Translating a gene expression signature for multiple myeloma prognosis into a robust high-throughput assay for clinical use
Full text
Figure
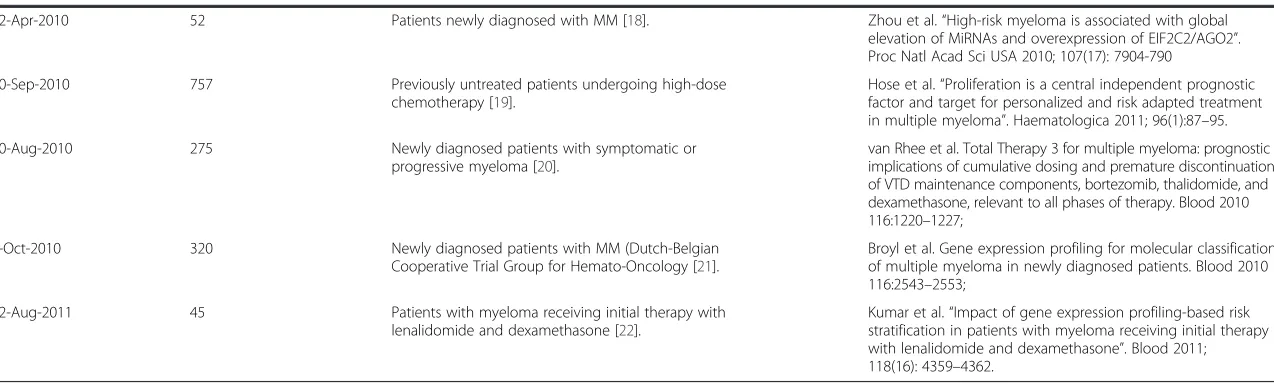
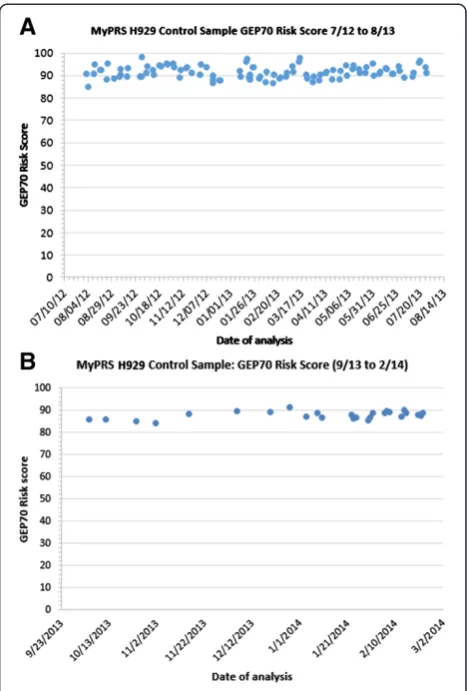
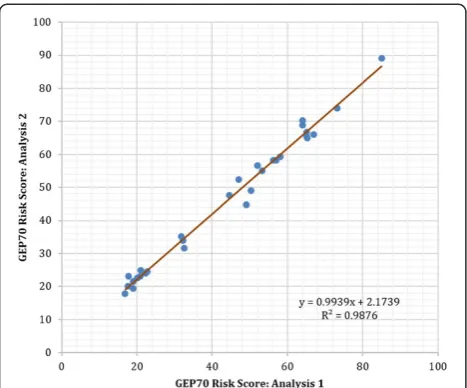
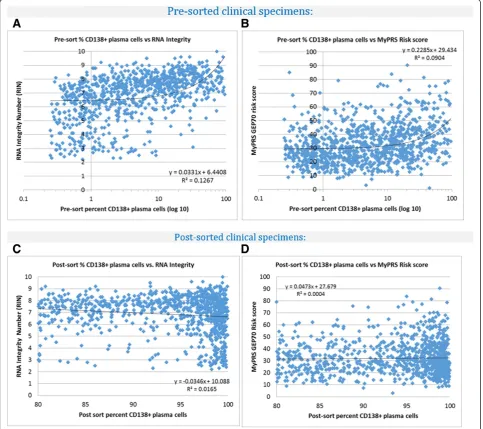
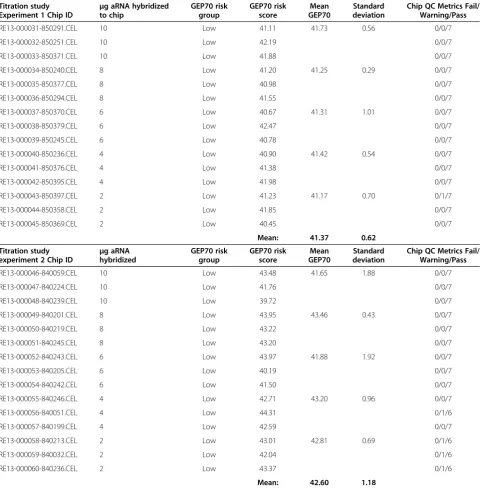
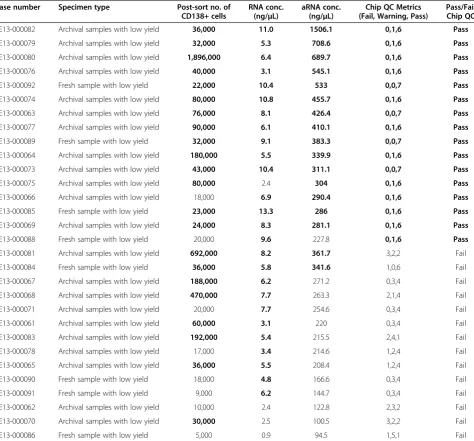
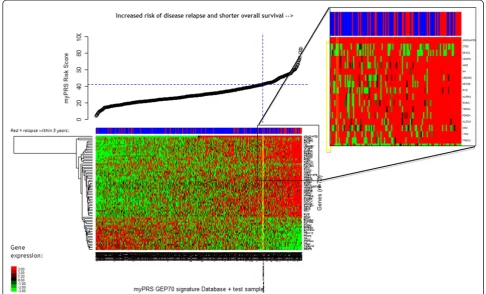
Related documents
The purpose of the signal cancellation circuit is to suppress the reference signal from the main power amplifier output signal leaving only amplifier distortion, both linear
keep records on fatal accidents involving cell phones..
Pressure independent flow provided by a positive displacement diaphragm pump ensures that critical parameters do not vary during the concentration step Fully automated system
> Putting your backup software on a Windows server means that you are at risk for all those computer viruses against which you’re trying to protect your environment.. >
We have used concatenated sequences of seven house-keeping loci from 770 strains of 11 named Neisseria species, and phylogenetic trees, to investigate whether genotypic clusters
Table 6-28: Cost Efficiency scale elasticities estimates for Jordan, Egypt, Saudi Arabia and Bahrain Banks over 1992-2000 — Individual Bank Estimates 309 Table 6-29: Standard
If a child enters the family by adoption, however, the very substantial expenses involved (typically $10,000 to $25,000), including all medical costs associated with the birth of
• Lack of independent RD&D • Lack of Incentives & Regulation • Globally the new frontier is infrastructure and software energy efficiency / consumption • Many
