INTRODUCTION
The maintenance of adult tissue mass can be controlled by the differentiation of adult stem cells or by the replication of differentiated cells in the tissue. In the case of pancreatic beta cells, recent studies have shown that tissue homeostasis relies on the replication of differentiated, insulin-expressing beta cells, rather than stem cells (Brennand et al., 2007; Dor et al., 2004; Georgia and Bhushan, 2004; Meier et al., 2008; Nir et al., 2007; Teta et al., 2007). Furthermore, it has been shown that the rate of beta cell proliferation responds to certain physiological conditions, such as pregnancy and the destruction of most beta cells (Cano et al., 2008; Gupta et al., 2007; Nir et al., 2007; Parsons et al., 1992). Despite these findings, it remains unclear what factors govern adult beta cell proliferation and to what extent beta cell mass can be expanded in vitro and in vivo.
Understanding the processes that regulate beta cell replication might have potential therapeutic value for type 1 and type 2 diabetes, diseases that are characterized by insufficient beta cell mass (Butler et al., 2007). Although transplantation of cadaveric human islets can normalize blood glucose levels in type 1 diabetes patients, the scarcity of donors limits this therapy (Shapiro et al., 2006). Expanding donor islets in vitro or in vivo by activating beta
cell replication would have a significant impact on the utility of clinical islet transplantation. A second therapeutic solution involves the use of external stimuli to induce beta cell regeneration in vivo. To succeed, both approaches require significant beta cell expansion, probably necessitating numerous divisions of each individual beta cell. However, it is unclear whether a replicating beta cell has the capacity to divide again and what might control this decision.
Several studies have shown that all beta cells have a similar replicative potential, with no sub-population of replication-privileged cells (Brennand et al., 2007; Teta et al., 2007). This suggests that replicated beta cells are no more likely to divide again than undivided beta cells. Further analysis suggested that, post-division, beta cells become dramatically less likely to re-enter the cell cycle, as they re-enter a prolonged ‘refractory period’, estimated in months, during which they cannot divide again (Teta et al., 2007). This idea was supported by a recent study suggesting that a single beta cell undergoes only two or three replications over the course of its life time (Desgraz and Herrera, 2009).
The limited replicative capability of a single beta cell contrasts with reports that indicate a dramatic increase in beta cell mass from birth to maturity (Dor et al., 2004; Finegood et al., 1995). Additionally, beta cell mass is known to undergo a several-fold expansion during regeneration and in response to physiological demand (Cano et al., 2008; Kulkarni et al., 2004; Nir et al., 2007). To understand the mechanisms of beta cell expansion under conditions of both normal and compensatory beta cell division, it is crucial to characterize the proliferative capacity of a single beta cell, and to investigate the nature of the beta cell replication refractory period (Teta et al., 2007).
Here we describe a novel and broadly applicable pulse-chase assay designed to study the post-replication dynamics of cells in vivo. Using this assay we can identify and study cells that have replicated, exited mitosis and returned to cycle (‘re-entered cells’). We report that dividing beta cells in adult mice quickly re-enter the Development 137, 3205-3213 (2010) doi:10.1242/dev.054304
© 2010. Published by The Company of Biologists Ltd
1Department of Developmental Biology and Cancer Research and Molecular Biology, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel. 2Department of Systems Biology, Harvard Medical School, Boston, MA 02115, USA. 3Cavendish Laboratory, Department of Physics, J. J. Thomson Avenue, University of Cambridge, Cambridge CB3 0HE, UK. 4Department of Stem Cell and Regenerative Biology, Howard Hughes Medical Institute, Harvard Stem Cell Institute, Harvard University, 7 Divinity Avenue, Cambridge, MA 02138, USA. 5Department of Metabolic and Vascular Diseases, Hoffmann-La Roche, Nutley, NJ 07110, USA.
*Present address: Developmental Biology Program, Sloan-Kettering Institute, 1275 York Avenue, New York, NY 10021, USA.
†Author for correspondence (yuvald@ekmd.huji.ac.il)
Accepted 23 July 2010
SUMMARY
Pancreatic beta cell proliferation has emerged as the principal mechanism for homeostatic maintenance of beta cell mass during adult life. This underscores the importance of understanding the mechanisms of beta cell replication and suggests novel approaches for regenerative therapy to treat diabetes. Here we use an in vivo pulse-chase labeling assay to investigate the replication dynamics of adult mouse beta cells. We find that replicated beta cells are able to re-enter the cell division cycle shortly after mitosis and regain their normal proliferative potential after a short quiescence period of several days. This quiescence period is lengthened with advanced age, but shortened during injury-driven beta cell regeneration and following treatment with a pharmacological activator of glucokinase, providing strong evidence that metabolic demand is a key determinant of cell cycle re-entry. Lastly, we show that cyclin D2, a crucial factor in beta cell replication, is downregulated during cell division, and is slowly upregulated post-mitosis by a glucose-sensitive mechanism. These results demonstrate that beta cells quickly regain their capacity to re-enter the cell cycle post-mitosis and implicate glucose control of cyclin D2 expression in the regulation of this process.
KEY WORDS: Beta cells, Regeneration, Diabetes, Mouse
Glucose and aging control the quiescence period that
follows pancreatic beta cell replication
Seth J. Salpeter1, Allon M. Klein2,3, Danwei Huangfu4,*, Joseph Grimsby5and Yuval Dor1,†
D
E
V
E
LO
P
M
E
N
pool of replication-competent cells and are able to divide again a few days after mitosis. Clonal analysis of beta cells independently supports this finding. We show that organismal aging lengthens the time needed for replicated beta cells to return to the cell cycle; by contrast, beta cell injury and glucose metabolism shorten the post-mitosis quiescence period of beta cells and increase the likelihood of individual beta cells to divide again.
MATERIALS AND METHODS Mice
Animal care and experiments were approved by the Institutional Animal Care and Use Committee of the Hebrew University. We used either ICR mice from Harlan Israel or Insulin-rtTA; TET-DTA mice (Nir et al., 2007) (bDTA) that were given doxycycline for 7 days in the drinking water [200
g/ml doxycycline in 2% (w/v) sucrose] at 4 weeks of age.
BrdU labeling was performed at 4-5 or 6 weeks of age in ICR orbDTA mice, respectively. In the experiement on older mice, 3-month-old ICR mice were used. BrdU (Sigma-Aldrich) was dissolved in PBS and injected intraperitoneally (i.p.) (1 mg/g body weight) three times a day every 3 hours for 2 days. Mice were sacrificed 16 hours, 2 days, 5 days, 1 week and 2 weeks after the last injection. In experiments with a single pulse, BrdU was injected once and mice were sacrificed after 4, 28 and 48 hours. Glucokinase activator (GKA) (Grimsby et al., 2003) was diluted in saline containing 20% DMSO and 1% Tween 80 and injected i.p. at 0.04 mg/g body weight.
Immunostaining
Pancreas was fixed with 4% buffered formaldehyde for 4 hours. Paraffin sections (5 m) were rehydrated and antigen retrieval was performed using a Biocare pressure cooker and citrate buffer. The following primary antibodies were used: guinea pig insulin (1:500, DAKO), rabbit anti-Ki67 (1:200, Neomarkers), mouse anti-BrdU (1:300, Amersham/GE Healthcare), rabbit cyclin D2 (1:400, Santa Cruz) and rabbit anti-pHH3 (1:100, Cell Signaling). For DNA counterstaining on acinar tissue, Sytox (1:500, Invitrogen) was used. Secondary antibodies were from Jackson ImmunoResearch: anti-guinea pig Cy2 (1:200), anti-mouse Cy3 (1:500), anti-rabbit Cy5 (1:500). Immunofluorescence images were captured using a Nikon C1 confocal microscope. Four mice were used for each data point. Approximately 30 islets or 3000 beta cells were counted per 1-month-old animal and 50 islets or 8000-10,000 cells in 3-month-old animals using ImageJ.
Statistics
Statistical analyses were performed using a two-tailed Student’s t-test (P<0.05). Results are reported with a mean standard deviation.
Modeling
Predictions for the likelihood of cell cycle re-entry (Fig. 1B,C) were generated by simulation of an expanding cell population, implemented in MATLAB (MathWorks). The predictions were further confirmed by theoretical analysis (see Fig. S6 in the supplementary material). In the simulation, each cell is assigned a life time between birth and mitosis that is drawn at random from a cell cycle distribution with a refractory period that is either short (days) or long (months) compared with the average cell division rate (see Fig. S6 in the supplementary material). To mimic the action of BrdU labeling, all cell divisions occurring within a short period of time during the simulation are ‘labeled’. The likelihood of cell cycle re-entry is then calculated as the ratio of the division rates (i.e. the number of cell divisions per cell per unit time) of the labeled and total cell populations. The simulations assume no cell loss and a steady decline in the average cell division rate.
RESULTS
An assay for studying cell cycle re-entry of recently replicated cells
To identify previously divided cells that have returned to the cell cycle, we developed a pulse-chase protocol based on the thymidine analog BrdU. One-month-old mice were injected with BrdU (six
injections over 48 hours – the pulse period) to label replicating cells, and sacrificed at one of several subsequent time points (the chase period). Beta cells that replicated during the pulse period should stain for BrdU, whereas beta cells that were dividing at the time of sacrifice should stain for the general cell cycle marker Ki67 (Gerdes et al., 1991; Key et al., 1993). The fraction of recently replicated beta cells that re-entered the cell cycle can be determined by staining beta cells for BrdU and Ki67 at the chase time periods and calculating the percentage of BrdU+Ki67+cells among the BrdU-labeled population (Fig. 1A). By comparing the likelihood of replication among the general beta cell population and among pulse-labeled beta cells, this novel pulse-chase assay can distinguish between models of beta cell replication dynamics. Moreover, it can provide an accurate estimate of the duration of the post-replication refractory period of beta cells and, therefore, of the time necessary for a replicated cell to re-enter the pool of replication-competent cells. We generated a simple model (Fig. 1B) to demonstrate the expected behavior of beta cells if there were no refractory period, if there were a short refractory period, or if there were a prolonged refractory period, as has been suggested (see Introduction). To distinguish between these possibilities, we can compare the fraction of all replicating beta cells (percentage Ki67+ beta cells; here termed the normal cell division rate) with the fraction of currently replicating beta cells among the population of BrdU+beta cells (percentage BrdU+Ki67+ among BrdU+ beta cells; here termed the replicated cell division rate). If, at a given time after pulse, the replicated cell division rate is less than the normal cell division rate, this means that replicated beta cells exhibit a replication refractory period. The first chase time when the two are equal represents the point at which the likelihood of a replicated beta cell to re-enter the cell cycle equals the likelihood of a non-divided cell to divide, and therefore represents the end of the refractory period (Fig. 1C).
We performed control experiments to establish the validity of our assay. We first assessed whether Ki67 is indeed expressed and then downregulated post-mitosis in BrdU-labeled beta cells. Four hours after a single injection of BrdU, all BrdU+cells were Ki67+ (Fig. 1D,F). Twenty-eight hours after the injection, 15% of BrdU+ cells expressed Ki67 and 48 hours after the injection only 1% co-stained for BrdU and Ki67. This demonstrates that Ki67 is downregulated in BrdU-labeled cells post-mitosis. Therefore, a subsequent increase in the percentage of co-stained cells must represent cells that have re-entered the cell cycle. Furthermore, to quantify exactly when BrdU-labeled beta cells exited the cell cycle, we co-stained for BrdU+ and phospho-histone H3 (pHH3), a replication marker that is immediately downregulated post-mitosis (see Fig. S1A,B in the supplementary material). Three hours after a single injection of BrdU, nearly all BrdU+beta cells co-stained for pHH3, whereas at 12 hours post-injection only 40% of BrdU+ beta cells co-stained for pHH3. By 24 hours post-injection, almost no BrdU+cells co-stained for pHH3, indicating that by this time all BrdU+cells have exited the cell cycle.
In addition, we verified that beta cells incorporating BrdU undergo productive mitosis. We counted the percentage of BrdU+ beta cells present at 4, 28 and 48 hours after a single pulse. If most BrdU-labeled cells divide, the percentage of BrdU+beta cells should roughly double after mitosis, when one cell becomes two. Indeed, at 4 hours we found 2.5% BrdU+beta cells, whereas by 48 hours we detected 4.2% BrdU+ beta cells (Fig. 1E), suggesting that most BrdU-labeled beta cells undergo mitosis. Furthermore, we quantified the percentage of BrdU+cells found in doublets 4 hours and 48 hours after a BrdU pulse (see Fig. S2
D
E
V
E
LO
P
M
E
N
in the supplementary material). We performed our analysis by confocal microscopy on paraffin sections stained for insulin and BrdU. Theoretically, we expected to observe about one-third of BrdU+cells in doublets, reflecting cell divisions that fall within the confocal optical plane, but not above or below the plane. By 48 hours post-injection, 40% of BrdU+ cells were found in doublets (see Fig. S2 in the supplementary material). These data further confirm that most BrdU-labeled beta cells undergo productive mitosis.
Replicated beta cells regain replicative potential after a short period of quiescence
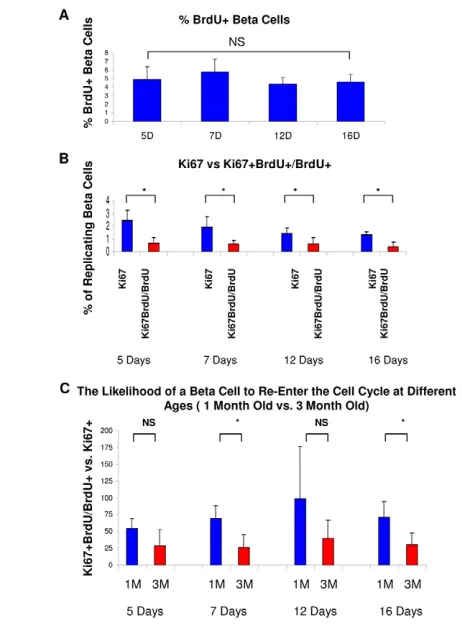
After performing the 2-day pulse protocol, we analyzed the percentage of BrdU+beta cells at the chase time periods to confirm the labeling of a large group of replicated beta cells. Two days after the last BrdU injection, over 11% of the beta cell population were BrdU+, representing an increase in BrdU labeling over day 0 and indicating that although almost all beta cells have exited the cell cycle by day 0, there is still a small amount of residual mitosis (Fig. 2A). At subsequent time points, the percentage of BrdU+cells
slightly declined, which we attribute both to the expansion of the total beta cell population and to the BrdU dilution effect caused by cells that returned to cycle.
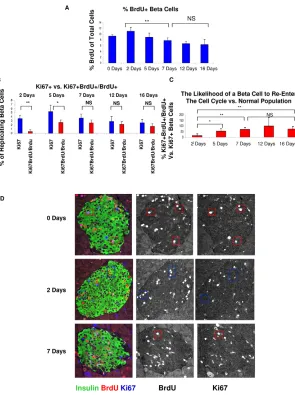
[image:3.612.53.328.58.504.2]To establish the percentage of ‘re-entered cells’ at each time point and the beta cell replication refractory period, we counted the percentage of Ki67+cells among the BrdU+beta cell population (replicated cell division rate) and compared it with the percentage of total beta cells expressing Ki67 (normal cell division rate) (Fig. 2B,D). Two days after the last BrdU injection, 0.5% of BrdU-labeled beta cells expressed Ki67, in contrast to the total population in which 3.5% of beta cells expressed Ki67, representing a significant difference and supporting the idea of a refractory period of beta cells. By day 5, the percentage of the BrdU+population expressing Ki67 increased to 2.5%, while the total beta cell population expressed Ki67 in 5% of cells, still yielding a significant difference between the two groups. However, by day 7, and again on days 12 and 16, there was no significant difference between the replicated cell division rate and the normal cell division rate. When the replicating cell division rate was compared with the normal cell division rate, on day 2 replicated beta cells were only 15% as likely
Fig. 1. An assay for measuring the beta cell post-replication quiescence period.(A)Outline of the pulse-chase experiment for analyzing the return of beta cells to the cell cycle. BrdU was injected a total of six times (three times every day for 2 days) and mice were sacrificed at the
indicated chase periods. (B)A computer-generated model
shows possible beta cell behaviors after replication. Green shading represents the normal population, in which beta cell replication declines with age. Black line represents a similar behavior of post-replicating beta cells and hence no quiescence/refractory period. Red line represents a short refractory period and the blue line represents a long refractory period, as proposed by Teta et al. (Teta et al.,
2007). (C)In our assay, the percentage of BrdU+Ki67+out
of the total BrdU+population (the division rate of replicated
cells) represents the likelihood of a replicated cell to divide again. When the replicated cell division rate immediately equals the normal rate (black, represented at 100%) there is no refractory period, when it recovers quickly there is a short refractory period (red), and when there is a long lag
there is a long refractory period (blue). (D)After mitosis,
dividing cells retain BrdU but lose expression of Ki67. Beta cells were pulsed with a single injection of BrdU. At 4 hours
all BrdU+beta cells expressed Ki67, but by 48 hours only
1% co-expressed BrdU and Ki67. (E)The percentage of
BrdU+cells in the total population approximately doubles
between 4 and 48 hours, consistent with productive cytokinesis of cells that incorporated BrdU.
(F)Representative images of beta cells 4 and 48 hours after
injection of BrdU, stained for insulin (green), BrdU (red) and
Ki67 (blue). The boxed regions indicate BrdU+cells and their
Ki67 expression **, P<0.01; NS, not significant.
D
E
V
E
LO
P
M
E
N
to replicate, yet by day 7 they were 70% as likely to replicate, and by day 12 they were equally as likely to replicate (Fig. 2C). Indeed, although there was a significant increase in the likelihood of cell cycle re-entry from days 2 to 5 and 5 to 7, there was no statistically significant increase in the likelihood from 7 days onward. These results indicate that within ~1 week after mitosis, a beta cell regains its normal potential to divide. These data are consistent with a short refractory period (Fig. 1B,C), after which previously divided beta cells re-enter the pool of beta cells that can respond to mitogenic stimuli.
To confirm these findings using an additional marker of cell cycle entry, we compared the replicated cell division rate with the normal cell division rate using pHH3 instead of Ki67. At day 7 post-BrdU, 0.75% of BrdU+beta cells were pHH3+, whereas 0.8% of the total beta cell population was pHH3+(see Fig. S3 in the supplementary material). This suggests that at day 7, BrdU+beta cells were 90% as likely to replicate as normal beta cells. These results again demonstrate that divided beta cells regain their normal replication rate after ~1 week.
To compare the beta cell re-entry behavior with that of a different cell population, we performed the pulse-chase assay analysis on pancreatic acinar cells. Two days after the last BrdU injection, the normal cell division rate and the replicated cell division rate were nearly identical at 1.5%, demonstrating that after
only 2 days replicated acinar cells are equally as likely to replicate as the normal acinar cell population (see Fig. S4 in the supplementary material). These results differ dramatically from the re-entry rate of the beta cell, which is only 15% 2 days after mitosis, showing that replicated acinar cells more rapidly regain their potential to divide.
Clonal analysis confirms a short quiescence period To further examine the ability of beta cells to re-enter the cell cycle, we analyzed the size of pulse-labeled clones derived from single beta cells at 1-2 months post-labeling (Fig. 3A). If the duration of the refractory period extends beyond the clone age of 1-2 months, then pulse-labeled cells should give rise only to one-cell and two-cell clones. By contrast, a shorter refractory period would allow successive divisions, leading to larger clones (Fig. 3B).
[image:4.612.55.350.58.453.2]For clonal analysis, we analyzed in detail the clone fate data originally presented by Brennand et al. (Brennand et al., 2007). This system indelibly labels individual beta cells with both GFP and RFP at extremely low efficiencies (0.1-0.5%) using a tamoxifen-dependent Cre-lox system. Mice were labeled at 4 to 8 weeks of age. At 4 days post-labeling, 90% of the islets containing labeled cells had only one labeled cell, and the remaining 10% contained two labeled cells. Therefore, given the low labeling efficiency, single cell-derived clones were defined as clusters of labeled cells within a single islet. Fig. 2. Beta cells can re-enter the cell cycle shortly after mitosis and proliferate normally after 1 week.(A)Beta cells were pulsed with six injections of
BrdU and the percentage of BrdU+beta cells was
analyzed at each time point. (B)The percentage of
Ki67+beta cells in the general population (blue bars)
and in the BrdU+pulse-labeled population (red bars).
Whereas at 2 and 5 days there was a significant difference in these two populations, by 7 days and
onwards there was no difference. (C)The percentage
BrdU+Ki67+/BrdU+population was divided by the
percentage Ki67+at each time point. Whereas on day
2 a replicated cell was only 15% as likely to replicate as the general population, by day 7 it was 70% as likely to replicate, and by day 12 it was equally as
likely. (D)Representative images demonstrating beta
cells pulse chased with BrdU and stained for insulin (green), BrdU (red) and Ki67 (blue) at 0, 2 and 7 days.
Red boxes indicate Ki67+BrdU+cells; blue boxes
point to BrdU+cells that are Ki67–(i.e. that were
quiescent at the time of sacrifice). The boxed regions
indicate BrdU+cells and their Ki67 expression.
*, P<0.05; **, P<0.01; NS, not significant.
D
E
V
E
LO
P
M
E
N
Following a 1- to 2-month chase period, clones were sampled and their size scored from single random sections (Fig. 3C) (Brennand et al., 2007). The results show that only 16 of 45 clones (35%) contained 1-2 cells following a 1-month chase period, and only 3 of 40 clones (8%) contained 1-2 cells after 2 months. Therefore, it appears that the majority of cells are capable of dividing at least twice within a period of 1 month. Remarkably, the clonal analysis also revealed the presence of larger clones, indicating that cells may divide repeatedly within the chase period. At 1 month post-labeling, 5 out of 45 clones (11%) contained at least 10 cells, and at 2 months post-labeling 6 out of 40 clones (15%) contained at least 16 cells. The largest clones at 1 month contained 26 cells, suggesting that a single cell might occasionally divide four or five times within this time period, or approximately once per week. Taken together, the clone fate data provide independent support for the notion that replicated beta cells rapidly regain their ability to re-enter the cell cycle. Furthermore, these results dramatically demonstrate that beta cells in adult mice are capable of multiple divisions.
The post-replication quiescence period is prolonged with age
The rate of beta cell replication drops dramatically with age in rodents and humans (Chen et al., 2009; Dhawan et al., 2009; Finegood et al., 1995; Krishnamurthy et al., 2006; Meier et al.,
[image:5.612.309.540.58.371.2]2008; Rankin and Kushner, 2009; Teta et al., 2005; Tschen et al., 2009; Wong et al., 2009). However, it is not known how age affects the likelihood of an individual beta cell to divide again after it had completed mitosis. To examine the role of aging in the regulation of cell cycle re-entry of beta cells, we performed our pulse-chase assay on older mice. Given the necessity of labeling a significant population of replicating beta cells with BrdU, we chose 3-month-old mice, which still maintain a considerable rate of beta cell replication. Mice were injected with BrdU according to our standard protocol (Fig. 1A) and sacrificed at 5, 7, 12 and 16 days. At 5 days, 5% of all beta cells were BrdU+(Fig. 4A). Interestingly, the percentage of BrdU-labeled cells did not significantly decline during the remainder of the chase period, supporting the assertion that the decline in BrdU+cells found in the 1-month chase period resulted from a significant beta cell expansion while the BrdU+ cells were in the refractory period.
Fig. 3. Clonal analysis using RipCreER-MADM mice supports multiple divisions of the same beta cell and a short post-replication quiescence period.(A)Outline of the pulse-chase labeling experiment. Mice were activated with Tamoxifen between 4 and 8
weeks and chased for 1 and 2 months. (B)A model of short- and
long-term quiescence periods. Short quiescence periods should quickly yield
numerous labeled cells. (C)Distribution of clonal sizes at 1 and 2
months. At both time points there were large clones (more than 10 cells), strongly suggesting that beta cells can undergo several sequential divisions.
Fig. 4. Older beta cells have a longer quiescence period.
(A)Three-month-old mice were pulsed with six injections of BrdU
and analyzed at the same time points as 1-month-old mice. (B)The
percentage of Ki67+beta cells in the general population (blue bars)
and in the BrdU+pulse-labeled population (red bars). In contrast to
1-month-old mice, in which at 2 and 5 days there was a significant difference between these two populations but after 7 days there was not, in 3-month-old mice there was a significant difference between the normal population and replicated population at all time points.
(C)The percentage BrdU+Ki67+/BrdU+population was divided by the
percentage Ki67+at each time point for 1- and 3-month-old mice.
Whereas replicated cells in young mice were 70% as likely to replicate on day 7 and equally as likely by day 12, replicated beta cells in older mice were 25% as likely to replicate on day 7 and 40%
as likely to replicate on day 12. *, P<0.05; NS, not significant.
D
E
V
E
LO
P
M
E
N
[image:5.612.53.264.58.341.2]We then quantified the replicated cell division rate (the percentage of Ki67+cells among the BrdU+population) and the normal cell division rate (the percentage of Ki67+among the total beta cell population), at each of the chase time periods (Fig. 4B). Whereas in 1-month-old mice at 7 days of chase there was no significant difference between the two rates, in 3-month-old mice the replicated cells showed significantly lower cell division throughout the entire chase period. Furthermore, whereas replicated cells in young mice were 70% as likely to replicate on day 7 and equally as likely by day 12, replicated beta cells in older mice were 25% as likely to replicate on day 7, 40% as likely to replicate on day 12, and 30% as likely to replicate on day 16, as compared with the general population (Fig. 5C). Thus, although beta cells in older mice can, in principle, re-enter the cell cycle rapidly after cell division, they are less likely to do so than beta cells in younger mice.
Regenerating and glucose-stimulated beta cells have a shortened quiescence period
We next investigated whether the duration of the beta cell post-replication quiescence period is under physiological control, and specifically whether it could be shortened in response to specific stimuli. First, using a transgenic mouse model for conditional ablation of beta cells (Nir et al., 2007), we examined the beta cell quiescence period in regenerating islets. Treatment of 4-week-old Insulin-rtTA; TET-DTA mice with doxycycline for 1 week caused
hyperglycemia (blood glucose of 550 mg/dl on average, compared with 140 mg/dl on average in control littermates) and ablation of the majority of beta cells. Mice were then injected with BrdU as described above (Fig. 1A) and sacrificed 2 and 5 days after the last injection. Surprisingly, after just 2 days the replicated cell division rate (the percentage of Ki67+cells among the BrdU+population) equaled the normal cell division rate (the percentage of Ki67+cells among the total beta cell population), each proliferating at 6% (Fig. 5A,C). This phenotype continued on day 5, when both groups again proliferated at 6%. When the replicating cell division rate was compared with the normal cell division rate, on day 2 and again on day 5 the replicated cells were equally as likely as normal cells to enter the cell cycle (Fig. 5B). These results indicate that in contrast to the normal adult beta cell, which requires 7 days to regain its normal proliferative capacity, regenerating beta cells almost immediately recover from replication, essentially eliminating the quiescence period.
[image:6.612.56.365.55.436.2]We hypothesized that the shortened beta cell post-replication quiescence period might be a result of increased glucose signaling due to hyperglycemia in Insulin-rtTA; TET-DTA mice. To determine whether higher glucose levels can shorten the beta cell post-replication quiescence period, we treated mice with a pharmacological activator of glucokinase, glucokinase activator (GKA), shown previously to reduce the S0.5 for glucose and increase the Vmax of glucokinase in beta cells (Grimsby et al., 2003). This drug causes mild systemic hypoglycemia (due to
Fig. 5. Beta cell regeneration and glucose stimulation shorten the beta cell quiescence period. (A)The percentage of beta cells expressing Ki67 in the general population was compared with the
percentage of Ki67+beta cells among the BrdU+
population at each time point. In beta cells of Insulin-rtTA; TET-DTA mice, there was no evidence for a
quiescence period at 2 or 5 days after division. (B) The
percentage BrdU+Ki67+/BrdU+population was divided
by the percentage Ki67+at both time points,
demonstrating that in regenerating mice a replicated beta cell regains its normal proliferation rate already
after 2 days. (C)Representative images from islets of
Insulin-rtTA; TET-DTA mice pulsed with BrdU and chased for 2 days. Insulin (green), BrdU (red), Ki67 (blue). The
boxed regions indicate BrdU+cells and their Ki67
expression. (D) Glucokinase activator (GKA) transiently
reduces blood glucose levels due to enhanced glycolysis
in beta cells and increases insulin secretion. (E)The
percentage of Ki67+beta cells among the BrdU+
population increases after GKA injection. (F)GKA
injection restores the ability to re-enter the cell cycle in
beta cells that completed mitosis. **, P<0.01; NS, not
significant.
D
E
V
E
LO
P
M
E
N
increased insulin secretion), but within the beta cell it increases glycolysis and mimics the effect of systemic hyperglycemia. After six injections of BrdU over 2 days, mice were injected with vehicle or GKA and sacrificed 24 hours later. As expected, GKA treatment lowered the average blood glucose levels from 115 to 45 mg/dl (Fig. 5D). Mice receiving the vehicle demonstrated a 0.25% replicated cell division rate and a 3.5% normal cell division rate (comparable to values for wild-type mice not receiving vehicle). By contrast, mice receiving GKA exhibited a 2.8% replicated cell division rate and a 4% normal cell division rate (Fig. 5E). When compared as a ratio, vehicle-treated replicated beta cells were 5% as likely to divide as normal beta cells, whereas replicated beta cells of mice receiving GKA were 70% as likely to divide again compared with the normal beta cell population (Fig. 5F). Similar results were obtained when mice were sacrificed 2 days after GKA administration, when the glucose-lowering effect had vanished (see Fig. S5 in the supplementary material).
These results show that an increased rate of glucose metabolism shortens the post-replication quiescence period of beta cells. Furthermore, they suggest that the dramatic shortening of the quiescence period in regenerating beta cells in the Insulin-rtTA; TET-DTA model is a result of hyperglycemia.
Cyclin D2 is downregulated during beta cell replication and slowly returns to basal levels by a glucose-sensitive mechanism
We considered possible molecular mechanisms that could account for the regulation of the quiescence period. Cyclin D2 is essential for beta cell proliferation (Georgia and Bhushan, 2004; Kushner, 2006; Kushner et al., 2005). Although the dynamics of cyclin D2 expression during cell division have not been studied before, cyclin D1 is known to be downregulated in vitro during the S/G2 phases of the cell cycle in other cell types (Baldin et al., 1993; Lukas et al., 1994). We hypothesized that such a phenomenon could occur in replicating beta cells in vivo, and that low levels of cyclin D2 might persist after mitosis. This in turn could account for the quiescence period by preventing beta cells from returning to the cell cycle until cyclin D2 is re-expressed.
To examine cyclin D2 expression during the cell cycle of beta cells in vivo, we injected 1-month-old ICR mice with BrdU and sacrificed after 2 hours. Beta cells that incorporated BrdU co-stained for cyclin D2 25% of the time, as compared with the quiescent beta cell population which stained positive 80% of the time, indicating that cyclin D2 is downregulated during the S phase of replicating beta cells in vivo (Fig. 6A,B).To further investigate the regulation of cyclin D2 during the cell cycle and post-mitosis, we injected mice with BrdU and sacrificed at later time points up to 350 hours post-BrdU (Fig. 6B). Strikingly, replicating beta cells remained largely cyclin D2-negative even after mitosis had ended, and very slowly started to re-express the gene. At 7 and 23 hours post-labeling, only ~20% of BrdU-positive beta cells expressed cyclin D2; 30 hours after labeling 40% of BrdU-positive cells expressed cyclin D2, and at 150 hours post-labeling 70% of BrdU-positive cells co-stained for cyclin D2. As beta cells need cyclin D2 to replicate, these results suggest that the decrease in beta cell cyclin D2 during cell division and immediately after mitosis prevents cells from re-entering the cell cycle. This assertion is strengthened by the correlation between the time necessary for the replicated beta cell population to regain its normal ability to replicate (~7 days) and the time necessary for the replicated beta cells to express levels of cyclin D2 found in quiescent cells (150 hours, or 6 days).
[image:7.612.327.531.56.398.2]If cyclin D2 is indeed a key factor in the control of beta cell cycle re-entry, our models with a shortened beta cell quiescence period should show a faster post-mitotic return of normal cyclin D2 levels. By contrast, beta cells in older mice (having a lengthened refractory period) should exhibit a slower return of cyclin D2. To investigate whether hyperglycemia induces faster upregulation of cyclin D2, we compared vehicle- and GKA-treated mice on day zero (16 hours after the last BrdU injection). Control mice expressed cyclin D2 in just 28% of BrdU+cells, in contrast to the quiescent beta cell population in which 80% of the cells expressed cyclin D2 (Fig. 6C). By contrast, on day zero, 52% of the BrdU+ beta cells in regenerating Insulin-rtTA; TET-DTA mice were cyclin D2+. Similarly, in mice injected with GKA 24 hours before sacrifice 58% of BrdU+cells expressed cyclin D2. In both Insulin-rtTA; TET-DTA and GKA models, the fraction of BrdU+cells that expressed cyclin D2 was significantly higher than in untreated mice. To examine whether beta cells in older mice upregulated cyclin D2 at a slower rate after mitosis, we compared the Fig. 6. Cyclin D2 is downregulated during the S/G2 phases and slowly returns post-mitosis via a glucose-regulated pathway.
(A)Pancreas sections stained for insulin (green), cyclin D2 (red) and BrdU
(blue) after a 2-hour pulse with BrdU. The boxed regions indicate BrdU+
cells and their cyclin D2 expression. (B) Beta cells were pulsed with a
single injection of BrdU and the percentage of cyclin D2+cells among
BrdU+beta cells was measured at each time point. (C)Replicated (BrdU+)
beta cells in Insulin-rtTA; TET-DTA mice and in GKA-treated mice regain
cyclin D2 expression faster than BrdU+beta cells in control mice.
(D)Replicated (BrdU+) beta cells in 1- and 3-month-old mice have similar
dynamics of cyclin D2 expression. **, P<0.01; NS, not significant.
D
E
V
E
LO
P
M
E
N
percentage of cyclin D2+BrdU+cells in 1- and 3-month-old beta cells on day 5 (Fig. 6D). At both ages, beta cells expressed cyclin D2 in 70% of Brdu+cells, suggesting that the return of cyclin D2 levels post-mitosis occurs at a similar rate in each.
These data suggest that metabolic demand might control the replication refractory period of beta cells via the rate of cyclin D2 return post-mitosis. Further work will be required to test this idea. However, aging is likely to affect the replication refractory period via other mechanisms.
DISCUSSION
We conclude that adult beta cells are capable of multiple sequential divisions and, post-mitosis, quickly regain their potential to replicate. Using our pulse-chase assay and clonal analysis, we show that post-mitotic beta cells re-enter the pool of replication-competent cells after a short quiescence period lasting ~1 week. This recovery period is lengthened in older beta cells, but can be shortened by physiological conditions that increase the rate of glucose metabolism, possibly by upregulating cyclin D2.
Previous reports have demonstrated that replication is the primary method for beta cell expansion and regeneration (Brennand et al., 2007; Dor et al., 2004; Georgia and Bhushan, 2004; Meier et al., 2008; Nir et al., 2007; Teta et al., 2007), but the replicative potential of a single beta cell has remained unclear. The large clusters of labeled beta cells in our pulse-chase clonal analysis demonstrate that single beta cells are capable of numerous divisions. Furthermore, in BrdU pulse-chase experiments, the appearance of post-mitotic cells that have re-entered the cell cycle demonstrates the ability of a beta cell to replicate multiple times. These findings are consistent with previous studies showing dramatic beta cell mass expansion from birth to maturity, necessitating multiple divisions of individual beta cells (Finegood et al., 1995). Very recently, a report employing clonal analysis of Ngn3+endocrine progenitor cells suggested that beta cells replicate only once or twice throughout the life time of a mouse (Desgraz and Herrera, 2009). This estimation differs considerably from our findings, and is difficult to reconcile with the significant expansion and turnover of beta cells during postnatal life. The reason for the differences between the two studies remains to be determined.
Our study shows that after a short period of quiescence lasting ~7 days, replicated beta cells from 1-month-old mice are equally as likely to divide again when compared with the normal beta cell population. Although we do not find any significant difference in the likelihood of re-entry after day 7, we cannot rule out the possibility of a slight increase in the likelihood beyond day 7. Additionally, there might be a small population of cells with a longer refractory period that is difficult to detect with our assay. Indeed, clonal analysis showed multiple very small clones that might be representative of cells that remain in an extended period of quiescence. However, the presence of small clones is equally consistent with a normal distribution of cells dividing at random. Furthermore, even though there is a possibility that some cells have entered a long refractory period, they would represent a very small proportion of the beta cell population given that the overall likelihood of a beta cell to re-enter the cell cycle returns to normal after ~1 week.
Recently, using a novel assay of serial thymidine analog labeling, Teta et al. demonstrated that all beta cells maintain equal proliferative potential and suggested that, post-replication, beta cells enter a long refractory period of several months (Teta et al., 2007). Our results are consistent with their study, as we have demonstrated a short refractory period in 1-month-old mice,
whereas their studies were performed on 3-month-old mice. Indeed, at 3 months of age we also see an extended refractory period. However, we believe that our method, which directly measures the time to cell cycle re-entry and relies on two very different epitopes (BrdU and Ki67, in contrast to CldU and IdU in Teta et al.), is a more straightforward assay for quantifying the duration of the refractory period, as the continuous labeling assay might lead to an overestimate of the true period length (see Fig. S6 in the supplementary material).
The lengthened quiescence period in older mice and the shortened quiescence period in response to beta cell ablation and GKA stimulation provide a novel insight into the mechanisms of beta cell proliferation. Previous studies have demonstrated that ablation can increase the overall rate of beta cell division (Cano et al., 2008; Nir et al., 2007), whereas aging decreases the rate of beta cell replication (Finegood et al., 1995; Krishnamurthy et al., 2006; Meier et al., 2008; Rankin and Kushner, 2009; Tschen et al., 2009). Our work suggests that beyond the direct replicative effect that glucose, regeneration and aging have on beta cells, these factors might also regulate the pool of cells that can replicate by controlling the recovery time of quiescent post-mitotic cells. More generally, our results raise the idea that the duration of a post-mitosis refractory period could be a control point in tissue dynamics. For example, it would be interesting to examine whether tumor cells differ from their cell of origin in their post-replication quiescence period. If such differences exist, the underlying molecular mechanisms might provide an interesting target for intervention.
Notably, we found that beta cell regeneration in the Insulin-rtTA; TET-DTA model, and simulation of glycolysis with GKA, considerably shorten the quiescence period, potentially by upregulating the expression of cyclin D2 from the low levels present during and after replication. Previous studies have demonstrated that cyclin D2 is important for beta cell replication, explaining why recently replicated beta cells lacking this protein might be unable to re-enter the cell cycle. Interestingly, cyclin D2 is not necessary for acinar cell replication, suggesting a possible explanation for why acinar cells do not maintain a detectable refractory period. Several studies have demonstrated that the activation of pathways that upregulate cyclin D2 increases beta cell proliferation. Most notable among these pathways is high glucose, which has been shown to increase beta cell cyclin D2 levels (Alonso et al., 2007), supporting our assertion that both beta cell ablation and GKA directly increase cyclin D2 and allow beta cells to rapidly re-enter the proliferative pool. The molecular mechanism by which glucose regulates the levels of cyclin D2 in beta cells is currently under investigation. More experiments are necessary to understand why cyclin D2 is downregulated during S phase and what regulatory factors cause its slow return after mitosis.
Interestingly, we did not observe a difference in the post-mitotic regulation of cyclin D2 in young and older mice. Previous work has demonstrated that as beta cells age, they express higher levels of cell cycle inhibitors such as INK4A/p16 (Cdkn2a) (Krishnamurthy et al., 2006). We speculate that the higher level of CDK inhibitors is involved in the delay of cell cycle re-entry with age. Thus, the post-replication quiescence period of beta cells might be regulated by modulators of CDK activity: cyclin D2 in the context of glucose stimulation and CDK inhibitors in the context of aging. Indeed, it is likely that several levels of cell cycle regulation are involved in the re-entry of beta cells, as cyclin D2 returns at a faster rate than the termination of the refractory period. Although the absence of cyclin D2 could prevent replicated beta
D
E
V
E
LO
P
M
E
N
cells from returning to the pool of dividing cells, it does not necessarily control the process alone. Further work will be necessary to determine the role of CDK inhibitors and other cell cycle proteins in the regulation of the refractory period. We also acknowledge that the current work provides only a correlation between cyclin D2 levels and the duration of the refractory period. Demonstration of a causal role for cyclin D2 in the refractory period will require direct manipulation of the protein. Our BrdU/Ki67 pulse-chase assay provides a new tool for identifying cells returning to the cell cycle and for establishing the likelihood of cell cycle re-entry. Further investigation will be necessary to examine re-entry in other adult tissues and in beta cells under different physiological stimuli, such as pregnancy or a high-fat diet. Finally, these results might have several clinical implications. First, the capacity of beta cells to undergo multiple divisions in vivo further supports the theoretical feasibility of regenerative therapy as a treatment for diabetes. Recent reports have demonstrated the existence of beta cell proliferation in normal and diabetic patients, suggesting the opportunity for further beta cell expansion (Kassem et al., 2000; Meier et al., 2008; Meier et al., 2006). Second, although islet transplantation is an effective treatment for type 1 diabetes, the scarcity of donor islets significantly limits its therapeutic scope. Our results suggest that existing donor islets might be induced into significant proliferation in vitro, thereby increasing the number of available donor islets. Lastly, our ability to regulate the post-mitotic quiescent period using a pharmacological activator suggests interesting new directions for the expansion of beta cell mass in vivo.
Acknowledgements
We thank Doug Melton and Ittai Ben-Porath for critical reading of the manuscript and Michal Maatouf, Avigail Dreazen and Judith Magenheim for assistance. This work was supported by grants from the Beta Cell Biology Consortium (NIH), Juvenile Diabetes Research Foundation, Israel Science Foundation, the EU FP6, FP7 BetaCellTherapy consortium and the Helmsley Foundation (to Y.D.). A.M.K. is supported by an EPSRC LSI fellowship (F043325/1. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at
http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.054304/-/DC1
References
Alonso, L. C., Yokoe, T., Zhang, P., Scott, D. K., Kim, S. K., O’Donnell, C. P. and Garcia-Ocana, A.(2007). Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes56, 1792-1801.
Baldin, V., Lukas, J., Marcote, M. J., Pagano, M. and Draetta, G.(1993). Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev. 7, 812-821.
Brennand, K., Huangfu, D. and Melton, D.(2007). All beta cells contribute equally to islet growth and maintenance. PLoS Biol. 5, e163.
Butler, P. C., Meier, J. J., Butler, A. E. and Bhushan, A.(2007). The replication of beta cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab. 3, 758-768.
Cano, D. A., Rulifson, I. C., Heiser, P. W., Swigart, L. B., Pelengaris, S., German, M., Evan, G. I., Bluestone, J. A. and Hebrok, M.(2008). Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes57, 958-966. Chen, H., Gu, X., Su, I. H., Bottino, R., Contreras, J. L., Tarakhovsky, A. and
Kim, S. K.(2009). Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 23, 975-985. Desgraz, R. and Herrera, P. L.(2009). Pancreatic neurogenin 3-expressing cells
are unipotent islet precursors. Development136, 3567-3574.
Dhawan, S., Tschen, S. I. and Bhushan, A.(2009). Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 23, 906-911.
Dor, Y., Brown, J., Martinez, O. I. and Melton, D. A.(2004). Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature429, 41-46.
Finegood, D. T., Scaglia, L. and Bonner-Weir, S.(1995). Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes44, 249-256.
Georgia, S. and Bhushan, A.(2004). Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 114, 963-968.
Gerdes, J., Li, L., Schlueter, C., Duchrow, M., Wohlenberg, C., Gerlach, C., Stahmer, I., Kloth, S., Brandt, E. and Flad, H. D.(1991). Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am. J. Pathol. 138, 867-873.
Grimsby, J., Sarabu, R., Corbett, W. L., Haynes, N. E., Bizzarro, F. T., Coffey, J. W., Guertin, K. R., Hilliard, D. W., Kester, R. F., Mahaney, P. E. et al.(2003). Allosteric activators of glucokinase: potential role in diabetes therapy. Science 301, 370-373.
Gupta, R. K., Gao, N., Gorski, R. K., White, P., Hardy, O. T., Rafiq, K., Brestelli, J. E., Chen, G., Stoeckert, C. J., Jr and Kaestner, K. H.(2007). Expansion of adult beta-cell mass in response to increased metabolic demand is dependent on HNF-4alpha. Genes Dev. 21, 756-769.
Kassem, S. A., Ariel, I., Thornton, P. S., Scheimberg, I. and Glaser, B.(2000). Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes49, 1325-1333.
Key, G., Petersen, J. L., Becker, M. H., Duchrow, M., Schluter, C., Askaa, J. and Gerdes, J.(1993). New antiserum against Ki-67 antigen suitable for double immunostaining of paraffin wax sections. J. Clin. Pathol. 46, 1080-1084. Krishnamurthy, J., Ramsey, M. R., Ligon, K. L., Torrice, C., Koh, A.,
Bonner-Weir, S. and Sharpless, N. E.(2006). p16INK4a induces an age-dependent decline in islet regenerative potential. Nature443, 453-457.
Kulkarni, R. N., Jhala, U. S., Winnay, J. N., Krajewski, S., Montminy, M. and Kahn, C. R.(2004). PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Invest. 114, 828-836.
Kushner, J. A.(2006). Beta-cell growth: an unusual paradigm of organogenesis that is cyclin D2/Cdk4 dependent. Cell Cycle5, 234-237.
Kushner, J. A., Ciemerych, M. A., Sicinska, E., Wartschow, L. M., Teta, M., Long, S. Y., Sicinski, P. and White, M. F.(2005). Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol. Cell. Biol. 25, 3752-3762.
Lukas, J., Pagano, M., Staskova, Z., Draetta, G. and Bartek, J.(1994). Cyclin D1 protein oscillates and is essential for cell cycle progression in human tumour cell lines. Oncogene9, 707-718.
Meier, J. J., Lin, J. C., Butler, A. E., Galasso, R., Martinez, D. S. and Butler, P. C.(2006). Direct evidence of attempted beta cell regeneration in an 89-year-old patient with recent-onset type 1 diabetes. Diabetologia49, 1838-1844. Meier, J. J., Butler, A. E., Saisho, Y., Monchamp, T., Galasso, R., Bhushan, A.,
Rizza, R. A. and Butler, P. C.(2008). Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes57, 1584-1594.
Nir, T., Melton, D. A. and Dor, Y.(2007). Recovery from diabetes in mice by beta cell regeneration. J. Clin. Invest. 117, 2553-2561.
Parsons, J. A., Brelje, T. C. and Sorenson, R. L.(1992). Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology130, 1459-1466.
Rankin, M. M. and Kushner, J. A.(2009). Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes58, 1365-1372. Shapiro, A. M., Ricordi, C., Hering, B. J., Auchincloss, H., Lindblad, R.,
Robertson, R. P., Secchi, A., Brendel, M. D., Berney, T., Brennan, D. C. et al. (2006). International trial of the Edmonton protocol for islet transplantation. New Engl. J. Med. 355, 1318-1330.
Teta, M., Long, S. Y., Wartschow, L. M., Rankin, M. M. and Kushner, J. A. (2005). Very slow turnover of beta-cells in aged adult mice. Diabetes54, 2557-2567.
Teta, M., Rankin, M. M., Long, S. Y., Stein, G. M. and Kushner, J. A.(2007). Growth and regeneration of adult Beta cells does not involve specialized progenitors. Dev. Cell12, 817-826.
Tschen, S. I., Dhawan, S., Gurlo, T. and Bhushan, A.(2009). Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes58, 1312-1320.
Wong, E. S., Le Guezennec, X., Demidov, O. N., Marshall, N. T., Wang, S. T., Krishnamurthy, J., Sharpless, N. E., Dunn, N. R. and Bulavin, D. V.(2009). p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev. Cell17, 142-149.