for Correlated Wavefunctions
Thesis by
Jason D. Goodpaster
In Partial Fulfillment of the Requirements
for the Degree of
Doctor of Philosophy
California Institute of Technology
Pasadena, California
2014
c
2014
Jason D. Goodpaster
Acknowledgments
It is with immense gratitude that I acknowledge the support and help of my thesis
advisor Professor Thomas F. Miller III. His encouragement, enthusiasm, and guidance
was invaluable throughout my Ph.D. He has taught me how to develop interesting
questions and how to approach answering them. He has made me a better scientist,
theorist, and communicator. I truly cannot imagine a better advisor and mentor for
my graduate studies.
Additionally, I would like to thank the faculty of the Division of Chemistry and
Chemical Engineering. In particular, I thank my thesis committee, Dr. William
Goddard III, Dr. Zhen-Gang Wang, and Dr. Mark Davis for their support, scientific
discussions, and guidance in my future career. I am also deeply grateful to the
late Aron Kuppermann for his elegant presentation of quantum mechanics, constant
encouragement, and his friendly late night discussions while we were “burning the
midnight oil”.
I also must thank the division administrative staff, Priscilla Boon for all the
sup-port she has provided to myself and the Miller Group, Kathy Bubash for her
ad-ministrative expertise, and Tom Dunn, Zailo Leite, and Naveed Near-Ansari for their
technical expertise, specifically with computing.
I thank all the members of the Miller group: Bin, Nick, Nandini, Romelia, Artur,
Josh, Connie, Taylor, Mike, Fran, Kuba, Frank, Michiel, Joonho, Mark, and Matt.
Thank you all for your scientific discussion, help with coding and computing, and
encouragement. It was truly an honor to work with you all.
through-out graduate school; you were always there for me. I thank my friends for their
understanding, encouragement, and for making my graduate school experience more
enjoyable.
Lastly, thank you Julia. You challenge me to be a better scientist and person, you
support me in all my endeavors, and you encourage me to strive above and beyond.
Abstract
Methods that exploit the intrinsic locality of molecular interactions show significant
promise in making tractable the electronic structure calculation of large-scale
sys-tems. In particular, embedded density functional theory (e-DFT) offers a formally
exact approach to electronic structure calculations in which the interactions between
subsystems are evaluated in terms of their electronic density. In the following
disser-tation, methodological advances of embedded density functional theory are described,
numerically tested, and applied to real chemical systems.
First, we describe an e-DFT protocol in which the non-additive kinetic energy
component of the embedding potential is treated exactly. Then, we present a general
implementation of the exact calculation of the non-additive kinetic potential (NAKP)
and apply it to molecular systems. We demonstrate that the implementation using
the exact NAKP is in excellent agreement with reference Kohn-Sham calculations,
whereas the approximate functionals lead to qualitative failures in the calculated
energies and equilibrium structures.
Next, we introduce density-embedding techniques to enable the accurate and
sta-ble calculation of correlated wavefunction (CW) in complex environments.
Embed-ding potentials calculated using e-DFT introduce the effect of the environment on
a subsystem for CW calculations (DFT). We demonstrate that
WFT-in-DFT calculations are in good agreement with CW calculations performed on the full
complex.
We significantly improve the numerics of the algorithm by enforcing orthogonality
projection-based embedding scheme, we rigorously analyze the sources of error in quantum
embedding calculations in which an active subsystem is treated using CWs, and the
remainder using density functional theory. We show that the embedding potential
felt by the electrons in the active subsystem makes only a small contribution to the
error of the method, whereas the error in the nonadditive exchange-correlation energy
dominates. We develop an algorithm which corrects this term and demonstrate the
Contents
Acknowledgments iv
Abstract vi
Summary 1
1 Exact non-additive kinetic potentials for embedded density
func-tional theory 6
1.1 Introduction . . . 6
1.2 Orbital-Free Embedded DFT . . . 7
1.3 The Exact Non-Additive Kinetic Potential . . . 10
1.3.1 Step 1: The Levy Constrained Search . . . 10
1.3.2 Step 2: Exact Kinetic Potentials from KS Orbitals . . . 11
1.3.3 Computational Details . . . 14
1.3.3.1 Basis Sets . . . 15
1.3.3.2 DFT Implementation Details . . . 15
1.3.3.3 ZMP Extrapolation . . . 16
1.4 Results . . . 17
1.5 Extension to Larger Systems . . . 24
1.6 Conclusions . . . 26
1.7 Appendix: Unrestricted Open-Shell e-DFT . . . 27
2 Embedded density functional theory for covalently bonded and strongly
2.1 Introduction . . . 33
2.2 Theory . . . 34
2.2.1 Orbital-Free Embedded DFT . . . 34
2.2.2 Exact Calculations of NAKP . . . 36
2.3 Implementation Details . . . 38
2.3.1 Supermolecular vs. Monomolecular Basis Sets . . . 38
2.3.2 ZMP Step . . . 38
2.3.3 NAKP Numerics for Regions of Weak Density Overlap . . . . 39
2.4 Results: Small Systems . . . 41
2.4.1 Calculation Details . . . 41
2.4.2 Water Dimer . . . 43
2.4.3 Li+-Be . . . 45
2.4.4 CH3-CF3 . . . 47
2.5 Results: Extension to Larger Systems . . . 48
2.5.1 Pairwise Treatment of the NAKP . . . 48
2.5.2 Water Trimer Application: Testing Pairwise Additivity in the NAKP . . . 50
2.5.3 Parallel Scaling of e-DFT-EE . . . 52
2.6 Conclusions . . . 54
3 Density functional theory embedding for correlated wavefunctions: Improved methods for open-shell systems and transition metal com-plexes 61 3.1 Introduction . . . 61
3.2 Theory . . . 62
3.2.1 DFT-in-DFT Embedding . . . 62
3.3 Methods of Implementation . . . 66
3.3.1 Embedding for Open-Shell Systems . . . 67
3.3.1.1 Open-Shell DFT-in-DFT Embedding . . . 67
3.3.1.2 Open-Shell WFT-in-DFT Embedding . . . 70
3.3.2 Optimized Effective Potential . . . 71
3.3.3 Orbital-Occupation Freezing . . . 74
3.3.4 Computational Details . . . 75
3.4 Results . . . 78
3.4.1 The Ethylene-Propylene Dimer: WFT-in-DFT Embedding . . 78
3.4.2 The Hexaaquairon(II) Cation . . . 79
3.4.2.1 DFT-in-DFT Embedding . . . 79
3.4.2.2 WFT-in-DFT Embedding . . . 82
3.5 Conclusion . . . 87
4 Accurate and systematically improvable density functional theory embedding for correlated wavefunctions 94 4.1 Introduction . . . 94
4.2 Projector-Based Embedding . . . 95
4.3 Results I: Sources of error in WFT-in-DFT embedding . . . 98
4.3.1 Term-By-Term Comparison with LCSSD(T) . . . 98
4.3.2 Calculation Details . . . 100
4.3.3 Sources of Error in WFT-in-DFT embedding . . . 103
4.3.3.1 Error from the Embedding Potential . . . 103
4.3.3.2 Error from Use of DFT for Subsystem B . . . 104
4.3.3.3 Error from the Nonadditive Exchange-Correlation En-ergy . . . 105
4.4 Results II: Continuity, Convergence, and Conjugation in WFT-in-DFT
embedding . . . 108
4.4.1 Potential Energy Surfaces . . . 108
4.4.2 WFT-in-DFT Embedding of Conjugated Systems . . . 109
4.5 Conclusions . . . 115
4.6 Accurate and Systematically Improvable Density Functional Theory Embedding for Correlated Wavefunctions: Supplemental Information 117 4.6.1 Potential Energy Surfaces . . . 117
4.7 Data Set Computational Details . . . 118
4.7.1 Symmetric SN2 Reaction Barrier . . . 118
4.7.2 Acid Hydrolysis Reaction Energy . . . 120
4.7.3 Phenol Deprotonation . . . 121
4.7.4 Ring Closing . . . 122
4.7.5 Diels Alder . . . 124
List of Figures
1.1 Accuracy in the extrapolation of kinetic energy from the ZMP method 15
1.2 The 2s electron density for an isoelectronic series of atoms calculated
using KS-DFT and e-DFT . . . 17
1.3 The KSCED effective potential for an isoelectronic series of atoms . . . 21
1.4 The NAKP, KSCED effective potential, and the 2s electron density for the modified NAKP . . . 25
2.1 The water dimer dissociation curve obtained using KS-DFT and e-DFT 43 2.2 Basis set dependence of the water dimer dissociation curve obtained using KS-DFT and e-DFT . . . 45
2.3 The Li+-Be dissociation curve obtained using KS-DFT and e-DFT . . 46
2.4 The CH3-CF3dissociation curve for heterolytic cleavage of the C-C bond obtained using KS-DFT and e-DFT . . . 48
2.5 Symmetric dissociation curves for the water trimer, illustrating the pair-wise additivity of the NAKP. . . 51
2.6 Wall-clock timings for lattices of hydrogen molecules . . . 53
2.7 Error in the total energy of e-DFT relative to KS-DFT for lattices of hydrogen molecules . . . 54
3.1 An illustrative Newton step in the OEP calculation . . . 75
3.2 WFT-in-DFT embedding for the ethylene-propylene dimer. . . 77
3.3 DFT-in-DFT embedding for the hexaaquairon(II) cation. . . 80
3.5 CCSD(T)-in-DFT embedding for the hexaaquairon(II) cation. . . 85
4.1 Contributions to the total WFT-in-DFT error . . . 103
4.2 MP2 corrected WFT-in-DFT errors . . . 106
4.3 Potential energy surface for 1-penten-1-one . . . 108
4.4 Error in dissociation of F− from alkane chain . . . 110
4.5 Error in dissociation of F− from alkene chain . . . 112
4.6 Error in exchange reaction energy for alkane and alkene chains . . . 114
List of Tables
1.1 Total energy and ionization energy obtained using KS-DFT and e-DFT. 20
1.2 Total energy and ionization energy obtained using e-DFT with the NAKP
switching function . . . 24
3.1 High-spin/low-spin splitting energies in cm−1 for the hexaaquairon(II) cation using MP2 theory and MP2-in-DFT . . . 84
3.2 High-spin/low-spin splitting energies in cm−1 for the hexaaquairon(II) cation using CCSD(T) theory and CCSD(T)-in-DFT . . . 86
4.1 CCSD(T) reaction energies and barriers in the test set . . . 102
4.2 Change in the dipole moment for dissociation of F− from the alkane and
Summary
Quantum-chemistry calculations invariably feature a compromise between
computa-tional efficiency and accuracy. Kohn-Sham Density Funccomputa-tional Theory (KS-DFT) is
one of the most commonly used methods because it is tractable for systems
contain-ing up to a thousand atoms. However, DFT methods, which generally employ
ap-proximate descriptions of electron correlation, often fail to even qualitatively predict
reaction barriers, which are critical in determining reaction rates and mechanisms.
More rigorous correlated wavefunction (CW) methods can provide an accurate and
systematically improvable description of reaction barriers, but poor scaling of these
methods can lead to impractical computation costs for systems with even tens of
atoms.
Embedded density functional theory (e-DFT), is a formally exact approach to
electronic structure, which exploits the locality of molecular interactions. In the
e-DFT approach, a system is divided into smaller subsystems. Then, the interactions
between subsystems are evaluated in terms of their electronic density. This approach
naturally leads to two different partitioning schemes. One, a large system can be
sep-arated into many smaller subsystems, allowing for significant computational savings
as this leads to a naturally parallelizable algorithm. Two, a large system is
sepa-rated into a ‘high-level’ (accurate) and a ‘low-level’ (approximate) regions; therefore,
if higher accuracy is required for a region of system, a higher level of theory can
be seamlessly embedded into a DFT environment and still remain computationally
tractable as only a handful of atoms are treated at the CW level. The objectives of the
frag-mentation schemes; however, e-DFT avoids the uncontrolled approximations (such as
link atoms) and errors associated with subsystem interfaces that fundamentally limit
other widely used methods.
This dissertation is focused on the development of new methods that make e-DFT
an accurate, practical, and scalable method for the description of complex systems.
The following key contributions are discussed: (i) the development of numerically
exact methods for obtaining subsystem embedding potentials in e-DFT, which reduce
embedding errors by orders of magnitude in comparison with previously available
approximate methods, (ii) the development of parallelization algorithms that enable
the description of large systems with sub-linear scaling of the required computational
time, (iii) the combination of e-DFT potentials with correlated wavefunction theory
(WFT) methods to enable seamless WFT-in-DFT embedding for general systems,
and (iv) the development of systematically improvable methods for WFT-in-DFT to
allow for a hierarchy of embedding methods with increasing accuracy.
In Chapter 1, we describe an embedded density functional theory (DFT)
proto-col in which the non-additive kinetic energy component of the embedding potential
is treated exactly. At each iteration of the Kohn-Sham equations for constrained
electron density, the Zhao-Morrison-Parr constrained search method for constructing
Kohn-Sham orbitals is combined with the King-Handy expression for the exact kinetic
potential. We use this exact embedding protocol to calculate ionization energies for
a series of 3- and 4-electron systems, and the results are compared to embedded DFT
calculations that utilize the Thomas-Fermi (TF) and the Thomas-Fermi-von
Wei-sacker approximations to the kinetic energy functional. These calculations illustrate
a breakdown due to the TF approximation for the non-additive kinetic potential, with
errors of 30-80% in the calculated ionization energies; by contrast, the exact protocol
N. Ananth, F. R. Manby, and T. F. Miller III, “Exact non-additive kinetic potentials
for embedded density functional theory,” J. Chem. Phys., 133, 084103 (2010). In Chapter 2, we present a general implementation of the exact calculation of
the non-additive kinetic potential (NAKP). Potential energy curves are computed
for the dissociation of Li+-Be, CH3-CF3, and hydrogen-bonded water clusters, and
e-DFT results obtained using this exact treatment of the NAKP are compared with
those obtained using approximate kinetic energy functionals. In all cases, the exact
NAKP is in excellent agreement with reference Kohn-Sham calculations, whereas the
approximate functionals lead to qualitative failures in the calculated energies and
equilibrium structures. We also demonstrate an accurate pairwise approximation to
the NAKP that allows for efficient parallelization of the method in large systems;
benchmark calculations on molecular crystals reveal ideal, size-independent scaling
of wall-clock time with increasing system size. This work has been published as J.
D. Goodpaster, T. A. Barnes, and T. F. Miller III, “Embedded density functional
theory for covalently bonded and strongly interacting subsystems,” J. Chem. Phys.,
134, 164108 (2011).
In Chapter 3, we introduce density embedding techniques to enable the
accu-rate and stable calculation of CWs in complex environments. Embedding potentials
calculated using e-DFT introduce the effect of the environment on a subsystem for
wavefunction calculations (WFT-in-DFT). These methods are demonstrated for
cal-culating the potential energy curve of the dispersion-bound ethene-propene dimer
and for the hexaaquairon(II) ion (HA). The potential energy curve for the
ethene-propene dimer reveals that benchmark CCSD(T) calculations performed over the full
complex can be reproduced within 0.05 kcal/mol using this method, illustrating that
small energy differences can be accurately calculated while embedding across a
cova-lent bond. e-DFT calculations on HA that employ these new techniques demonstrate
the high-spin states. The ability of different exchange-correlation (XC) functionals to
describe the energy differences between the low-spin and the high-spin states (∆ELH)
and to describe the ligation energy in HA is studied. KS-DFT calculations of ∆ELH
demonstrate a strong dependency on XC functionals where WFT-in-DFT
calcula-tions reveal a significantly diminished dependency and that ∆ELH is in good
agree-ment withab initio calculations performed on the full complex. The ligation energies
have a small dependency on the XC functional and are near identical for KS-DFT
and WFT-in-DFT demonstrating that the interactions energies between subsystems
remain at DFT level accuracy. This work has been published as J. D. Goodpaster, T.
A. Barnes, F. R. Manby, and T. F. Miller III, “Density functional theory embedding
for correlated wavefunctions: Improved methods for open-shell systems and transition
metal complexes,” J. Chem. Phys., 137, 224113 (2012).
In Chapter 4, we present continuing work on projection based embedding. The
original formulation has been published as F. R. Manby, M. Stella, J. D. Goodpaster,
and T. F. Miller III, “A simple, exact density-functional-theory embedding scheme,”
J. Chem. Theory Comput., 8, 2564 (2012) and T. A. Barnes, J. D. Goodpaster,
F. R. Manby, and T. F. Miller III, “Accurate basis set truncation for wavefunction
embedding,” J. Chem. Phys., 139, 024103 (2013). Building upon that work, we analyze the sources of error in quantum embedding calculations in which an active
subsystem is treated using CW methods, and the remainder using DFT. We show
that the embedding potential felt by the electrons in the active subsystem makes only
a small contribution to the error of the method whereas the error in the nonadditive
exchange-correlation energy dominates. We test an MP2 correction for this term
and demonstrate that the corrected embedding scheme accurately reproduces the
full wavefunction calculations for a series of chemical reactions. This work has been
“Accurate and systematically improvable density functional theory embedding for
Chapter 1
Exact non-additive kinetic potentials for embedded
density functional theory
1.1
Introduction
Orbital-free embedded density functional theory (e-DFT) is an appealing method
for calculating the electronic structure of complex molecular systems. It provides a
formally exact framework for dividing the total electronic density of a system into
subsystem densities that can be separately calculated.1–4This feature of e-DFT allows for the development of multiscale strategies in which the electronic density for regions
of central interest is calculated with high accuracy, while the electronic density for
surrounding regions is obtained using more approximate techniques.5–7
However, in addition to the usual approximations for the basis set and the
exchange-correlation functional that appear in Kohn-Sham (KS) DFT,8 e-DFT requires the evaluation of a non-additive contribution to the kinetic energy from the subsystem
densities.4 This term, which is generally largest for cases in which the subsystem densities are strongly overlapping,9 is a significant source of error in many e-DFT calculations, and it currently limits the method to applications in which the
subsys-tem densities involve non-bonded or weakly interacting molecular groups.4,9Although encouraging progress towards the accurate calculation of the non-additive kinetic
energy contribution in e-DFT calculations, and we report calculations in which the
protocol is applied to 3- and 4-electron systems that exhibit strongly overlapping
subsystem densities. Although, in its numerically demonstrated form, this protocol
does not offer any computational speed-up over the KS calculation for the full system,
it suggests new methods to systematically, efficiently, and accurately perform e-DFT
calculations for large systems, which we discuss.
1.2
Orbital-Free Embedded DFT
Suppose that the entire electronic density ρAB for a closed-shell system is divided
into two subsystems, ρA and ρB, such that ρAB=ρA+ρB. The one-electron orbitals
that give rise to these subsystem electronic densities obey the coupled Kohn-Sham
equations for constrained electron density (KSCED),4
−1
2∇
2+VKSCED
eff [ρA, ρB;r]
φAi (r) = Ai φAi (r), i= 1, ..., NA
2 , (1.1)
−1
2∇
2+VKSCED
eff [ρB, ρA;r]
φBi (r) =Bi φBi (r), i= 1, ...,NB
2 , (1.2)
where NA and NB are the number of electrons in the respective subsystems,
ρA(r) = 2
NA/2
X i=1
|φA
i (r)|
2, and (1.3)
ρB(r) = 2
NB/2
X i=1
|φB
i (r)|
2. (1.4)
In these coupled equations,VeffKSCED[ρA, ρB;r] is the KS effective potential for
VeffKSCED[ρA, ρB;r] =vne(r) +vJ[ρAB;r] +vxc[ρAB;r] +vnad[ρA, ρB;r], (1.5)
and VeffKSCED[ρB, ρA;r] is the similarly defined KS effective potential for subsystem B
embedded in subsystem A. The contributions to the KS effective potential include
vne(r) = −
Nnuc
X i
Zi |r−Ri|
, (1.6)
vJ[ρAB;r] =
Z ρ
AB(r0)
|r0−r|dr
0, and (1.7)
vxc[ρAB;r] =
δExc[ρ]
δρ
ρ=ρAB
(r), (1.8)
which are the usual nuclear-electron Coulomb potential, Hartree potential, and
exchange-correlation potential, respectively, and Nnuc is the number of nuclei in the system.
The final term in VeffKSCED[ρB, ρA;r] is the non-additive kinetic potential (NAKP)
vnad[ρA, ρB;r] =
δTnad
s [ρA, ρB]
δρA
(r) = δTs[ρ]
δρ
ρ=ρAB
(r)−δTs[ρ] δρ
ρ=ρA
(r), (1.9)
which is obtained from the functional derivative of the non-additive component of the
non-interacting kinetic energy
Tsnad[ρA, ρB] =Ts[ρAB]−Ts[ρA]−Ts[ρB]. (1.10)
The total energy functional for the embedded system is
where the last three terms on the right hand side (RHS) are the nuclear-electron
Coulomb energy, Hartree energy, and exchange-correlation energy for the total
den-sity.
Two aspects of the orbital-free embedding DFT formulation are worth
emphasiz-ing. Firstly, like conventional KS-DFT, it is a theory that is exact in principle, but
practical calculations must employ an approximate form for the unknown
exchange-correlation functional. Secondly, unlike conventional KS-DFT calculations, the
em-bedding formulation introduces an NAKP because the KS orbitals for subsystem A
are not necessarily orthogonal to those of subsystem B. Without knowledge of the
exact functional for the non-interacting kinetic energy, this creates a second source of
approximation in the e-DFT approach. The significance of the NAKP is system
de-pendent, with the most severe cases including those for which the subsystem densities
ρA and ρB greatly overlap.4,9,14,15
The non-interacting kinetic energy for the density corresponding to a set of N
closed-shell orbitals is
Ts[ρ] = 2
N
X i=1
hφi| −
1 2∇
2|φ
ii. (1.12)
Standard approximations to this kinetic energy functional include the Thomas-Fermi
(TF) result for the homogenous electron gas,16,17
TTF[ρ] =CTF
Z
ρ5/3(r)dr (1.13)
where CTF =
3 10(3π
2)2/3, and the von Weizs¨acker (vW) result for the limit of a
one-electron density,
TvW[ρ] =
1 8
Z |∇ρ
(r)|2
ρ(r) dr. (1.14)
Other approximate kinetic energy functionals can be constructed using the
PW91k kinetic energy functional10,11 employs the analytical form of the Perdew-Wang (PW91) exchange functional,19 and the TW02 functional13 and the PBE2, PBE3, and PBE4 functionals9 utilizes the form suggested by Becke.20 These func-tionals have been shown to successfully describe weakly interacting systems and
co-ordination compounds.9 Furthermore, kinetic energy functionals that have been de-veloped using linear response corrections to the homogeneous electron gas and have
been shown to work well for metals.3,21–23 However, no approximate kinetic energy functional has been demonstrated to yield accurate results for embedded subsystems
that are connected by a covalent bond.9,15,24
1.3
The Exact Non-Additive Kinetic Potential
For each iteration of the KSCED equations (Eqs. 1.1 and 1.2), {φA
i } and {φBi } (and
thusρAand ρB) are known from either the previous iteration or the initial guess, and
the NAKP must be calculated. We employ a two-step protocol to obtain the exact
NAKP. In the first step, a Levy constrained search25 (LCS) or equivalent method is used to determine the full set of orthogonal KS orbitals, {φAB
i }, that correspond to
the total density ρAB. In the second step, the NAKP is calculated from the orbital
sets {φAB
i }, {φ
A
i }, and {φ
B
i }.
1.3.1
Step 1: The Levy Constrained Search
Given a total electron density ρAB, the fully orthogonal KS orbitals {φABi } can be
calculated from an LCS, in which the non-interacting kinetic energy is minimized with
respect to one-electron orbitals that are constrained to yieldρAB.25 Alternatively, we
of KS orbitals are obtained by solving the one-electron equations " −1 2∇ 2− Nnuc X i Zi |r−Ri|
+Vcλ(r)
#
φiAB,λ(r) =iφiAB,λ(r), i= 1, ..., NAB
2 , (1.15)
where NAB=NA+NB,
Vcλ(r) =λ Z ρ
AB(r0)−ρ˜AB(r0)
|r0−r| dr
0, (1.16)
˜
ρAB(r) = 2
NAB/2
X i=1
|φAB
i (r)|
2, and Vλ
c (r) is a potential energy function that restrains
the ˜ρAB(r) to the target density ρAB(r). Solution of Eq. 1.15 in the limit λ → ∞ is
equivalent to performing the LCS.26–28
In practice, Eq. 1.15 is solved for six large, but finite, values of λ, and the KS orbitals and eigenvalues are obtained via extrapolation.26–28 For each value of λ, the
{λi}, {φABi ,λ}, and {∇2φABi ,λ} are calculated and stored on a spatial grid. For the orbitals, extrapolation to λ→ ∞ is performed via expansion to third order in 1
λ,
φABi ,λ(r) =φABi (r) + 1
λa
(1)
i (r) +
1
λ2a (2)
i (r) +
1
λ3a (3)
i (r), (1.17)
with a linear least-squares fit of the expansion coefficients{φAB
i (r), a
(1)
i (r), a
(2)
i (r), a
(3)
i (r)}
at each value ofr. The{∇2φAB
i }are similarly obtained via extrapolation at each value
ofr, while eachi requires only a single extrapolation. With the{φABi }and{∇
2φAB
i }
obtained on the spatial grid, the non-interacting kinetic energy for the total system
can be calculated via numerical integration using Eq. 1.12.
1.3.2
Step 2: Exact Kinetic Potentials from KS Orbitals
To calculate the NAKP from the orbital sets {φAB
i }, {φ
A
i }, and {φ
B
i }, we extend the
Minimization of the electronic energy with respect to the total electron density
ρAB yields the stationary condition8
δTs[ρ]
δρ
ρ=ρAB
(r) +vne(r) +vJ[ρAB;r] +vxc[ρAB;r] =µAB (1.18)
where µAB is a Lagrange multiplier that imposes the constraint
Z
ρAB(r)dr= NAB.
Furthermore, rearrangement of the usual KS equations yields
2
ρAB(r)
NAB/2
X i −1 2φ AB
i (r)∇
2φAB
i (r)−iφABi (r)
2
+vne(r) +vJ[ρAB;r] +vxc[ρAB;r] = 0.
(1.19)
Comparison of these two results leads to an exact expression for the total kinetic
potential,29
δTs[ρ]
δρ
ρ=ρAB
(r) = 2
ρAB(r)
NAB/2
X i −1 2φ AB
i (r)∇
2φAB
i (r)−iφABi (r)
2
+µAB. (1.20)
Analogous results can be derived for each of the embedded subsystems.
Specifi-cally, the electron density for subsystem A also obeys a stationary condition,2
δTs[ρ]
δρ
ρ=ρA
(r) +vne(r) +vJ[ρAB;r] +vxc[ρAB;r] +vnad[ρA, ρB;r] =µA, (1.21)
where µA is the Lagrange multiplier that imposes the constraint
Z
ρA(r)dr = NA.
Combination of Eq. 1.21 with Eq. 1.1 results in an exact expression for the subsystem
kinetic potential,
δTs[ρ]
δρ
ρ=ρA
(r) = 2
ρA(r)
NA/2
X i −1 2φ A
i (r)∇
2
φAi (r)−Ai φAi (r)2
+µA, (1.22)
which can be compared with kinetic potential for the total system in Eq. 1.20.
arbitrarily chosen subsystem, we likewise obtain µAB=µB, or
µA=µB. (1.23)
This result has a simple physical interpretation. In the zero temperature limit, the
Lagrange multipliers µA andµB, are equivalent to the chemical potential for the
sub-system electronic densities.8 Solution to the KSCED equations thus yields densities that are in equilibrium with respect to the number of electrons in each subsystem.
Finally, insertion of Eq. 1.20 and 1.22 into Eq. 1.9 yields the desired expression
for the NAKP,
vnad[ρA, ρB;r] =
2
ρAB(r)
NAB/2
X i −1 2φ AB
i (r)∇
2φAB
i (r)−iφABi (r)
2
− 2
ρA(r)
NA/2
X i −1 2φ A
i (r)∇
2φA
i (r)−
A
i φ
A
i (r)
2
. (1.24)
Note that the ZMP protocol generally yields a constant shift in the calculated set of
KS eigenenergies,{λ i};
26in Eq. 1.24, we see that this leads only to a constant shift in
vnad[ρA, ρB;r] and causes no change in any calculated observables. Throughout this
study, the NAKP is shifted such that it approaches zero at large distances.
Visscher and coworkers previously observed that the NAKP can be expressed in
terms of the stationary condition for the total system (Eq. 1.18) and a subsystem
(Eq. 1.21).30 They suggested a strategy in which the ZMP method is used to ob-tain the external potentials for both the full system and the subsystem, so that the
difference between those potentials is equal to the NAKP, but this strategy has not
been implemented to our knowledge. The approach presented here directly expresses
the NAKP in terms of the KS orbitals for the total system and the subsystem (Eq.
1.24); it avoids performing the ZMP method for the subsystem, and it avoids using
is straightforward to show that Eq. 1.24 recovers the limit for weakly overlapping
subsystem densities that is reported in Ref. 30.
In another approach that does not utilize the exact framework of the KSCED
equations, Aguado and coworkers employ an embedding strategy in which the ZMP
formalism is used to constrain the sum of the subsystem densities to that of the total
density.31,32 This approach has been pursued as a useful, but approximate, strategy for partitioning a total density into subsystems.
Other e-DFT strategies also express the kinetic potential in terms of the KS
or-bitals, as we have done here. For example, Huang and Carter report an explicit
expression for the kinetic potential in terms of the KS orbitals, using the assumption
that the non-interacting kinetic energy is a linear functional of the density; an
em-pirical parameter is included in their result to account for non-linear effects.33 The approach presented here involves no adjustable parameters and no assumptions about
the linearity of the kinetic energy functional.
1.3.3
Computational Details
Calculations are performed on four atomic systems: Li, Ne7+, Q−2.05.5 and Be, where Q−2.05.5 is a model 3-electron atom that has a nuclear charge of +2.5. In all e-DFT calculations, we take ρA to be the density for a single 2s electron, and ρB includes
all other electrons. The KSCED equations for each system were solved with ρB fixed
at the density obtained from the corresponding orbitals of an unrestricted KS-DFT
calculation on the full system; this is justified for the cases studied here because
solution of the KSCED equations for ρA subject to a fixed ρB ≤ρ0 (at all r), where
ρ0 is the exact ground state density for the full system, ensures the exact calculation
1.3.3.1 Basis Sets
All calculations were performed using the fully uncontracted cc-pVTZ basis set of
Gaussian-type orbitals (GTOs),34 with only the s-type orbitals included. For calcu-lations on Q−2.05.5, the Li basis set was used. For Ne7+, the most diffuse s-orbital was removed to facilitate convergence. Although not reported, all calculations were also
repeated with slater-type orbitals, which led to somewhat improved convergence but
very similar numerical accuracy.
[image:29.612.206.446.311.484.2]1.3.3.2 DFT Implementation Details
Figure 1.1: The difference between the non-interacting kinetic energy Ts[ρ] from
KS-DFT and from the ZMP method, plotted as a function of γ. The extrapolation is performed using {λ}={γ−jτ}, j = 5,4, . . . ,0, and using τ of 10 (red), 20 (green), and 40 (blue). See text for details.
For all applications considered here, ρA is an open shell system, and the
calcula-tions were performed using the unrestricted KS formalism. Details for the unrestricted
KSCED equations are given in the Appendix. All calculations employ the Slater
ex-change functional35 and the Vosko, Wilk, and Nusair correlation functional.36 In calculating VeffKSCED[ρA, ρB;r], a uniform radial grid is used to evaluate the
KSCED equations, the same radial grid is used to evaluate the exchange-correlation
energy and to numerically integrate the kinetic energy. For Be and Li the grid
ex-tends from r = 0 to 15, while for Q2−.05.5, r = 0 to 20 and Ne7+, r = 0 to 2. For Be, Li and Q the grid density is 2000 points/a0 and for Ne7+, 20000 points/a0. We note
that future applications that employ either a non-uniform37 or variational38 mesh will require fewer gridpoints to achieve the same level of accuracy. Unless otherwise
stated, the iterative solution of the KSCED equations were deemed converged when
the total energy of the system changed by less than 10−8 Hartrees between successive iterations.
1.3.3.3 ZMP Extrapolation
To examine the extrapolation error associated with the ZMP method, convergence
tests were performed for the Li atom system. The total density for the system, ρAB,
and the reference value for the non-interacting kinetic energy were calculated from a
full KS calculation. This ρAB was used to define the restraint potential (Eq. 1.16),
and the ZMP extrapolation was performed using six equally spaced values of λ (i.e.,
{λ} = {γ −jτ}, where j = 5,4, . . . ,0). For a given pair of parameters γ and τ,
the non-interacting kinetic energy was numerically integrated, and the extrapolation
error was taken to be the difference between this result and the reference value from
the full KS calculation. Fig. 1.1 presents this calculated error as a function γ and for various values of τ. These results indicate that the extrapolation error decreases to within 0.1 mH for γ >500, and the spacing parameter τ has only a small effect. The error decreases to within 1 µH for larger values of γ. Results reported hereafter employγ = 600 andτ = 10. The orbitals from Eq. 1.15 were deemed converged when all occupied orbital coefficients changed less than 10−7 between successive iterations. The ZMP extrapolation scheme used here does not constrain the normalization of
then 0.01%, and it was found that normalizing the orbitals after extrapolation led to
less than 0.1 mH change in the total energy. The results reported here do not include
a posteriori orbital normalization.
[image:31.612.244.405.220.592.2]1.4
Results
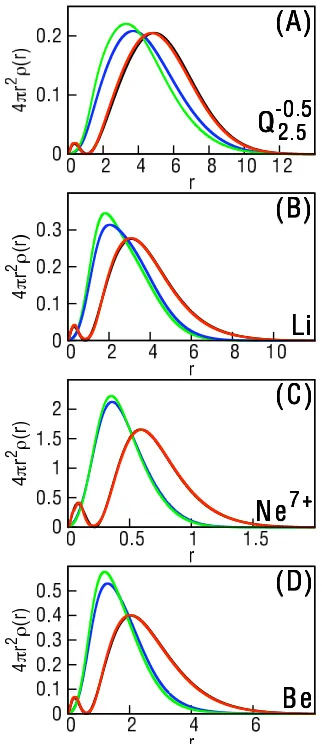
Figure 1.2: The 2s electron density (ρA) for (A) the Q2−.50.5 ion, (B) the Li atom,
(C) the Ne+7 ion, and(D) the Be atom. Calculations performed using e-DFT with the non-additive kinetic energy calculated using our exact protocol (red), the TF functional (blue), and the TFvW functional (green). The black curve, which is nearly indistinguishable from the exact protocol, presents the results from the full KS-DFT calculation.
as well as the four-electron Be atom. For each application, ρA was chosen to include
a single 2s electron, and the remaining electrons were included in ρB. In addition
to using the exact embedding protocol described here, the NAKP in the embedding
calculations was treated using the approximate TF kinetic energy functional (TTF[ρ] ,
Eq. 1.13) and the TFvW functional with the standard 1/9 mixing parameter (TTF[ρ]+
1
9TvW[ρ]).
Fig. 1.2 presents the ρA obtained in these e-DFT calculations. For reference,
Fig. 1.2 also includes the 2s orbital density from the full KS-DFT calculation.
Ab-solute agreement between the KS-DFT results and the e-DFT results would only
be expected if all results were obtained with the exact exchange-correlation
func-tional. Nonetheless, since all calculations in this study employ the same approximate
exchange-correlation functional, comparison of the e-DFT and KS-DFT results
pro-vides a significant test of the accuracy of the various NAKP descriptions.
Fig. 1.2 clearly demonstrates the sensitivity of e-DFT calculations to the method
of treating the NAKP. In comparison to KS-DFT, the e-DFT results from the
approx-imate TF and TFvW functionals are peaked at significantly shorter radial distances,
and they qualitatively fail to capture the nodal structure. Interestingly, the vW
correction to the TF functional actually worsens the agreement with the KS-DFT
reference. The exact embedding protocol describe here, however, is graphically
indis-tinguishable from the KS-DFT result.
Further evaluation of the e-DFT methods can be obtained by comparing the
cal-culated one-electron ionization energies (IEs) for the various methods. The e-DFT
IE is calculated from the difference between the total electron energy from Eq. 3.6
and the energy from a full KS-DFT calculation performed on the ionized (N-1
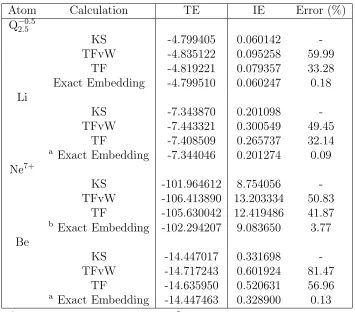
elec-tron) system. These results are presented in Table 1.1, which again illustrates the
qualitative shortcomings of the approximate NAKP treatments. For the approximate
result for the IEs ranges from 30-60% for 3-electron systems, and up to 80% for Be.
As has been observed previously,40 including the vW gradient correction decreases the accuracy of the IE calculation. The exact embedding protocol almost completely
eliminates these differences with the reference calculation, with errors of less than
0.2% for Q−2.05.5, Li, and Be and with an error of less 4% for Ne7+.
The lower accuracy of our embedding protocol for the case of Ne7+ arises from the description of the nuclear cusp. The KSCED equations converged slowly for this
case, and the convergence threshold had to be raised to 10−5 hartrees. By changing from GTOs to Slater-type orbitals (results not shown), the convergence problem was
removed, and it was found that for all four applications, the IEs obtained using our
e-DFT protocol were within 1% of the full KS-DFT result. Below, we describe how
the use of a simple switching function for the NAKP in the cusp region also removes
these convergence problems for the GTOs, while preserving the accuracy of the IE
calculation.
We note that the ionization of the closed shell Be atom presents an electronic
structure challenge that is similar to the homolytic cleavage of a covalent bond. From
the perspective of the NAKP, this atomic system is especially challenging since both
electrons in the 2s “bond” are co-localized on a single attractive center. The difficulty
of this particular case is confirmed by the especially poor description provided by the
TF and TFvW functionals for the IE of the Be atom. The excellent accuracy of the
new embedding protocol for this case suggests that the method will allow for accurate
e-DFT calculations in which the subsystems are linked by covalent bonds.
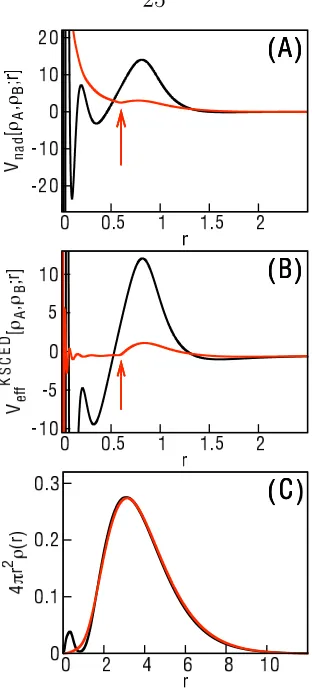
Fig. 1.3 illustrates the KSCED potentials, VeffKSCED[ρA, ρB;r], and the
correspond-ing NAKPs,vnad[ρA, ρB;r], that are obtained from the exact embedding calculations.
For each system, the similarity between these two potentials illustrates the
domi-nance of the NAKP at short distances. However, the NAKP decays rapidly, and the
Table 1.1: Total energy (TE) and ionization energy (IE) obtained using KS-DFT and e-DFT.
Atom Calculation TE IE Error (%)
Q−2.05.5
KS -4.799405 0.060142
-TFvW -4.835122 0.095258 59.99
TF -4.819221 0.079357 33.28
Exact Embedding -4.799510 0.060247 0.18 Li
KS -7.343870 0.201098
-TFvW -7.443321 0.300549 49.45
TF -7.408509 0.265737 32.14
a
Exact Embedding -7.344046 0.201274 0.09 Ne7+
KS -101.964612 8.754056
-TFvW -106.413890 13.203334 50.83
TF -105.630042 12.419486 41.87
b
Exact Embedding -102.294207 9.083650 3.77 Be
KS -14.447017 0.331698
-TFvW -14.717243 0.601924 81.47
TF -14.635950 0.520631 56.96
a Exact Embedding -14.447463 0.328900 0.13
a KSCED equations converged to 10−7 Hartree
Although it is not visible from the scale of the plots in Fig. 1.3, the vnad[ρA, ρB;r]
term comprises less than 1% of theVeffKSCED[ρA, ρB;r] for distances greater than 3 a.u.
[image:35.612.202.444.149.434.2]for all cases. (For Ne7+, this regime is reached at 0.43 a.u.)
Figure 1.3: The KSCED effective potential, VeffKSCED[ρA, ρB;r], for (A) the Q−2.05.5
ion, (B) the Li atom, (C) the Ne+7 ion, and (D) the Be atom and the NAKP,
vnad[ρA, ρB;r], for (E) the Q2−.05.5 ion, (F) the Li atom, (G) the Ne
+7 ion, and (H)
the Be atom using the e-DFT protocol presented here.
Comparison of the NAKPs in Fig. 1.3E-H with the densities in Fig. 1.2 illustrates
that the nodal structure in the 2s electron density is enforced by the NAKP. For each
system, the large outer peak in the NAKP coincides with the nodal feature in the
2s density. Unlike the KS-DFT results, we note that the densities obtained using
e-DFT in Fig. 1.2 do not exhibit a genuine radial node, since ρA corresponds to the
ground-state eigenvector of Eq. 1.1. A large peak in the e-DFT effective potential
is therefore essential to achieve the correct 2s shell structure. The NAKPs obtained
(not shown), which leads to the poor descriptions for the 2s electron density (Fig.
1.2) and the IE (Table 1.1).
In addition to the pronounced outer-most peak for each NAKP in Fig. 1.3E-H,
oscillations at short distances are observed. This oscillatory behavior is sensitive to
the basis set representation. Small changes in the orbital coefficients for regions of low
density give rise to large changes in the kinetic potential (Eq. 1.24), resulting in slow
convergence of the KSCED equations. (These oscillations are not observed when the
density vanishes at large distances since the basis set expansion is dominated by only
the slowest-decaying function in that regime.) Using STOs rather than GTOs, the
NAKP oscillations at short distances were diminished (not shown), and the iterative
convergence was improved. In future applications of the exact embedding protocol
with GTOs, the use of the convergence acceleration algorithms such as DIIS39 may prove beneficial. However, we now demonstrate that the problems associated with
poor convergence and NAKP oscillations can be alleviated with a simple modification
of Eq. 1.24.
As ρAvanishes close to the nucleus, evaluation of the second term in Eq. 1.24
be-comes unstable, leading to the aforementioned convergence problems. This is avoided
by introducing a switching function that changes from the exact expression for the
kinetic potential of subsystem A to the corresponding TF approximation near the
nucleus,
vnad[ρA, ρB;r] =
2
ρAB(r)
NAB/2
X i=1 −1 2φ AB
i (r)∇2φABi (r)−iφABi (r)2
(1.25)
− 2
ρA(r)
NA/2
X i=1 −1 2φ A
i (r)∇2φAi (r)−Ai φAi (r)2
× (1.26)
(1−f[ρB;r])−
5 3CTF ρ
2/3 A
where f[ρB;r] is the smooth switching function
f[ρB;r] =
1
eκ(−ρB(r)+ρ0B)+ 1
. (1.27)
Previous work used a similar function to switch between approximate expressions for
the NAKP in the vicinity of the nuclear cusp.40 The parameters ρ0B and κ determine the radial distance and the abruptness with which switching occurs, respectively. The
parameterρ0Bwas related to the integrated electron density in the cusp region, setting
ρ0B =ρB(r0), where
ξ = 4π Z r0
0
r2ρB(r)dr. (1.28)
e-DFT results obtained using range of values for κand ξ were compared to deter-mine robust parameters for the switching function. Setting κ = 50, we varied ξ over the range from 0.4 to 0.8 for Li and Ne7+, which led to changes in the total calculated energy of less than 0.4 mH and 5 mH, respectively. Similarly, setting ξ = 0.6 and
varying κ over the range from 50 to 500 led to differences of less than 0.1 mH for both Li and Ne.
Using the NAKP expression in Eq. 1.27 with ξ= 0.6 andκ= 50, our e-DFT pro-tocol was applied to all four systems, and the results are presented in Table 1.2. All
calculations reached full 10−8 convergence within 80 iterations of the KSCED equa-tions, in contrast with the calculations using Eq. 1.24, which was difficult to converge
in some cases even with 2000 iterations. Furthermore, the e-DFT calculations with
the modified NAKP expression in Eq. 1.27 yields excellent accuracy in comparison
to the full KS-DFT equations, with less than 1.5% error in the IE for all cases.
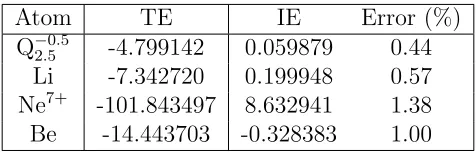
For the Li atom, Fig. 1.4 compares the NAKP, the KSCED effective potential,
and the 2s electron density obtained by solving the KSCED equations using either
Eq. 1.24 (black) or Eq. 1.27 (red) for the NAKP. The black curves in this figure are
Table 1.2: Total energy (TE) and ionization energy (IE) obtained using e-DFT with the NAKP switching function (Eq. 1.27).
Atom TE IE Error (%)
Q−2.05.5 -4.799142 0.059879 0.44 Li -7.342720 0.199948 0.57 Ne7+ -101.843497 8.632941 1.38 Be -14.443703 -0.328383 1.00
distances, the switching function produces a relatively featureless, repulsive NAKP
due to the TF approximation; the arrow in this figure indicates the radial distancer0
that corresponds to the parameter ξ = 0.6. Fig. 1.4B illustrates that the repulsive NAKP largely cancels the attractive electron-nuclear Coulomb term in the KSCED
effective potential (Eq. 1.5). As ρA vanishes at the nucleus, the KSCED effective
potential must also approach zero.30 The remaining oscillations at radial distances in Fig. 1.4B are an artifact of the finite basis set. Finally, Fig. 1.4C demonstrates
that the 2s electron density that is obtained using the switching function does not
reproduce the features of the radial node, but it recovers the exact embedding result
for distances beyond 1 a.u. This close agreement at large distances is expected41 from the accuracy of the IE calculations in Table 1.2. In light of the much improved
convergence efficiency, use of the NAKP expression in Eq. 1.27 compares favorably
with exact embedding via Eq. 1.24.
1.5
Extension to Larger Systems
The calculations reported here demonstrate a proof-of-principle for the exact
calcula-tion of the NAKP. However, for applicacalcula-tions of e-DFT to large systems, performance
of the ZMP extrapolation at each iteration of the KSCED equations is impractical.
Nonetheless, the short-ranged nature of the NAKP (see Fig. 1.3E-H) suggests several
strategies for employing our e-DFT protocol in larger systems.
Figure 1.4: (A) The NAKP, (B) the KSCED effective potential, and (C) the 2s electron density (ρA) for the Li atom, obtained using exact embedding (black) and
using the modified NAKP in Eq. 1.27 (red). The arrow indicates the radial distance at which switching occurs.
. . ., Bf), and consider the sum of the NAKP terms due to the individual fragments,
vnad[ρA, ρB;r]≈
f X
i=1
δTs[ρ]
δρ
ρ=ρA+ρBi
− δTs[ρ] δρ
ρ=ρA
. (1.29)
This equation is exact in the limit of one fragment, and its implementation with our
protocol will avoid ZMP extrapolation for anything larger than the union of subsystem
A with a single fragment.
The assumption in Eq. 1.29 that the NAKP is additive among the fragments
corrected using the TF (or other) approximate kinetic energy functional,
vnad[ρA, ρB;r] ≈
δTs[ρ]
δρ
(appr)
ρ=ρA+ρB
− δTs[ρ] δρ
(appr)
ρ=ρA
!
− f X
i=1
δTs[ρ]
δρ
(appr)
ρ=ρA+ρBi
− δTs[ρ] δρ
(appr)
ρ=ρA
!
+
f X
i=1
δTs[ρ]
δρ
(exact)
ρ=ρA+ρBi
− δTs[ρ] δρ
(exact)
ρ=ρA
!
. (1.30)
Here, the first term on the RHS corresponds to the NAKP obtained from the TF
functional for the full system. In the second term, the contribution due to each of the
fragments using the TF approximation is removed, and in the third term, each of the
fragment contributions is replaced using the exact protocol. The short-ranged nature
of the NAKP suggests that distance-based cutoffs can be employed with summations
in Eqs. 1.29 and 1.30, allowing for significant computational savings.
1.6
Conclusions
We have described a general and exact protocol for treating the non-additive kinetic
potential in embedded density functional theory calculations. In applications to a
series of three- and four-electron systems, we have numerically demonstrated the
ap-proach, and we have illustrated the qualitative failures that can arise from the use
of approximate kinetic energy functionals. We have also shown that improved
con-vergence of the KSCED equations can be obtained with appropriate switching of the
NAKP in the vicinity of the nuclear cusps, and we have described possible strategies
for the scalable implementation of our embedding protocol in large systems.
Natu-ral applications of exact embedding include the rigorous calculation of one-electron
pseudopotentials, the calculation of DFT embedding potentials for use with high-level
of the “molecular embedding” strategy in which each molecule of a large system is
assigned to a different embedded subsystem.44
1.7
Appendix: Unrestricted Open-Shell e-DFT
For unrestricted open-shell e-DFT calculations, the density of each subsystem is
fur-ther partitioned intoα andβ spin densities, such thatρAB=ραA+ρ
β
A+ρ
α
B+ρ
β
B. This
leads to the KSCED equations
−1
2∇
2+VKSCED eff [ρ
α
A, ρ
β
A, ρ
α
B, ρ
β
B;r]
φA,αi (r) = A,αi φiA,α(r) i = 1, ..., NAα, (1.31)
−1
2∇
2+VKSCED eff [ρ
β
A, ρ
α
A, ρ
β
B, ρ
α
B;r]
φA,βi (r) = A,βi φiA,β(r) i = 1, ..., NAβ, (1.32)
−1
2∇
2+VKSCED eff [ρ
α
B, ρ
β
B, ρ
α
A, ρ
β
A;r]
φB,αi (r) =B,αi φiB,α(r) i = 1, ..., NBα, (1.33)
−1
2∇
2+VKSCED eff [ρ
β
B, ρ
α
B, ρ
β
A, ρ
α
A;r]
φB,βi (r) =B,βi φiB,β(r) i = 1, ..., NBβ. (1.34)
Here, Nµν is the number of electrons in each subsystem, and ρνµ(r) =
Nν µ X
i=1
|φµ,νi (r)|2,
whereµ∈ {A,B}andν ∈ {α, β}. The KSCED effective potential,VeffKSCED[ραA, ρβA, ραB, ρβB;r], is
VeffKSCED[ραA, ρβA, ραB, ρβB;r] =vne(r)+vJ[ρAB;r]+vxc[(ραA+ρ
α
B),(ρ
β
A+ρ
β
B);r]+vnad[ραA, ρ
α
B;r]
(1.35)
where vne(r) and vJ[ρAB;r] are unchanged from Eq. 1.5, vxc[(ραA+ρ
α
B),(ρ
β
A+ρ
β
B);r]
is the usual open-shell exchange-correlation potential for the total system,8 and the NAKP is discussed below.
The kinetic energy functional is separable into two different spin contributions8
Ts[ραµ, ρ β
where
Ts[ραµ,0] = Nα
µ X
i=1
hφµ,αi | − 1
2∇
2|φµ,α
i i (1.37)
and likewise for Ts[0, ρβ]. Therefore, the NAKP depends only on spin densities
cor-responding to the same spin, such that
vnad[ραA, ρ
α
B;r] =
δTs[ρα,0]
δρα
ρα=ρα
A+ραB
(r)− δTs[ρ α,0] δρα
ρα=ρα
A
(r), and (1.38)
vnad[ρβA, ρ
β
B;r] =
δTs[0, ρβ]
δρβ
ρβ=ρβ
A+ρ
β
B
(r)− δTs[0, ρ β]
δρβ
ρβ=ρβ
A
(r). (1.39)
The ZMP extrapolation is used to calculate the KS spin orbitals {φABi ,ν} and eigenvalues{ABi ,ν}for the full system, exactly as is described in the text, except that the total spin density is employed instead of the total electron density. Finally, our
exact expression for the NAKP for open-shell systems is modified from Eq. 1.24 as
follows:
vnad[ρνA, ρ
ν
B;r] =
1
ρν
AB(r)
NAν+NBν X i −1 2φ AB,ν i (r)∇
2φAB,ν
i (r)−
AB,ν
i φ
AB,ν i (r)
2
− 1
ρν
A(r)
Nν A X i −1 2φ A,ν i (r)∇
2
φA,νi (r)−A,νi φA,νi (r)2
. (1.40)
The TF approximation for the non-additive kinetic energy in an open-shell
calcu-lation is
TTFnad[ρνA, ρνB] = 22/3CTF
Z
ρνAB5/3(r)−ρνA5/3(r)−ρνB5/3(r)dr, (1.41)
and corresponding result for the TFvW functional is
TTFvWnad [ρνA, ρνB] =TTFnad[ρνA, ρνB] + 1 72
Z |∇ρν
AB(r)|2
ρν
AB(r)
− |∇ρ ν
A(r)|2
ρν
A(r)
−|∇ρ ν
B(r)|2
ρν
B(r)
dr.
Bibliography
[1] P. Cortona, Phys. Rev. B44, 8454 (1991).
[2] T. A. Wesolowski and A. Warshel, J. Phys. Chem. 97, 8050 (1993).
[3] N. Govind, Y. A. Yang, A. J. R. da Silva, and E. A. Carter, Chem. Phys. Lett.
295, 129 (1998).
[4] T. A. Wesolowski,Computational Chemistry: Reviews of Current Trends, edited
by J. Leszczynski (World Scientific, Singapore, 2006), Vol. 10, 1-82.
[5] T. Kl¨uner, N. Govind, Y. A. Yang, and E. A. Carter, J. Chem. Phys. 116, 42 (2001).
[6] G. Hong, M. Strajbl, T. A. Wesolowski, and A. Warshel, J. Comp. Chem. 21, 234110 (2000).
[7] M. Hodak, W. Lu, and J. Bernholc, J. Chem. Phys. 128, 014101 (2008). [8] R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules
(Oxford University Press, New York, 1989).
[9] A. W. G¨otz, S. M. Beyhan, and L. Visscher, J. Chem. Theory Comput.5, 3161 (2009).
[10] T. A. Wesolowski, J. Chem. Phys.106, 8516 (1997).
[12] V. V. Karasiev, S. B. Trickey, and F. E. Harris, J. Comput.-Aided Mater. Des.
13, 111 (2006).
[13] F. Tran and T. A. Wesolowski, Int. J. Quantum Chem. 89, 441 (2002). [14] C. R. Jacob and L. Visscher, J. Phys. Chem. 128, 155102 (2008).
[15] S. M. Beyhan, A. W. G¨otz, C. R. Jacob, and L. Visscher, J. Chem. Phys. 132, 044114 (2010).
[16] L. H. Thomas, Proc. Camb. Phil. Soc. 23, 542 (1927). [17] E. Fermi, Z. Phys. 48, 73 (1928).
[18] C. F. von Weizs¨acker, Z. Phys. 96, 431 (1935).
[19] J. P. Perdew, J. Chevary, S. Vosko, K. A. Jackson, M. R. Pederson, D. Singh,
and C. Fiolhais, Phys. Rev. B. 46, 6671 (1992). [20] A. D. Becke, J. Chem. Phys. 84, 8 (1986).
[21] Y. A. Wang, N. Govind, and E. A. Carter, Phys. Rev. B 60, 16350 (1999). [22] J. A. Alonso and L. A Girifalco, Phys. Rev. B17, 3735-3743 (1978).
[23] P. Garc´ıa-Gonz´alez, J. E. Alvarellos, and E. Chac´on, Phys. Rev. B53, 9509-9512 (1996).
[24] S. Fux, K. Kiewish, C. R. Jacob, J. Neugebauer, and M. Reiher, Chem. Phys.
Lett. 461, 353 (2008).
[28] Q. S. Zhao, R. C. Morrison, and R. G Parr, Phys. Rev. A 50, 2138 (1994). [29] R. A. King and N. C Handy, Phys. Chem. Chem. Phys. 2, 5049-5056 (2000). [30] C. R. Jacob, S. M. Beyhan, and L. Visscher, J. Chem. Phys.126, 234116 (2007). [31] O. Roncero, M. P. de Lara-Castells, P. Villarreal, F. Flores, J. Ortega, M.
Pani-agua, and A. Aguado, J. Chem. Phys. 129, 184104 (2008).
[32] O. Roncero, A. Zanchet, P. Villarreal, and A. Aguado, J. Chem. Phys. 131, 234110 (2009).
[33] P. Huang and E. A. Carter, J. Chem. Phys. 125, 084102 (2006). [34] T. H. Dunning, J. Chem. Phys. 90, 1007 (1989).
[35] J. C. Slater, Phys. Rev. 81, 385-390 (1951).
[36] S. H. Vosko, L. Wilk, and M. Nusair, Can. J. Phys.58, 1200-1211 (1980). [37] B. N. Papas and H. F. Schaefer, III, J. Mol. Struc.: THEOCHEM 768, 175
(2006).
[38] M. R. Pederson and K. A. Jackson, Phys. Rev. B 41, 7453 (1990). [39] P. Pulay, Chem. Phys. Lett.72, 393 (1980).
[40] J. M. L. Lastra, J. W. Kaminski, and T. A. Wesolowski, J. Chem. Phys. 129, 074107 (2008).
[41] D. J. Tozer and H. C. Handy, Molec. Phys. 101, 2669 (2003).
[42] P. Huang and E. A. Carter, Annu. Rev. Phys. Chem. 59, 261 (2008).
Chapter 2
Embedded density functional theory for covalently
bonded and strongly interacting subsystems
2.1
Introduction
Important methodological challenges in electronic structure theory include the
long-timescale simulation ofab initio molecular dynamics and the seamless combination of
high- and low-level electronic structure methods in complex systems. Methods that
exploit the intrinsic locality of molecular interactions have demonstrated encouraging
progress towards these goals.1–17
In particular, orbital-free embedded DFT (e-DFT) offers a formally exact
ap-proach to electronic structure theory in which the interactions between subsystems
are evaluated in terms of their electronic densities.1–4 Recent work has demonstrated that constructing the embedded subsystems from individual molecules leads to a
linear-scaling electronic structure approach that maps naturally onto
distributed-memory parallel computers,13,18 and it provides a systematic framework for calculat-ing electronic excited states in condensed phase systems.19,20 However, approximate treatments of the non-additive kinetic potential (NAKP) limit the accuracy of this
approach in applications involving strongly interacting subsystems.21 For example, severe artifacts in the structure of liquid water, including the complete absence of a
second peak in the oxygen-oxygen radial distribution function, have been predicted
co-valently bonded embedded subsystems have also been shown to qualitatively fail.21–23 The development of improved methods to address the NAKP problem will open new
doors for on-the-fly, massively parallel electronic structure calculations in general,
condensed-phase systems.
In this paper, we describe progress towards the development of accurate, scalable
treatments for the NAKP in e-DFT. We provide the first molecular applications of
our recently developed Exact Embedding (EE) method,24 demonstrating that it suc-cessfully describes the breaking of covalent bonds and hydrogen bonds with chemical
accuracy. Additionally, we introduce and numerically demonstrate a pairwise
ap-proximation to the NAKP, which allows for the scalable implementation of the EE
method in large systems. Benchmark calculations are presented for systems with up
to 125 molecules, demonstrating that parallel implementation of the method enables
constant system-size scaling of the wall-clock calculation time.
2.2
Theory
2.2.1
Orbital-Free Embedded DFT
We utilize the orbital-free e-DFT formulation of Cortona1and Wesolowski and cowork-ers.2,3 For the case in which the total electronic density ρAB is partitioned into two
subsystems, ρAB =ρA+ρB, the corresponding one-electron orbitals obey the
Kohn-Sham Equations with Constrained Electron Density (KSCED),3
−1
2∇
2+v
eff[ρA, ρAB;r]
φAi (r) =Ai φAi (r) (2.1)
−1
2∇
2
+veff[ρB, ρAB;r]
where i= 1, . . . , NA, j = 1, . . . , NB, and NA and NB are the number of electrons in the respective subsystems. veff is the effective potential for the coupled one-electron
equations, such that
veff[ρA, ρAB;r] = vne(r) +vJ[ρAB;r] +vxc[ρAB;r]
+ vnad[ρA, ρAB;r], (2.3)
where the Nnuc nuclei occupy positions {Ri},
vne(r) = −
Nnuc
X i
Zi |r−Ri|
, (2.4)
vJ[ρ;r] =
Z ρ(r0) |r0 −r|dr
0
, (2.5)
vxc[ρ;r] =
δExc[ρ]
δρ
(r), (2.6)
and Exc[ρ] is the exchange-correlation functional. vnad[ρA, ρAB;r] is the potential due
to the non-additive kinetic energy for non-interacting electrons, such that
vnad[ρA, ρAB;r] =
δTnad
s [ρA, ρB]
δρA
(r), (2.7)
where Tsnad[ρA, ρB] ≡ Ts[ρAB]−Ts[ρA]−Ts[ρB]. The subsystem densities are
con-structed from the corresponding KS orbitals, using ρA(r) =
NA X
i=1
|φA
i (r)|2 and ρB(r) =
NB
X j=1
|φB
j(r)|
2. Eqs. 3.1-2.7 are easily generalized for the e-DFT description of multiple
embedded subsystems.1,18
Two aspects of e-DFT are worth emphasizing. Firstly, like conventional
Kohn-Sham (KS)-DFT, it is a theory that is exact in principle, but practical calculations
must employ approximations to the unknown exchange-correlation functional.
intro-duces the NAKP, vnad[ρA, ρAB;r], since the one-electron orbitals for subsystem A
are not necessarily orthogonal to those of subsystem B. Without knowledge of the
exact functional for the non-interacting kinetic energy, this creates a second source
of approximation in e-DFT calculations. The significance of the NAKP is
system-dependent, with the most severe cases including those for which the subsystem
den-sities greatly overlap; no approximate kinetic energy functional has been previously
demonstrated to yield accurate results for embedded subsystems that are connected
by covalent bonds.3,21,22,25,26
2.2.2
Exact Calculations of NAKP
We have recently developed the Exact Embedding (EE) method to calculate the
NAKP.24 The general method can be summarized for two embedded subsystems as follows: A Levy constrained search (LCS)27 or equivalent technique is first used to determine the full set of orthogonal KS orbitals, {φAB
i }, that correspond to the total
density ρAB from the latest iteration of Eqs. 3.1-2.3. Then, from the KS orbitals
{φAB
i },{φ
A
i }, and{φ
B
i}, the corresponding kinetic potentials are calculated using the
exact result of King and Handy,28
vTs(r) =
Pn i=1(−
1
2φi(r)∇ 2φ
i(r))−iφi(r)2)
ρ(r) +µ, (2.8)
where n is the number of occupied orbitals, i is the KS eigenvalue corresponding to
orbital φi, and µ is a constant. Finally, the NAKP needed for the next iteration of
Eqs. 3.1-2.3 is calculated directly from the difference
vnad[ρA, ρAB;r] =vTABs (r)−v
A
where the superscripts in this equation indicate the orbital set to which each kinetic
potential corresponds.
Rather than explicitly performing the LCS, we use the equivalent protocol of Zhao,
Morrison, and Parr (ZMP)29–31to obtain the exact non-interacting kinetic energy and the KS orbitals {φABi }. This requires solution of the following one-electron equations
−1
2∇
2
+vext(r) +vcλ(r)
φABi,λ(r) =ABi,λφABi,λ(r) (2.10)
in the limit λ→ ∞, wherei= 1, . . . ,(NA+NB), and
vcλ(r) = λ Z
ρ(r0)−ρAB(r)
|r0−r| dr 0
. (2.11)
vext(r) corresponds to any well-behaved external potential,30,31 and various choices
for this potential are described in Sec. III B. In practice, Eq. 2.10 is solved at several
large, finite values ofλ, and the KS orbitals and eigenvalues, as well as the final non-interacting kinetic energy, are obtained via extrapolation.29–31 In Sec. V, we discuss a technique to robustly implement the ZMP step for NAKP calculations in large
systems.
The EE method outlined in Eqs. 2.8 - 2.11 is unique in that it allows for the
for-mally exact calculation of the total electronic density within the e-DFT framework,
using integer orbital occupancies and without approximations to the NAKP. The
method was previously demonstrated for atomic systems with strongly overlapping
subsystem densities,24and the current paper presents its first molecular applications. We note that several other groups have also used density inversion techniques to
cal-culate the NAKP, assuming that the total electron density is already available from
![Figure 1.1: The difference between the non-interacting kinetic energy Ts[ρ] fromKS-DFT and from the ZMP method, plotted as a function of γ](https://thumb-us.123doks.com/thumbv2/123dok_us/8816261.920515/29.612.206.446.311.484/figure-dierence-interacting-kinetic-energy-fromks-plotted-function.webp)
![Figure 1.3: The KSCED effective potential, V KSCEDeff[ρA, ρB; r], for (A) the Q−0.52.5ion, (B) the Li atom, (C) the Ne+7 ion, and (D) the Be atom and the NAKP,vnad[ρA, ρB; r], for (E) the Q−0.52.5ion, (F) the Li atom, (G) the Ne+7 ion, and (H)the Be atom using the e-DFT protocol presented here.](https://thumb-us.123doks.com/thumbv2/123dok_us/8816261.920515/35.612.202.444.149.434/figure-ksced-eective-potential-kscede-using-protocol-presented.webp)