also, in 8 the Re-N1 bond length (2.2 Å) is longer than Re-N3 bond length (1.92 Å).
Scheme 2.4. Synthesis of DAmA rhenium(III) dichloride complex 8.
Re N
N N
C6F5 CO
C6F5
OAc excess HCl N Re
N N
C6F5 CO
C6F5 Cl
Cl H
CH2Cl2, 24 h
Figure 2.5. Thermal ellipsoid plot of 8. Thermal ellipsoids are at 50%. H atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C1, 1.862(3); N1, 2.200(2); Re1-N2, 2.262(2); Re1-N3, 1.920(2); Re1-Cl1, 2.4361(6); Re1-Cl2, 2.3685(6); N1-Re1-N3, 93.97(8); Re1-N2, 78.18(7); Re1-C1, 98.15(9); N2-Re1-C1, 174.44(9); Re1-Cl1, 80.01(6); N1-Re1- Cl2, 163.79(5); N1-Re1-N1-C8, 107.95(14); N1-Re1-N3-C12, 120.44(16).
Figure 2.6. 1H NMR of DAmA rhenium(III) dichloride complex (8) in acetonitrile-d3.
2.3.2 DAAm Rhenium(III) Complexes for the Catalytic Hydrosilylation of Aldehydes
The catalytic competency of complexes 6a, 6b, and 8 for the hydrosilylation of benzaldehyde with dimethylphenylsilane were examined according to Equation 1. Reactions were conducted at different temperatures and catalyst loadings in acetonitrile. The results are summarized in Table 2.1.
Re N
N N C6F5 CO
CH3 C6F5 Cl
Cl H
H
H H H
CH2 CH
2
Table 2.1. Catalytic hydrosilylation of benzaldehyde according to equation 1.
Entrya [Re] mol% T (°C) time (h) %Conversionb TON
1c 8 1 80 95 79 79
2d 6a 0.1 rt 165 95 1125
3d 6a 0.1 40 118 41 513
4d 6a 0.1 60 48 46 575
5d 6a 0.1 80 48 49 613
6e 6a 0.5 rt 8 95 190
7d 6b 0.1 rt 24 NR -
8d 6b 0.1 40 34 NR -
aGeneral Reaction Conditions: Under a N2 atmosphere, 2 mg of catalyst, 1.2 equiv silane, and 0.5 equiv 2-bromomesitylene (internal standard) in 0.35 mL CH3CN. bCalculated by 1H NMR spectroscopy of the crude reaction against 2-bromomesitylene as the internal standard. cRe (0.003 mmol), and aldehyde (0.3 mmol). dRe (0.003 mmol), and aldehyde (3 mmol). eRe (0.003 mmol), and aldehyde (0.6 mmol). TON = turnover number. NR = no reaction. rt = room temperature.
Complex 6a is the most efficient catalyst in the series, 95% conversion was attained at room temperature with 0.1 mol% catalyst loading achieving a turnover number (TON) of 1125 (Entry 2). Higher temperatures did not improve the catalytic competency of 6a (Entries 3-5). The low percent conversion at high temperatures is attributable to catalyst decomposition. Complex 8 is not as active as 6a (Entry 1) and the reaction must be heated due to the poor solubility of the complex in the solvent. This suggests that changing the hybridization in one of the amido nitrogens in the ligand framework has a notable effect on the reactivity of the complex. Hydrosilylation was not observed when 6b was utilized as catalyst (Entries 7 and 8). The results demonstrate that the electronics of the diamidoamine ligand dramatically affects the reactivity of the complex. We hypothesized that electron-donating groups decrease the electrophilicity and stabilize the metal
H O
1.2 equiv HSiMe2Ph, CH3CN, T x mol% [Re]
H
OSiMe2Ph
center making it less susceptible to react and activate the silane reagent.
The effect of various silane substrates in the catalytic hydrosilylation of benzaldehyde was investigated according to Equation 2. Reactions were conducted at room temperature in acetonitrile. The results are summarized in Table 2.2.
Table 2.2. Catalytic hydrosilylation of benzaldehyde with various silane substrates.
Entrya Silane mol% time (h) %Conversionb
1c HSiMe2Ph 0.1 165 95
2c HSiPh3 0.1 114 NR
3d HSiEt3 0.5 48 51
4d HSiPh3 0.5 46 12
aGeneral Reaction Conditions: Under a N2 atmosphere, 2 mg of catalyst, and 0.5 equiv 2-bromomesitylene (internal standard) in 0.35 mL CH3CN. bCalculated by 1H NMR spectroscopy of the crude reaction against 2-bromomesitylene as the internal standard. cRe (0.003 mmol), and aldehyde (3 mmol). dRe (0.0024 mmol), and aldehyde (0.554 mmol). NR = no reaction.
Data in Table 2.2 suggest that the catalytic hydrosilylation of benzaldehyde with 6a is more efficient with dimethylphenylsilane (Entry 1). Hydrosilylation was not observed when HSiPh3 was utilized as the substrate (Entry 2), suggesting that steric bulk present in the silane inhibited the activation of the substrate by 6a. Similar results using HSiPh3 were observed previously by our group.5 Increasing the catalyst loading in the presences of HSiPh3 does not have any notable improvement in the conversion of benzaldehyde to the silyl ether (Entry 4). The reaction in the presence of HSiEt3 just affords a 51% conversion (Entry 3). Even when the reaction was stirred for more than 48 h, no change in conversion was observed. The catalytic reactivity in the presences of HSiEt3 is slower and less efficient in comparison to the reactivity using HSiMe2Ph as the substrate. In previous reports by our group, comparison between HSiEt3 and HSiMe2Ph, had a similar trend.5
H O
1.2 equiv HSiR3, CH3CN, rt x mol% 6a
H OSiR3
The catalytic reactivity for the hydrosilylation of benzaldehyde with 6a was investigated and optimized under neat conditions according to Equation 3. Reactions were conducted at room temperature with varying catalyst loadings. The results are summarized in Table 2.3.
Table 2.3. Catalytic hydrosilylation of benzaldehyde according to equation 3.
Entrya mol% time (h) % Conversionb TON
1c 0.01 72 92 8,586
2d 0.03 24 100 3,000
3e 0.05 16 100 2326
4f 0.10 9 100 1163
5g 0.50 2 100 233
6h 1 1 100 115
aGeneral Reaction Conditions: 2 mg [Re], 1.2 equiv silane, under a N2 atmosphere, 0.5 equiv 2-bromomesitylene (internal standard), and under neat conditions. bCalculated by 1H NMR spectroscopy of the crude reaction against 2-bromomesitylene as the internal standard. cAldehyde (28 mmol) and Re (0.003 mmol). dAldehyde (9 mmol) and Re (0.003 mmol). eAldehyde (6 mmol) and Re (0.003 mmol). fAldehyde (3 mmol) and Re (0.003 mmol).gAldehyde (0.6 mmol) and Re (0.003 mmol). hAldehyde (0.7 mmol) and Re (0.007 mmol).
The catalytic efficiency of 6a drastically improved when the reaction was performed in dimethylphenylsilane as the solvent. Complex 6a quantitatively converts benzaldehyde to the silyl ether product in less than 24 h using low catalyst loadings at room temperature (Entries 2-6). Also, 6a is able to afford TONs of up to 8,586 (Entry 1) using 0.01 mol% catalyst loading without any additional solvent at room temperature. In contrast, the catalytic hydrosilylation reaction using complex 3a must be heated to afford comparable results. At room temperature 6a (0.03 mol%) is a more efficient hydrosilylation catalyst than its precursor 3a (0.01 mol%).5
H O
1.2 equiv HSiMe2Ph, neat, rt
x mol% 6a
H OSiMe2Ph
The effect of various silane substrates on the catalytic hydrosilylation of benzaldehyde was investigated according to Equation 4. The reaction was carried at room temperature under neat conditions at the same catalyst loading. The experimental results are summarized in Table 2.4.
Table 2.4. Catalytic hydrosilylation of benzaldehyde with various silane substrates under neat conditions.
Entrya Silane time (h) %Conversionb
1 HSiMe2Ph 24 100
2 HSi(OEt)3 18 100
3 HSiMePh2 240 71
4 HSi(Et)3 240 30
aGeneral Reaction Conditions: Re (0.003 mmol), and benzaldehyde (9 mmol); under a N2 atmosphere and 0.5 equiv 2-bromomesitylene (internal standard). bCalculated by 1H NMR spectroscopy of the crude reaction against 2-bromomesitylene as the internal standard.
Catalyst 6a performed efficiently for the hydrosilylation of benzaldehyde with HSiMe2Ph (Entry 1) and HSiOEt3 (Entry 2). The reaction with HSiMePh2 (Entry 3) was slower than the reactions using HSiMe2Ph (24 h) or HSiOEt3 (18h), and 71% conversion was observed over 240 h. We attribute this to steric effects of the two benzene rings in the substrate. Our group observed similar effects when experimental results for the reaction using HSiMePh2 were compared to HSiMe2Ph.5 Under neat conditions, hydrosilylation of benzaldehyde with HSiEt3 is slow and afforded low conversions (Entry 4). The different reactivity of HSiOEt3 (Entry 2) and HSiEt3 (Entry 4) is notable. This suggests that a more electron-donating silane is favorable for the hydrosilylation reaction.
We chose 0.03 mol% catalyst loading (Table 2.3, Entry 2) as the optimized conditions to investigate the catalytic reactivity of 6a towards different aldehyde substrates. The reaction was performed according to Equation 5. The results are summarized in Scheme 2.5. The results suggest that catalyst 6a performed efficient hydrosilylation with a variety of aldehydes bearing different steric and electronic effects.
H O
1.2 equiv HSiR3, neat, rt 0.03 mol% 6a
H OSiR3
Scheme 2.5. Substrate scope using the optimized conditions according to equation 5.
The optimized reaction conditions had to be adapted based on the solubility of the aldehydes in dimethylphenylsilane. In general, most liquid aldehydes were treated under neat conditions and most solid aldehydes were dissolved in a minimum amount of acetonitrile. The percent conversion was determined by 1H NMR spectroscopy of the crude reaction mixture using 2-bromomesitylene as the internal standard.
Hence, these results demonstrate that the cationic DAAm rhenium(III) complex 6a is a more effective hydrosilylation catalyst than the neutral DAAm rhenium(III) complex 3a and 3a DAAm rhenium(V) 1 and 2 precursors. Evidenced suggested that cationic DAAm rhenium(III) complex 6a even surpasses previous cationic rhenium(V) catalysts reported by the Abu-Omar2-4 group. Some of the specific benefits of this particular catalytic system include: (1) The reaction can be performed with low catalysts loading; (2) The reaction does not require high temperatures; (3) The reaction can be performed under neat conditions; (4) The catalyst precursor is air and moisture stable; and (5) The catalyst effectively performed hydrosilylation with variety aldehydes.
R H
O
1.2 equiv HSiMe2Ph, neat, rt, time 0.03 mol% 6a
R H
OSiMe2Ph
H (5) O H O H Cl O H O H O H O
H3CO
H O OCH3 O H O H O
(H3C)2N
H O Cl H O H
18 h, 100% 24 h, 100%
24 h, 100% 24 h, 100%
24 h, 92% 24 h, 100% 24 h, 65% 24 h, 100%
2.3.3 Kinetics Studies
Several kinetic experiments were performed in order to determine the dependences of each reagent involved in this reaction. The time profile for the hydrosilylation of benzaldehyde with dimethylphenysilane using catalyst 6a was investigated under pseudo-first order conditions (Figure 2.7). Product formation using 6a at different catalyst loadings (0.279 mM, 0.01 mol%; 0.829 mM, 0.03 mol%; 1.385 mM, 0.5 mol%; 2.770 mM, 1 mol%) was monitored with dimethylphenylsilane (3.324 M) and benzaldehyde (2.770 M). The reaction was performed under neat conditions at room temperature. The percent conversion of benzaldehyde was calculated by 1H NMR spectroscopy with 2-bromomesitylene as the internal standard.
The overall reaction dependence was obtained by monitoring product formation over time with 6a as the catalyst, and benzaldehyde and dimethylphenylsilane in approximately a 1:1 ratio. As shown in Figure 2.7, the reaction follows clean pseudo-first order kinetics with observed rate constants (kobs) of 6.2 x 10-6 s-1, 2.3 x 10-5 s-1, 4.31 x 10-5 s-1, and 1.0 x 10-4 s-1. The results suggest a rate law that is first-order overall.
Rate = k[cat]x[Benzaldehyde]y[HSiMe 2Ph]z
𝐔𝐧𝐝𝐞𝐫 𝐒𝐭𝐞𝐚𝐝𝐲 𝐒𝐭𝐚𝐭𝐞 𝐂𝐨𝐧𝐝𝐢𝐭𝐢𝐨𝐧𝐬 Rate = kobs[Benzaldehyde]y[HSiMe
2Ph]z
where kobs = k[cat]x
From the kinetic plot the reaction is1st order overall; i.e. Either y=1 and z=0 or y=0 and z=1
The order with respect to rhenium was determined by plotting the observed rate constants (kobs) at varying concentration of rhenium. As shown in Figure 2.8 a linear fit was obtained which suggests a first order dependence on [Re] i.e term x in the kobs expression above is 1.
Figure 2.8. Rate law dependence on rhenium concentration. Plot kobs (s-1) versus Rhenium concentration (mM). Reaction conditions: [benzaldehyde] = 2.770 M; [silane] = 3.324 M; [2-bromomesitylene] = 1.385 M; [6a] = 0.279 mM (0.01 mol%), 0.829 mM (0.03 mol%), 1.385 mM (0.5 mol%), 2.770 (1 mol%). Reactions were performed in a vial under stirring at room temperature in a N2 atmosphere. At a fixed time, a representative aliquot from the reaction mixture was dissolved in CDCl3. The percent conversion was determined by 1H NMR spectroscopy by integrating the product peak against the internal standard.
The rate law obtained from the experiments in Figure 2.7 is first order overall. This means that the rate exhibits zero order dependences with respect to one of the substrates (benzaldehyde or silane) and a first order dependence with respect to the other substrate. The order with respect to benzaldehyde concentration was determined by the method of flooding. The decay in [benzaldehyde] (0.713 M) with 6a (0.713 mM) as the catalyst was monitored in an excess of dimethylphenylsilane (5.71 M, solvent). The reaction mixture was divided in 10 vials and each one was stirred at room temperature. At a fixed period of time benzaldehyde decay was measured by GC-MS, utilizing 2-bromomesitylene as the internal standard. A linear decay over time was observed which suggests a pseudo-zeroth order dependence with respect to [benzaldehyde] (Figure 2.9).
0 4 10-5 8 10-5 1.2 10-4
0 1 2 3
k obs
/ s
-1
[Re] / mM rate = 3.8(1) x 10-5 mM-1s-1
R2 = 0.9964 Plot: k
Figure 2.9. Kinetic plot of the concentration of benzaldehyde vs reaction time (s). Reaction conditions: [Rhenium] = 0.921 mM; [benzaldehyde] = 0.921 M; [silane] = 5.48 M. Benzaldehyde concentration was determined by GC-MS by integrating the benzaldehyde peak versus the internal standard, 2-bromomesitylene (0.438 M). The reaction was performed under neat conditions at room temperature in 10 different 4 mL screw cap vials for a fixed period of time.
Given that the overall reaction is first order overall (Figure 2.7), a zeroth order dependence on benzaldehyde (Figure 2.9), suggests that the reaction is first order with respect to dimethylphenylsilane i.e. in the equation above, y = 0 and z = 1. Thus, the rate equation for the catalytic reaction is:
Rate= kobs[HSiMe2Ph]
where kobs= k[cat]1
d[PhCH2OSiMe2Ph]
dt = k[cat][HSiMe2Ph]
0.2 0.6 1
0 10000 20000 30000
Plot: Benzaldehyde Decar Over Time
[Be
nza
ld
eh
yd
e]
/
M
Time / s
Rate = -3.3(3) x 10-5 Ms-1
2.3.4 Reaction Mechanism 2.3.4.1 Mechanistic Studies
An investigation of the mechanism of the hydrosilylation of benzaldehyde with dimethylphenylsilane with 6a began with the examination of stoichiometric reactions between the cationic complex and the substrates. Attempts to identify and isolate any intermediates of substrate activation started with the stoichiometric reaction of 6a with dimethylphenylsilane in the absence of benzaldehyde (Scheme 2.6).
Scheme 2.6. Stoichiometric reaction of 6a and dimethylphenylsilane.
The reaction of 6a with dimethylphenylsilane described in Scheme 2.6 resulted in the formation of a DAAm dirhenium(II) complex (9). The formation of complex 9 has been reported and characterized by our group in the stoichiometric reaction of 3a with dimethylphenylsilane at 80 °C.5 This result suggests that dimethylphenylsilane is activated, in the absence of benzaldehyde, in a similar manner by 6a and 3a. The formation of 9 was confirmed by X-ray crystallography.
The catalytic activity of 9 in hydrosilylation reactions was reported by our group. When 9 was used as a catalyst benzaldehyde was converted to the silyl ether product in < 5% yield in 5 h.5 A mechanism for the formation of 9 from 3a was proposed (Scheme 2.7). In this mechanism, complex 3a activates dimethylphenylsilane to produce the rhenium(III) hydride complex 10. In the absence of benzaldehyde, two molecules of 10 react to produce 9 and dihydrogen.
Re N
N N
C6F5 CO
C6F5 NCCH3
NCCH3 BF4
Re CO
Re(DAAm)
CO HSiMe2Ph
rt
6a 9
Scheme 2.7. Proposed mechanism for the formation of 9 from 3a by Ison group.
Identification of complex 9 in both investigations, lead us to propose a similar pathway for the formation of 9 from 6a (Scheme 2.8).
Scheme 2.8. Proposed mechanism for the formation of 9 from 6a.
On the other hand, consistent with kinetic data, no reactivity was observed in the stoichiometric reaction of 6a with benzaldehyde in the absence of dimethylphenylsilane (Scheme 2.9). The latter observation is consistent with studies reported by our group with 3a as the catalyst under similar reaction conditions.5
Re N
N N C6F5 CO
C6F5
OAc
3a
HSiMe2Ph
80 °C -Me2PhSiOAc
Re N
N N C6F5 CO
C6F5
H 10 (DAAm)Re Re(DAAm) CO CO 9 Re N N N C6F5 CO
C6F5 NCCH3
NCCH3 BF4 Re CO Re(DAAm) CO HSiMe2Ph
rt 6a 9 6a 10 Re N N N C6F5 CO
C6F5
H HSiMe2Ph
-BF4SiR3 -2 NCCH3
Re N
N N C6F5 CO
C6F5 NCCH3
NCCH3 BF4 rt Re N N N C6F5 CO
C6F5
Scheme 2.9. Stoichiometric reaction of 6a and benzaldehyde.
We also examined the effect on the catalytic reaction of changing the electronics of the aldehyde substrate. Competition experiments between benzaldehyde and the corresponding para -substituted benzaldehyde were performed according to Scheme 2.10. 1H NMR spectroscopy was used to calculated product formation after 1 d. From the Hammett plot (Figure 2.10), it was observed that electron-withdrawing substituents accelerate the product forming step while electron-donating substituents retard it. This is indicated by the positive ρ value (ρ = 0.36). The last observation is consistent with previous reports by Ison group (ρ = 0.52).5
Scheme 2.10. Competition experiments between para-substituted benzaldehyde.
6a
Re N
N N C6F5 CO
C6F5 NCCH3
NCCH3
BF4
H O
+ No Reaction
rt, 1 d
H O
0.5 equiv HSiMe2Ph, CH3CN, 1 d
0.5 mol% [6a]
H
OSiMe2Ph
H
H O
X
H
OSiMe2Ph
H
X X = OCH3, Cl, CF3, NO2
Figure 2.10. Hammett plot for the competition experiments between para-substituted benzaldehyde. Reaction was carried at room temperature in CD3CN. Reaction conditions: [Re] = 0.00241 mmol, [benzaldehyde] = 0.554 mmol, [p-benzaldehyde] = 0.554 mmol, [dimethylphenylsilane] = 0.277 mmol and [2-bromomesitylene] = 0.274 mmol. The 1H NMR integration against the internal standard (2-bromomesitylene) of the methylene protons of the
para-substituted silyl ether and the benzyloxydimethylphenylsilane were used to determine the product ratio value (PH/PX).
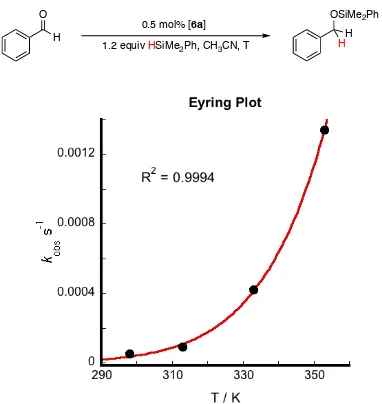
Variable temperature kinetic experiments were performed to investigate the activation parameters in a temperature range from 25 to 80 °C (Scheme 2.11). The enthalpy of activation (ΔH‡) was found to be 13.1(4) kcal/mol. The entropy of activation (ΔS‡) was found to be -34(1) cal/mol·K (Figure 2.11). The free energy of activation (ΔG‡) was calculated using the experimental activation parameters obtained as 23.5(4) kcal/mol.
0 0.2 0.4
-0.4 0 0.4 0.8
log
(P
x/P
H)
R
2= 0.9252
Hammet Plot
p-OCH
3
p-H
p-Cl
p-CF
3
p-NO
2
Scheme 2.11. Variable temperature kinetic experiments.
Figure 2.11. Eyring plot for the hydrosilylation of benzaldehyde with 6a. The rate of hydrosilylation was determined at different temperatures (25 - 80 °C). Reactions were performed in acetonitrile-d3.
2.3.4.2 Proposed Mechanism
The following mechanism has been proposed for the catalytic hydrosilylation of benzaldehyde by 6a based on kinetic data, the Hammett correlation, and mechanistic observations (Scheme 2.12).
H O
1.2 equiv HSiMe2Ph, CH3CN, T 0.5 mol% [6a]
H
OSiMe2Ph
Scheme 2.12. Proposed mechanism for the hydrosilylation reaction of aldehydes catalyzed by 6a.
Kinetic data suggest that the turnover-limiting step is the activation of the silane as no dependence was observed on [benzaldehyde] (Figure 2.9) and a first order dependence on [silane] was observed. Activation of the silane could occur via ƞ1 or ƞ2 coordination. Schubert previously discussed a possible reaction mechanism for the activation of a Si-H bond by an unsaturated metal center that can involve the generation of a M-H-Si bond as key intermediate. In the mechanism, coordination of the hydride to the metal center can lead to the formation of a ƞ1(H)-silane ligand. Then the coordination mode can change to a ƞ2-coordination which possess a stronger metal-silicon interaction compared to a ƞ1 coordination.11 Abu-Omar2,3 and Chan12 proposed activation of silane via ƞ2 coordination in their mechanistic studies for catalytic hydrosilylation. Attempts to isolate either a ƞ1 or a
ƞ2 coordination intermediate were unsuccessful. Theoretical DFT studies were conducted to propose a silane activation pathway and will be discuss in Chapter 3.
Re CO NCCH3 NCCH3 (DAAm) H OSiR3 H H OSiR3 Re CO NCCH3 (DAAm) NCCH3
HSiR3
Re CO SiR3 H (DAAm) NCCH3 Re CO H (DAAm) H O + 10 Re CO H
(DAAm) SiR3
NCCH3
or
RDS: Silane Activation
Rate = k[Re][HSiMe2Ph]
SN2-Si
In recent years, an ionic hydrosilylation mechanism has been proposed by Oestreich13,14 and computationally studied by Wei15-21 for catalysis by oxorhenium and oxomolybdenum complexes. In this mechanism aldehyde does not coordinate to the metal center; instead, silane is activated by coordination to rhenium. Our experimental data is consistent with these findings. The activation is followed by nucleophilic SN2-Si attack of the carbonyl substrate at the activated silicon leading to the cleavage of the Si-H bond and formation of an ion pair. Lastly, product formation results from hydride transfer from complex 10 to the silylcarbonium ion. Hydride transfer should be accelerated when electron-withdrawing groups are utilized in the para position of benzaldehyde. This is consistent with the Hammett data (Figure 2.10).
2.4 Conclusions
2.5 Experimental Section
General Considerations. 3a-b5 and [(3,5-(CF3)2C6H3)4B]-[H(OEt2)2]+ (HBArF4)22 were prepared according to previous procedures. All reactions were carried out in a nitrogen filled glove box unless otherwise noted. All reagents were purchased from commercial sources, placed in a nitrogen filled glove box and used as received without further purification. 1H, 13C and 19F spectra were acquired on a Varian Mercury 700 MHz, Varian Mercury 400 MHz or Varian Mercury 300 MHz spectrometer. NMR chemical shifts are listed in parts per million (ppm) and are referenced to residual protons or carbons of the deuterated solvents, respectively at room temperature unless otherwise noted. The FTIR spectra were obtained in KBr thin films on a JASCO FT/IR-4100 instrument. Gas Chromatography was performed on an Agilent 7820A GC/FIC using HP-5MS columns. Elemental analyses were performed by Atlantic Micro Laboratories Inc. X-ray crystallography was performed at the X-ray Structural Facility at North Carolina State University by Dr. Roger D. Sommer and Paul D. Boyle.
General Synthesis [(DAAm-aryl)Re(CO)(NCCH3)2][X] (aryl = C6F5, Mes) (X = OTf, BArF
4, BF4, PF6); 4a-b, 5a-b, 6a-b, 7a-b.
In a nitrogen filled glove box, the corresponding [(DAAm-aryl)Re(CO)(OAc)] (aryl = C6F5 (3a), Mes (3b)) (3a = 50 mg, 0.07 mmol; 3b = 50 mg, 0.08 mmol) was added to a screw cap vial and dissolved in 0.35 mL of acetonitrile. Then, the corresponding acid HX (X = HOTf, HBArF4, HBF4·OEt2, HPF6·H2O; 1 equiv) was added to the solution. The solvent was removed under pressure. The resulting oil was dissolved in diethyl ether and the corresponding bisacetonitrile product was precipitated with excess of hexanes. The final powder was collected via vacuum filtration.
[(DAAm-C6F5)Re(CO)(NCCH3)2][OTf], 4a. Following the general synthesis, complex
4a was synthetized in quantitative yield. 1H NMR (300 MHz, CD3CN) δ: 4.05 (m, 2H), 3.71 (m, 2H), 3.01 (m, 4H), 2.85 (s, 3H), 1.96 (s, 6H). 13C NMR (101 MHz, CD3CN) δ: 188.2, 172.9, 143.5, 142.7, 142.1, 141.3, 140.3, 139.0-138.2, 137.8, 137.5, 65.9, 59.1, 55.2. 19F NMR (376 MHz, CD3CN) δ: -79.4 (s, 3F), -149.8 (dd, J = 22.1, 5.7 Hz, 2F), -150.8 (dd,
[(DAAm-Mes)Re(CO)(NCCH3)2][OTf], 4b. Following the general synthesis, complex
4b was synthetized in quantitative yield. 1H NMR (400 MHz, CD3CN) δ: 6.87 (s, 2H), 6.76 (s, 2H), 4.03 (m, 2H), 3.56 (m, 2H), 3.07 (m, 2H), 2.91 (m, 5H), 2.33 (s, 6H), 2.25 (s, 6H), 1.96 (m, 6H), 1.84 (s, 6H). 13C NMR (126 MHz, CD3CN) δ: 193.4, 159.2, 135.3, 132.1, 131.6, 130.4, 129.7, 66.5, 60.2, 49.4, 20.4, 20.3, 20.0. 19F NMR (376 MHz, CD3CN) δ: -79.6 (s, 3F). IR (FTIR, cm-1): 𝑣(CO) 1890. Anal. Calc. for C29H39F3N5O4ReS·H2O: C, 43.71; N, 8.79; H, 4.93. Found: C, 42.50; N, 8.69; H, 5.00.
[(DAAm-C6F5)Re(CO)(NCCH3)2][BArF4], 5a. Following the general synthesis,
complex 5a was synthetized in quantitative yield. 1H NMR (300 MHz, CD3CN) δ: 7.69 (m, 12H), 4.03 (m, 2H), 3.72 (m, 2H), 2.99 (m, 4H), 2.84 (s, 3H), 1.96 (s, 6H). 13C NMR (175 MHz, CD3CN) δ: 188.1, 163.0, 162.7, 162.4, 162.1, 143.7, 142.9, 142.2, 141.4, 140.4, 139.2-138.6, 137.9, 137.6, 130.2, 130.0, 129.8, 129.6, 127.7, 126.2, 124.6, 123.1, 66.2, 59.3, 47.5. 19F NMR (376 MHz, CD3CN) δ: -63.25 (s, 24F), -149.8 (m, 2F), -150.8 (m, 2F), -162.9 (m, 2F), -165.8 (t, J = 20.7 Hz, 2F), -166.2 (m, 2F). IR (FTIR, cm-1): 𝑣(CO) 1925. Anal. Calc. for C54H29BF34N5ORe·H2O: C, 39.92; N, 4.31; H, 1.92. Found: C, 39.11; N, 4.80; H, 2.02.
[(DAAm-Mes)Re(CO)(NCCH3)2][BArF4], 5b. Following the general synthesis, complex
5b was synthetized in quantitative yield. 1H NMR (376 MHz, CD3CN) δ: 7.69 (m, 12H), 6.87 (s, 2H), 6.75 (s, 2H), 4.02 (m, 2H), 3.55 (m, 2H), 3.07 (m, 2H), 2.91 (m, 5H), 2.33 (s, 6H), 2.25 (s, 6H), 1.96 (m, 6H), 1.84 (s, 6H). 19F NMR (376 MHz, CD3CN) δ: -63.4 (s, 24F). IR (FTIR, cm-1): 𝑣(CO) 1890. The complex was not characterized by 13C NMR due to its very poor stability. Elemental Analysis was not attempted on this complex because of its instability.
[(DAAm-C6F5)Re(CO)(NCCH3)2][BF4], 6a. Following the general synthesis, complex
[(DAAm-Mes)Re(CO)(NCCH3)2][BF4], 6b. Following the general synthesis, complex
6b was synthetized in quantitative yield. 1H NMR (300 MHz, CD3CN) δ: 6.87 (s, 2H), 6.75 (s, 2H), 4.03 (m, 2H), 3.55 (m, 2H), 3.07 (m, 2H), 2.91 (m, 5H), 2.33 (s, 6H), 2.25 (s, 6H), 1.84 (d, 12H). 13C NMR (126 MHz, CD3CN) δ: 193.4, 159.3, 135.3, 132.1, 131.6, 130.4, 129.7, 66.5, 60.2, 49.42, 20.43, 20.30, 20.02. 19F NMR (376 MHz, CD3CN) δ: -151.3 (s, 4F). IR (FTIR, cm-1): 𝑣(CO) 1878. Anal. Calc. for C28H39BF4N5ORe·H2O: C, 44.68; N, 9.30; H, 5.49. Found: C, 43.80; N, 8.99; H, 5.38.
[(DAAm-C6F5)Re(CO)(NCCH3)2][PF6], 7a. Following the general synthesis, complex
7a was synthetized in quantitative yield. 1H NMR (300 MHz, CD3CN) δ: 4.07 (m, 2H), 3.74 (m, 2H), 3.05 (m, 4H), 2.87 (s, 3H), 1.99 (s, 6H). 13C NMR (101 MHz, CD3CN) δ: 188.1, 144.1, 143.3, 141.6, 140.8, 139.3, 138.5, 137.8, 136.9, 66.0, 59.2, 47.3. 19F NMR (376 MHz, CD3CN) δ: -72.0 (s, 3F), -73.9 (s, 3F), -150.0 (dd, J = 22.1, 5.6 Hz, 2F), -150.9 (dt, J = 16.1, 4.7 Hz, 2F), -163.0 (t, J = 20.9 Hz, 2F), -165.8 (t, J = 20.0 Hz, 2F), -166.2 (m, 2F). IR (FTIR, cm-1): 𝑣(CO) 1882. Anal. Calc. for C22H17F16N5OPRe: C, 29.74; N, 7.88; H, 1.93. Found: C, 29.62; N, 7.19; H, 2.03.
[(DAAm-Mes)Re(CO)(NCCH3)2][PF6], 7a. Following the general synthesis, complex
7b was synthetized in quantitative yield. 1H NMR (300 MHz, CD3CN, δ): 6.88 (s, 2H), 6.75 (s, 2H), 4.03 (m, 2H), 3.55 (m, 2H), 3.07 (m, 2H), 2.97 (m, 5H), 2.33 (s, 6H), 2.25 (s, 6H), 1.84 (m, 6H), 1.86 (s, 6H). 13C NMR (101 MHz, CD3CN, δ): 193.37, 159.31, 135.36, 132.15, 131.60, 130.40, 129.70, 66.52, 60.21, 49.47, 20.48, 20.34, 20.07. 19F NMR (376 MHz, CD3CN, δ): -71.96 (s, 3F), -73.84 (s, 3F). IR (FTIR, cm-1): 𝑣(CO) 1878. Elemental Analysis was not attempted on this complex because of its instability.
Synthesis of complex [(DAAm-C6F5)Re(CO)(Cl)2], 8.
characterized by 13C NMR due to its very poor solubility in acetonitrile-d3 or methylene chloride-d2. IR (FTIR, cm-1): 𝑣(CO) 1873. Anal. Calc. for C18H13Cl2F10N3ORe: C, 29.48; N, 5.73: H, 1.65. Found: C, 29.79; N, 6.01: H, 1.59.
General Procedure for Catalytic Hydrosilylation of Aldehydes.
In a nitrogen filled glove box, the corresponding [(DAAm-aryl)Re(CO)(OAc)] (aryl = C6F5 (3a), Mes (3b)) (3a = 2 mg, 0.00277 mmol; 3b = 2 mg, 0.00320 mmol) was added to a screw cap vial and dissolved in 0.35 mL of acetonitrile. Then HBF4·OEt2 (1 equiv) was added to the solution and mixed. The solvent was removed under reduced pressure to afford an oil. The aldehyde (27.7 mmol, 0.01 mol%; 9.23 mmol, 0.03 mol%; 5.54 mmol, 0.05 mol%; 2.77 mmol, 0.1 mol%; 0.554 mmol, 0.5 mol%; 0.277 mmol, 1 mol%), silane (1.2 equiv) and 2-bromomesitylene (0.5 equiv) were sequentially added to the vial. The reaction mixture was stirred at room temperature for the designated amount of time. Then, an aliquot of the crude reaction was dissolved in CDCl3 or CD3CN. The yield was determined by the proton NMR ratio of the product and starting material ((integration of methylene protons/2)/((integration of aldehyde peak) + (integration of methylene protons/2))) against 2-bromomesitylene as the internal standard.
General Procedure for Time Profile Experiments with 6a.
General Procedure to Obtain Order with Respect to Benzaldehyde by Gas Chromatography (GC).
In nitrogen filled glove box 6a (0.00241 mmol, 2.0 mg), benzaldehyde (2.77 mmol, 0.28 mL), dimethylphenylsilane (16.62 mmol, 2.5 mL), and 2-bromomesitylene (0.2 mL) were mixed in a small screw cap vial. The reaction mixture was divided into 10 small screw cap vials equipped with a stir bar. The reactions were stirred at room temperature for a designated amount of time. At that time, the ratio benzaldehyde:2-bromomesitylene was determined by GC using the calibration curve method. The concentration of benzaldehyde was calculated solving for the term x using the equation of the line, y = 1.25(5)x + 0.04(3). The calibration curve was obtained by calculating the ratio of benzaldehyde:2-bromomesitylene at various benzaldehyde concentrations (Figure 2.12).
General Procedure for Competition Reaction for Hammett Plot.
In nitrogen filled glove box 3a (2.0 mg, 0.00277 mmol), HBF4·OEt2 (0.37 µL, 0.00277 mmol), benzaldehyde (0.554 mmol, 56 µL), the indicated para-substituted benzaldehyde (0.554 mmol), dimethylphenylsilane (0.277 mmol, 0.5 equiv, 42 µL), and 2-bromomesitylene (0.274 mmol, 0.5 equiv, 42 µL) were sequentially added to a small screw cap vial. The reaction mixture was dissolved in CD3CN (0.35 mL) and stirred for 24 h at room temperature. An aliquot of the reaction mixture was placed in CD3CN. The 1H NMR integration against the internal standard (2-bromomesitylene) of the methylene protons of the para-substituted silyl ether and the benzyloxydimethylphenylsilane were used to determine the product ratio value (PH/PX).
General Procedure for Eyring Plot data.
In a nitrogen filled glove box 3a (2 mg, 0.00277 mmol) was added to a screw cap vial and dissolved in 0.35 mL of acetonitrile. Then HBF4·OEt2 (0.3 µL, 0.00277 mmol, 1 equiv) was added to the solution and mixed. The benzaldehyde (0.57 µL, 0.554 mmol), dimethylphenylsilane (0.1 mL, 0.665 mmol, 1.2 equiv) and 2-bromomesitylene (42 µL, 0.277 mmoles, 0.5 equiv) were sequentially added to the vial. The reaction was stirred at the respective temperature (25 – 80 °C). An aliquot at a fixed time was dissolved in either in CDCl3 or CD3CN. The yield was determined by the proton NMR ratio of the product and starting material ((integration of methylene protons/2)/((integration of aldehyde peak) + (integration of methylene protons/2))) against 2-bromomesitylene as the internal standard.
X-ray Crystallographic Procedures and Data General Procedure for X-ray Determination
![Figure 4.2 1H NMR (400 MHz, CD2Cl2) spectrum of [(DAAm)Re(O)(OC(CH3)OB(C6F5)3] (17). Residual unidentified impurity peaks observed at 2.68 ppm, 2.30 ppm, and 2.19 ppm](https://thumb-us.123doks.com/thumbv2/123dok_us/1742421.1223017/82.612.80.532.325.588/figure-spectrum-daam-residual-unidentified-impurity-peaks-observed.webp)
![Figure 4.6 1H NMR (300 MHz, C6D6) spectrum of [(DAAm-C6F5)Re(CO)(C6F5)] (19).](https://thumb-us.123doks.com/thumbv2/123dok_us/1742421.1223017/87.612.128.486.207.564/figure-h-nmr-mhz-spectrum-daam-re-co.webp)
