OpenCB development - A Big Data analytics and
visualisation platform for the Omics revolution
Abstract
The advent of Next Generation Sequencing (NGS) techniques in Computational Biology are revolutionising the practise of clinical medicine. These techniques produce vast amounts of data and when combined with other clinical data, drastically increases the amount of data processing power required to infer clinical meaning from the data. In fact, today the bottleneck in whole genome sequencing is not the sequencing itself but the analysis of the data. As time moves on, more varieties of medical data are becoming available, the problem grows and if we are able to realise the potential of the omics revolution to advance patient outcomes, new bioinformatics Big Data solutions are needed. The open-source software for Computational Biology project (OpenCB) is just such a new solution. It is an open-source collaborative initiative which is developing a High-Performance Computing (HPC) and Big Data software for storage, analysis, sharing and visualisation of big data in genomics. It provides a revolutionary new approach taking emerging technologies from the web-scale and Cloud industries utilising technologies such as Apache Hadoop or Spark together with other NoSQL databases.
With this new technology, significant functionality and performance for a broad range of genomics analysis and visualisation tasks have been achieved providing the breakthrough in performance that is needed to truly take advantage of the current NGS revolution. Some of the projects developed offer a 3x speedup such as NGS read aligner, while others such as the HBase implementation of OpenCGA Storage show a 12x when loading and indexing data. Depending on the tools used in different pipelines, the overall performance can be significantly improved. This performance increase, scalable architecture and commodity solution space provided by Dell and Intel mean that the data analysis bottleneck has now been removed, unlocking the potential of large scale next generation genomics data to drive improvements in patient health. The OpenCB platform consists of different projects that can be deployed independently such as CellBase or HPG BigData; or integrated together in major application called OpenCGA which allows for efficient processing, indexing and visualisation of hundreds of TBytes of genomic data. OpenCB development is being led out of the University of Cambridge with active participation of several other leading bioinformatics institutions. The source code is open and freely available in GitHub at
Introduction
Over the last few years, biology has experienced a revolution as a result of the introduction of new DNA sequencing technology, known as Next-Generation Sequencing (NGS), that makes it possible to sequence the whole genomic DNA or RNA transcriptome in days instead of years. These recent high-throughput sequencers produce data at unprecedented rates and scale, the decreasing costs and the increasing throughput have popularised their use in many fields of life sciences and clinics. Whole genome DNA re-sequencing allows us to find and catalogue genomic variants or mutations, helpful for discovering new disease-related mutations in clinical research. RNA sequencing (RNA-seq) has also arisen as a crucial analysis for biological and clinical research, as it can help to determine and quantify the expression of genes, the RNA transcripts that are activated or repressed in different diseases or phenotypes, therefore providing an unbiased profile of a transcriptome that helps to understand the etiology of a disease.
Current NGS technologies can sequence short DNA or RNA fragments, of length usually between 75 and 300 nucleotides (nts), some new sequencers with longer fragment sizes are being developed. Primary data produced by NGS sequencers consists of hundreds of millions or even billions of short DNA or RNA fragments which are called reads. The first step in NGS data processing in many comparative genomic experiments, including genome re-sequencing or RNA-seq, involves mapping the NGS reads onto a reference genome, in order to locate the genomic coordinates where these fragments emanate. The mapping process is particularly more difficult for RNA-seq, as the genes in eukaryotes may be split into small regions, called exons that are separated by intron zones composed of thousands of nucleotides. Once the exons are transcribed to RNA, they are brought together to form the transcripts in a splicing process. Thus, when mapping reads from RNA transcripts onto a reference genome, it must be taken into account that these reads may contain a splice junction and, therefore, involve different exons, so that in practice they may lie thousands of nucleotides apart, this situation is referred to as a gapped alignment. Mapping step constitutes a very expensive process from the computational point of view. Furthermore, sensitivity is also a serious concern at this point, given that natural variations or error sequencing may occur, yielding frequent mismatches between reads and the reference genome, which increase the computational complexity of the procedure. In order to ensure that such techniques become inexpensive, there is a need to optimise sequencing to take advantage of new processing techniques made available in modern day compute architectures. HPG Aligner project from OpenCB provides a High-Performance Computing (HPC) implementation of DNA and RNA-seq NGS aligners. This implementation is based on multi-threading and SIMD vectorization targeting Intel® Advanced Vector Extensions (AVX2)) - instruction set extension found in Intel processors and aims to provide a very high sensitivity and performance. HPG Aligner shows an excellent sensitivity, even with a high rate of mutations, and remarkable parallel performance for both short and long DNA and RNA-seq reads. In HPG Aligner, reads are aligned using a combination of mapping with Suffix Arrays (SA) and local alignment with the Smith-Waterman algorithm (SWA).
1 Interpreted here as the cost per operation halving every two years
The advances in high-throughput technologies have also produced an unprecedented growth in the number and size of public biological databases and repositories but unfortunately, the current status of many of these repositories is far from being optimal. For example, all this information is spread out in many small databases implementing different standards. Furthermore, data size is increasingly becoming an obstacle when accessing or storing biological data. All these issues make it very difficult to extract and integrate information from different sources, to analyse experiments or to access and query this information in a programmatic way. CellBase project from OpenCB provides a solution to the growing necessity of integration by easing the access to biological data. CellBase implements a set of RESTful web services that query a high-performance NoSQL database containing the most relevant biological data sources accounting for several TBytes.
Another step of NGS data processing include the variant calling, during this process hundreds of millions of genomic variants are identified from the mapped reads. Current genomic and clinical projects are sequencing and calling variants from thousands of samples, producing hundreds of TBs and making it extremely difficult, if not impossible, for researchers to store and analyse these big datasets with current bioinformatics tools. For example, Genomics England (GEL) project aims to sequence 100,000 rare diseases and cancer patients from NHS UK producing about 400TB of compressed data. For the analysis of these data, both variants and samples need to be highly annotated. OpenCGA project from OpenCB allows not only the storage and index of these big datasets using different NoSQL database or big data frameworks such as Hadoop but also the variant annotation from CellBase and the sample annotation using a built-in component called OpenCGA Catalog.
Other OpenCB projects include HPG BigData and Genome Maps. HPG BigData aims to provide a scalable solution for NGS in a Hadoop environment, most common data processing and analysis are being implemented using Hadoop MapReduce and Spark execution engines. Genome Maps is a high-performance web-based genome browser that can render CellBase data and render remote NGS experiments from OpenCGA.
Much of the existing software solutions in bioinformatics are not designed to work at these data volumes, thus inhibiting scalability and limiting efficiency and consequently making it very difficult for researchers to store, analyse, share and visualise data in a secure and collaborative manner. As time moves on this problem will get more severe since the genome cost is falling faster than Moore’s Law1 whereas the cost of sequencing a full genome is substantially quicker (see Figure below). In order to close this growing Omics – Moore’s law gap, new Omics analytics platforms are required that combine new computational methods and new leading edge hardware and software technologies.
Here we introduce the OpenCB initiative that offers a new capability in genomics analytics: to provide an extensible platform which aims to redress these challenges and provide a complete stack for big data in genomics. OpenCB is implemented using the state-of-the-art advanced High-Performance Computing (HPC), and Big Data technologies from Dell & Intel and is actively being developed at the University of Cambridge and other research institutes . With the OpenCB platform additional challenges relating to the integration of diverse “omics” data can be realised providing even more insight for clinicians and practitioners.
OpenCB Overview
OpenCB initiative was launched in 2012 by Ignacio Medina now Head of the Computational Biology Lab at UIS Cambridge to provide biological and clinical researchers a scalable ,high performance and high quality software environment for genome-scale data analysis. Cellbase is now used by many projects and research institutes. Currently it is being actively developed by more than 12 researchers from University of Cambridge, EMBL-EBI and Genomics England among others. More information can be found at
http://www.opencb.org/, OpenCB consists of different projects that solve different problems in current genomics, each of these projects constitutes a standalone solution than can be easily imported into existing projects The projects have been designed to provide a scalable and high-performance solution for storing, processing, analysing, sharing and visualizing big data in genomics and clinics in a secure and efficient manner. To achieve this, OpenCB uses the most advanced computing technologies in HPC (such as Task- and Data-parallelism with AVX2 or GPUs and Big Data (Hadoop MapReduce, Spark) for data processing and analysis; NoSQL databases (MongoDB, HBase) for data indexing or HTML5 for interactive data visualisation. An overview of all the projects can be seen at https://github.com/opencb
Figure 2 The increasing divide between Cost per Base of DNA vs. Moore’s Law Wetterstrand KA. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP) Available at: www.genome.gov/sequencingcosts. Accessed [Nov 2015].
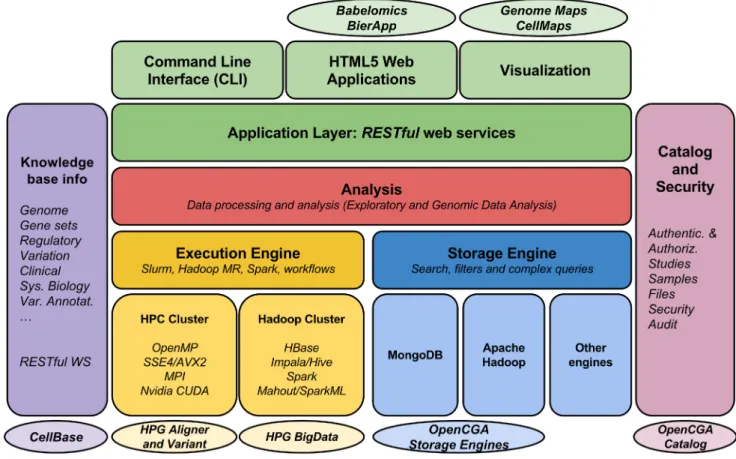
Fig 3 OpenCB architecture. Server side stores, indexes and executes all the analysis. Client side HTML5 applications and CLI use RESTful web services to interact with the server
OpenCB consists of a number of projects as listed below:
High-Performance Genomics (HPG)
HPG projects make use of standard HPC and big data technologies to provide a scalable and efficient solution for several genomic analysis. The main HPG subprojects are:
a) HPG Aligner (https://github.com/opencb/hpg-aligner) is a DNA and RNA-seq ultra-fast and sensitive HPCNGS read aligner. It combines advanced data structures and novel algorithms implemented with multi-threading and vectorization using AVX 2. Current work at Cambridge is being performed to explore the Intel® Xeon Phi™ Coprocessor as a platform.
b) HPG Variant (https://github.com/opencb/hpg-variant) is a HPC software to process and anlayse genomic variant data, several algorithms have been developed and implemented.
c) HPG BigData (https://github.com/opencb/hpg-bigdata) is a Hadoop MapReduce and Spark implementation of several genomic tools and analyses for working with genome-scale data.
CellBase
CellBase (https://github.com/opencb/cellbase) constitutes the knowledge-base database for all OpenCB projects. CellBase is a high-performance and scalable NoSQL database that integrates the most relevant biological repositories, among the most significant data we can find genomic features, proteins, gene expression, regulatory elements, functional annotation, genomic variation and systems biology information. Its knowledge base relies on the most relevant repositories such as ENSEMBL, Uniprot, ClinVar, COSMIC or IntAct among others. CellBase implements also a fast variant annotation built-in component that provides an Ensembl VEP compatible annotation. All data is available through a command line or by RESTful web services.
OpenCGA
OpenCGA (https://github.com/opencb/opencga) provides a scalable and high-performance platform for big data analysis and visualisation in a shared environment. OpenCGA integrates some of the OpenCB projects and implements, in addition, other components:
a) A Storage Engine framework to store and index alignments and genomic variants into different NoSQL such as MongoDB or Hadoop HBase - the current implementation can store efficiently thousands of gVCF files while remaining responsive when querying data.
b) A Catalog which keeps track of users, projects, files, samples annotations, etc and also provides authentication and authorization capabilities.
c) Analysis engine to execute genomic analysis in a traditional HPC cluster or in Hadoop. OpenCGA has implemented a command line and RESTful web services to manage and query all the data.
Visualisation with Genome Maps and CellMaps
Finally in OpenCB, a high-performance HTML5 web-based genome browser called Genome Maps (https://github.com/opencb/genome-maps) and a systems biology tool called CellMaps
(https://github.com/opencb/cell-maps) provide a Big Data scientific visualisation capability to OpenCB. Genome Maps can interactively display CellBase and OpenCGA indexed data such as BAM and VCF files. Users can also easily extend Genome Maps to display their own data and formats. In addition, OpenCB projects are compliant with the new GA4GH data models and formats.
Fig 4 An overview of main OpenCB components. Some OpenCB projects and tools in ovals
Who is using it
Many projects within research institutes around the world are using some OpenCB technologies demonstrating the success of this initiative. For instance ICGC, EMBL-EBI or Genomics England are using and contributing to some of this projects. Source code is open and it is freely available in GitHub at https://github.com/opencb
Benefits of moving to HPC and Hadoop
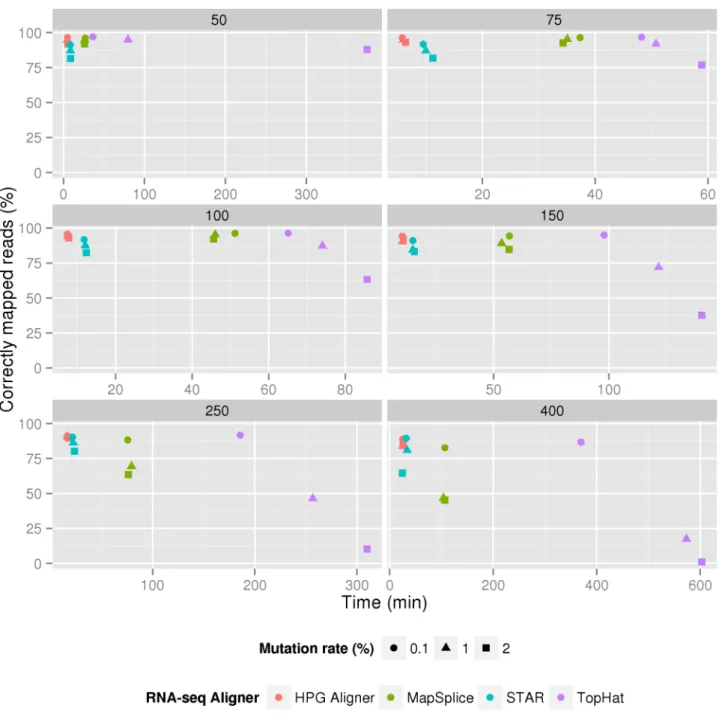
DNA and RNA-seq HPG Aligners implemented show a very high-performance while having the highest sensitivity in most scenarios such as short and longer reads or at different mutation rates. Results are especially good in the case of RNA-seq where HPG Aligner is the most sensitive and fast aligner at different read length or mutation rates when compared to reference RNA-seq aligners (Fig)
Fig 5. Wall clock times for different simulated read lengths ranging from 50 to 400 nucleotides. Three different mutations rates were studied for four different RNA-seq aligners. As can be seen HPG Aligner is the fastest and more sensitive in all scenarios.
HPG BigData implements most common NGS data processing and analysis tools. Some benchmarks have been run on a modest 8-node Hadoop cluster from Dell (see Appendix I) running Cloudera 5.4.3. Simulating a big variant data set of 500 million variants and 100 samples takes 40 min to generate few TBytes of data. Loading this variant data set into Hadoop HBase yields more than 500,000 variants/ second, this means a 12x speedup from similar implementation using other NoSQL databases like MongoDB, many of the HBase queries implemented run in sub-second scale when using the row key index and in a few seconds for table scans.
The current version of CellBase is based on a MongoDB NoSQL database and contains eleven collections accounting for about 1 TBytes of data, some of these collections have more than 100 million complex documents or a few billion data values. All data models and collections have been designed to offer a low latency and high-performance query execution. Most of the queries, even complex aggregations, perform in millisecond scale. A main component of CellBase is the built-in variant annotation tool, this has been implemented using a multithreaded and asynchronous approach to speedup performance, current version can annotate more than 1,200 genomic variants per second when connecting to MongoDB, and about 800 variants/second when using the RESTful web services.
OpenCGA integrates several projects from OpenCB and aims to provide a complete solution for genomic big data analysis and visualisation. The OpenCGA Catalog has been implemented using MongoDB and can load millions of file metadata and sample annotations in just a few minutes with complex queries and aggregations running in millisecond scale. The OpenCGA Storage built-in framework can normalize and transform data into binary formats using a multithreaded implementation, for example processing a gVCF file with about 400 million records takes less than 2 minutes in a standard server. Processed data can be loaded and indexed in MongoDB or HBase NoSQL databases, the performance of HBase is about 12x when compared with MongoDB reaching more than 500,000 variants loaded and indexed per second in a 8-node Hadoop cluster. Executing complex queries and aggregations perform in a seconds outperforming any other existing solution.
Future Developments
In order to ensure OpenCB maintains a cutting-edge platform for large-scale Genomics, processing access to state-of-the-art technologies is critical. At Cambridge, the OpenCB team is exploring future processing capabilities offered by Intel® Xeon Phi™ Coprocessors, FPGA, and GPU technologies. In addition new non-volatile RAM solutions are being investigated as a means to effect larger in-memory processing capability as well as enhanced MapReduce capability to ensure that scalability is maintained. These technologies will be coupled with enhanced statistical analysis techniques such as those provided by Dell Statistica to provide practitioners with even more insight into “omics”. The University of Cambridge under the auspices of the UIS is working with a number of industry partners in this respect. In particular the University will work with Dell and Intel to increase the performance of the solution and also shrink-wrap the solution onto a well-tested hardware platform to produce a turnkey Next Gen Sequence Analytics Appliance extending the specification detailed below in Appendix I.
Appendix I - Big Data Platform
The current OpenCB Development Platform consists of the following:
MongoDB solution
A replica set of three servers connected to a storage solution. Specifications:
Server Function Specification
Server 1-3 Database replica set Dell PowerEdge R630
2x Intel® Xeon® Processor E5-2560v3
256 Gbytes RDIMM
Storage Storage Dell MD3460
20x4TB SAS 7.2K RPM HDD
5x800GB SSD Read Intensive
Hadoop
A development cluster consisting of 8 nodes. Specifications:
Server Function Specification
Master Cloudera Manager Dell PowerEdge R720
2x Intel® Xeon® Processor E5-2560v2
128 Gbytes RDIMM
6x600GB SAS 15K RPM HDD
Server 1-8 Data node Dell PowerEdge R720xd
2x Intel® Xeon® Processor E5-2667v2
64 Gbytes RDIMM
24x500GB SAS 7.2K RPM HDD
Network Dell Force10 10GbE
Software Cloudera 5.4.5
Dell Statistica
Spark 1.3