ACKNOWLEDGEMENTS
TABLE OF CONTENTS CHAPTER
I. STUDY AIMS AND HYPOTHESES
In the current investigation, we examined the effect of acyl-CoA synthetase-1’s (ACSL1) location (mitochondria or endoplasmic reticulum) on function (directing fatty acids towards β -oxidation or triglyceride synthesis). ACSL1 is responsible for activating fatty acids to acyl-CoAs. We hypothesized that by manipulating the subcellular location of ACSL1, the production of lipid intermediates, triacylglycerol, and phospholipids would be altered. Our goal was to determine how and if ACSL1 is responsible for directing fatty acids after they are transported into the cell. Specifically, if ACSL1 were targeted to endoplasmic reticulum (ER) then we would see an increase in synthesis of lipids such as triglycerides, phospholipids, and cholesterol esters whereas if ACSL1 were targeted to mitochondria we would not see an increase in synthesis of complex lipids. ACSL1 targeted to mitochondria would increase oxidation of fatty acids in order to be used for energy production. The rationale behind this hypothesis was that the endoplasmic reticulum (ER) is the main site of glycerolipid synthesis. Increasing acyl-CoA synthesis at the ER would thus increase fatty acid esterification without increasing β-oxidation. In contrast, mitochondria are the major site of fatty acid oxidation. Increasing acyl-CoA synthesis at the mitochondria would increase entry of the acyl-CoAs into the mitochondria through carnitine palmitoyltransferase I (CPT-1) for oxidation. We therefore expected that by increasing oxidation, long chain fatty acids would be converted to acetyl-coA, which can then enter the citric acid cycle. Conversely, if our results do not present as predicted, it is possible that another protein
may be responsible for shuttling fatty acids either to oxidation or lipid synthesis. For example, fatty acid binding protein, or acyl-CoA binding protein may play a role in directly shuttling fatty acids through physical interactions.
multiplicity of infection (MOI) necessary to attain equal infection between wild-type, mitochondria, and ER targeted adeno-viruses. Equal infection of cells was achieved by
evaluating ACSL1 activity with an ACSL assay. In addition, this project focused on developing a protocol to isolate and purify mitochondrial and ER fractions in heart samples. Although
previous research shows that ACSL1 is located on the mitochondria in heart, it is not known whether ACSL1 is also located on the ER. Therefore, specifically heart tissue was used. Furthermore, it is necessary to obtain purified organelle fractions so that downstream functions of ACSL1 can be accurately compared.
SPECIFIC AIMS
AIM 1: Determine whether the subcellular location of ACSL1 is critical in directing fatty acids to either oxidation or complex lipid synthesis.
HYPOTHESIS: Mitochondria-targeted ACSL1 will direct fatty acids predominantly towards β -oxidation whereas ER-targeted ACSL1 will shuttle fatty acids primarily towards complex lipid synthesis
RATIONALE: Mitochondria are responsible for energy mobilization and therefore catabolism of molecules, whereas the ER is the main site of glycerolipid synthesis.
SUBAIM 1: Determine multiplicity of infection (MOI) that attains equal ACSL1 activity between wild-type, mitochondria, and ER targeted adeno-viruses.
SUBAIM 2: Develop an optimized protocol for subcellular fractionation that achieves purification of ER and mitochondria from heart.
EXPERIMENTAL DESIGN:
In this study, differentiated brown adipocytes and hepatocytes from control and Acsl1
II. INTRODUCTION
Diabetes is a highly prevalent disease in the United States. Currently, 8.3% of the population, or 25.8 million individuals are diagnosed with diabetes1. This number is projected to increase according to trends of diagnosed diabetes from the past 2 decades. The prevalence of individuals diagnosed with diabetes rose from 4.9% in 1990 to 6.5% in 1998, and 6.9% in 1999. In 2010, 1.9 million new cases of diabetes occurred2. Thus, it is clear that diabetes is a rising public health concern.
Diabetes is a group of diseases related to high blood glucose levels, which result from the body’s inability to produce or use insulin. Diabetes II occurs when an individual’s cells do not respond properly to insulin in order to maintain a fasting blood glucose level of ≤ 126 mg/dl3. High blood glucose levels can lead to a number of permanent complications including blindness,
kidney failure, neuropathy, and amputation1. Due to these consequences, the scientific
community has been pushed to find ways of addressing type II diabetes. Research studies have
focused on identifying and targeting risk factors for type II diabetes4. Factors that may be associated with developing type II diabetes include hypertension, hypercholesterolemia, and hyperlipidemia. Interestingly, while investigating risk factors for type II diabetes, researchers observed simultaneous presence of these risk factors in patients with type II diabetes.
Identification of such a relationship led to grouping of the individual risk factors under a single
name: Metabolic Syndrome.
Metabolic syndrome is a group of risk factors that increase the chance of developing coronary heart disease, stroke, and diabetes. Five main risk factors have been identified: waistline greater than 40 inches for men and greater than 35 inches for women, triglyceride level greater than 150mg/dl, a HDL cholesterol level less than 40 mg/dl, blood pressure greater than 130/85 mm Hg, and fasting blood glucose level greater than 100 mg/dl. Individuals with the metabolic syndrome (having it least 3 of the mentioned risk factors) have a 1.5 to 3 fold
II diabetes compared to individuals without the syndrome5. For this reason, diagnosing metabolic syndrome may be clinically important for introducing early treatment intervention in order to limit progression into clinical diabetes. However, metabolic syndrome is still relatively new and controversy remains over its clinical value6. A noteworthy concern is that treatment of metabolic syndrome is the same as treatment for the individual risk factors. However, it has been postulated that a distinguishing characteristic of metabolic syndrome may exist; a unifying link for the risk factors identified for metabolic syndrome. Whereas a single underlying cause for metabolic syndrome has not been determined, previous studies have suggested that obesity and insulin resistance play a role. If this is the case, then treatment for type II diabetes could potentially involve targeting insulin resistance as opposed to each individual risk factor.
Insulin resistance is defined as the diminished ability of the body’s cells to respond to insulin stimulation for glucose uptake, metabolism, or storage. It is further characterized by impaired suppression of hepatic glucose output. Although the exact mechanism underlying insulin resistance is unknown, it has been suggested that increased free fatty acids in the blood inhibit insulin-stimulated glucose uptake7. Free fatty acids and their metabolites including diacylglycerol have also been implicated as possible mediators of insulin resistance when allowed to remain in excess for extended periods of time within the cell8. Furthermore, sphingolipid ceramides have been shown to be an important intermediate in lipid metabolism due to ceramides role in linking fatty acids and inflammatory cytokines to insulin resistance22. It
is now clear that increased lipid intermediate content within the cell is due to an imbalance between fatty acid delivery and intracellular fatty acid oxidation and lipid storage9. For this reason, it is important to study fatty acid metabolism and understand how fatty acids are shuttled to various pathways after entering the cell.
key group of enzymes called acyl-CoA synthetases catalyze this reaction by esterifying fatty acids to Coenzyme A (RCOOH + CoASH + ATP ! RCO-SCoA + AMP + PPi). Once activated, acyl-CoAs enter various pathways such as β-oxidation and complex lipid formation. Of the five enzymes in the long-chain ACSL family, ACSL1 contributes 50-90% of total ACSL activity in liver, heart, adipose tissue, and skeletal muscle10. Previous studies have shown that fatty acid oxidation rates are lower in mice lacking the Acsl1 gene in heart, than in control mice11-12. Given that thioesterification of fatty acids is a necessary initial step for using fatty acids, it is not
surprising that ACSL1 is critical in utilizing fatty acids for either oxidation or lipid storage. However, whereas ACSL1 is required for β-oxidation to occur, overexpressing the enzyme has been shown to lead to incorporation of fatty acids into phosphatidylethanolamine, a critical product of diacylglycerol metabolism due to phosphatidylethanolamines potential role in insulin resistance13. Thus, it is a change in ACSL1 expression that greatly alters fatty acid metabolism. Therefore,understanding how ACSL1 directs fatty acids towards oxidative and synthetic pathways may help to determine a mechanism for production of lipid intermediates.
Furthermore, if a mechanism is determined, then potential targets for insulin resistance can be considered.
Research done in our lab shows that ACSL1 is located on the outer mitochondrial membrane and the endoplasmic reticulum (ER) in liver15. Although the ER is responsible for many processes, a primary function of the organelle is to regulate synthesis of various
macromolecules including cholesterol and phospholipids. Likewise, a major role of the
to mitochondria or ER, and downstream enzymes involved in oxidation and synthesis, it can be demonstrated whether ACSL localization affects the synthesis of various products in fatty acid metabolism. Due to the relevance of lipid intermediates to metabolic syndrome and insulin resistance, it is important to determine the effect of subcellular location on directing fatty acids towards production of lipid metabolites.
In order to determine the effect of subcellular location on directing FAs towards production of lipid metabolites, it is first necessary to confine ACSL1 either to the ER or mitochondria. Targeting ACSL1 to the ER was achieved with the use of a fusion protein that contained the active site of ACSL1 and the transmembrane domain of an ER located protein, fatty acid transport protein (FATP4). Targeting ACSL1 to the mitochondria was done in a similar way, except the transmembrane domain of an outer mitochondrial membrane protein,
translocase of the outer mitochondrial membrane 70 was used instead. Moreover, it is critical to
attain equal infection so that ACSL activity is equal between wild-type, mitochondria-, and ER-targeted ACSL1 adenoviruses. Equal ACSL1 activity guarantees that the same amount of fatty acyl-CoA is synthesized in each cell. If equal amounts of fatty acyl CoAs are initially present to be used for downstream pathways, then the presence of the resulting products can be attributed to the location of ACSL1 rather than to the amount of enzyme. For example, if the results
indicate that there is greater oxidation versus storage of fatty acyl CoAs, then this is due to a reason other than the amount of ACSL1 originally present within the cell.
Insulin function
Insulin maintains a normal blood glucose level (70-100 mg/dl) by activating glucose
transport into tissues via the insulin-signaling pathway and by stimulating glycogen and
triglyceride synthesis, therefore lowering high blood glucose levels. Insulin also inhibits the
Insulin Signaling
Insulin is released into the bloodstream by pancreatic islet β-cells when the blood glucose level is high. Glucose is absorbed into the cell through facilitated diffusion by hexose transporters. In muscles and adipose tissue, the major transporter of glucose is glucose transporter type 4 (GLUT4), and in liver and pancreas the major transporter is glucose transporter type 2 (GLUT2) (non-insulin dependent). Under basal conditions GLUT 4
transporters are present inside vesicles in the cell. However, when insulin levels are increased, insulin binds to insulin receptor tyrosine kinase located on the plasma membrane of cells, thus generating a cascade of events that lead to transport of GLUT4 onto the plasma membrane. This cascade begins with phosphorylation of insulin receptor substrates 1 and 2 (IRS1, 2). IRS
then activates phosphatidylinositol-4,5-bisphosphate 3-kinase
(
PI3 kinase) to synthesizephosphatidylinositol (3,4,5)-triphosphate (PIP3) by phosphorylating phosphatidylinositol
4,5-bisphosphate (PIP2). PIP2 activates protein kinase B (Akt), which then activates multiple
pathways involved in activation of transcription factors, glycogen synthesis and protein
phosphatase-1. GLUT4 can then transport glucose into the cell where glucose is either used for energy or stored for future use. However, impaired insulin signaling, specifically down regulation
of GLUT4 and impaired suppression of gluconeogenesis, may lead to insulin resistance19.
Obesity
It has been shown that in adipocytes of obese humans, IRS-expression can be
reduced16. This reduction may be due to increased expression of protein tyrosine phosphatases that dephosphorylate IRS16. Reduced IRS-expression may then lead to down regulation of GLUT4.
strongly linked to insulin resistance than are peripheral (gluteal/subcutaneous) fat depots. A leading hypothesis is that intra-abdominal adipocytes are more lipolytically active.”16 This may be because increased degradation of triglycerides in adipocytes leads to an increase in serum fatty acid levels. Increased free fatty acids in the blood inhibit insulin stimulated glucose uptake in muscle.
Insulin Resistance
Impaired skeletal muscle fatty acid oxidation has been proposed to contribute to insulin resistance. The idea behind this is related to evidence showing that excess intracellular
accumulation of fatty acid metabolites is involved in mediating insulin resistance. These fatty acid metabolites include diacylglycerol, ceramide, and long-chain acyl CoA and have been shown to be elevated in obesity and diabetes. In addition, genetic and pharmacological
manipulation to decrease fatty acid oxidation has been shown to improve insulin sensitivity. For these reasons, it has been proposed that increasing β-oxidation can decrease insulin
resistance. However, this remains a controversy20.
Carnitine palmitoyltransferase I (CPT-1) is an enzyme involved in the regulation of mitochondrial fatty acid oxidation. It is responsible for the formation of acyl carnitines by
transferring an acyl group from CoA to carnitine and is the rate-limiting enzyme for mitochondrial fatty acid uptake. Furthermore, inhibition of CPT-1 has been shown to effectively reduce fatty
oxidation actually improves insulin stimulated glucose uptake in mice with diet-induced obesity (shown with glucose tolerance test and insulin tolerance test results)20. In addition, inhibition of fatty acid oxidation did not cause accumulation of fatty acid intermediates20.
III. METHODS Samples
Cultured hepatocytes and pre-adipocytes from the brown adipose tissue depot were obtained from liver-specific knockout (KO) (Acsl1L-/-) and adipose-specific KO mice (Acsl1A-/-),
respectively. Pre-adipocytes were differentiated into brown adipocytes in culture media containing insulin, triiodothyronine (T3), and rosiglitazone. In the differentiation process, rosiglitazone binds to peroxisome proliferator-activated receptors (PPARs), which are nuclear receptors that regulate genes involved in fat cell differentiation. T3 is also responsible for regulation of genes involved in energy metabolism. In addition, insulin is a pro-growth hormone21.
Brown adipocytes and hepatocytes were infected with adenoviruses containing wild-type ACSL1, ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1), ACSL1 targeted to the ER-only (Ad-F4-Acsl1), or green fluorescent protein (Ad-GFP). To target ACSL1 to the mitochondria only, a fusion protein was made containing the transmembrane protein of an outer mitochondrial
membrane protein (translocase of the outer mitochondrial membrane 70 or T70). Specifically, the T70 transmembrane domain and the active site of ACSL1 were first amplified. Then the DNA fragments were subcloned to produce a single sequence. Finally, the full sequence was expressed in the chosen cell lines. To target ACSL1 to the ER only, a fusion protein was
demonstrated by immunofluorescence. Furthermore, all constructs contained a FLAG-tag on the C-terminus.
Cardiomyocytes were obtained from a rat cardiomyoblast cell line (H9c2). The cell line was grown to confluence and then allowed to differentiate for 4 days. The growth media contained 10% fetal bovine serum, penicillin-streptomycin, and 4 mM glutamine. The
differentiation media contained 1% horse media. The cells were then infected with adenovirus containing wild-type ACSL1, ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1), ACSL1 targeted to the ER-only (Ad-F4-Acsl1), or green fluorescent protein (Ad-GFP) for 24 or 48 hours.
ALT Assay
Alanine transaminase (ALT) is an enzyme that catalyzes the transfer of an amino group from L-alanine to alpha-ketoglutarate, to produce pyruvate and L-glutamate. The enzyme is normally found inside cells and in most types of tissues including heart and liver. Therefore, the presence of ALT in the plasma is indicative of cellular injury. Clinically, the serum level of ALT is
measured in order to determine tissue health. To determine the amount of ALT present in the media of cultured cells, an ALT kit containing reagent 1 (Tris buffer, L-alanine, lactate
dehydrogenase) and reagent 2 (a-ketoglutarate, NADH) was obtained from Stanbio. To make the working reagent, five parts of reagent 1 was mixed with 1 part of reagent 2, and warmed to 37°C in a water bath. To determine the amount of media to use, media volumes of 2.5 ul, 5 ul, 10 ul, 20 ul, and 50 ul were used from media with uninfected cells and media not incubated with
cells. These volumes were added to a 90-well plate. Water was used as a blank. Using a
multiple channel pipet, 200 ul of working reagent was added to each well. The plate was read at 340nm every minute for 3 minutes to determine the enzymatic rate.
ACSL Assay
To use the desired amount of protein, 2, 4, or 6µg, 20µL of homegenate cell sample was
[C14] palmitate) to a concentration of 0.1µg/µl, which is then diluted 10-fold in the final reaction. Master mix (1M Tris, pH 7.4, 160mM MgCl2, 100mM DTT, 100mM adenosine 5’-triphosphate (ATP), 2mM CoA, 0.5mM palmitate in 5mM Triton X-100 in EDTA) was prepared and 120µL was added to 13x100 glass tubes. To achieve a final reaction volume of 200µL, the appropriate volume of Medium 1 buffer was added, and in 30-second intervals, 20, 40, or 60µL of diluted sample, was measured into each tube. The samples were incubated for 10 minutes at room temperature. In order to stop the reaction, 1 mL of Dole’s reagent (80:20:1, heptane:
isopropanol: 1M sulfuric acid v/v/v) was added, and the samples vortexed. The fatty acyl-CoAs in solution were extracted by adding 2mL of heptane and 0.5mL of dH2O and allowing the phases to separate. The samples were then centrifuged at 1500 rpm for 5 minutes. The top organic layer was aspirated and discarded, and the extraction process was repeated. Then 0.6 mL of the bottom aqueous phase was counted to determine ACSL activity. Two blank samples containing no protein were used as the controls.
To collect cardiomyocytes, hepatocytes, and adipocytes from culture, media were removed, and cells were washed twice with 1 mL of 1X phosphate-buffered saline (8 g/L NaCl, 0.20 g/L KCl, 1.78 g/L Na2HPO4, 0.27 g/L KH2PO4, pH 7.4). The cells were placed on ice and 100 uL of Medium 1 buffer with DTT was added. The cells were then scraped, drawn up using a 25-gauge needle and syringe, and stored at -80°C.
Western Blot
incubated with 5% non-fat dry milk in Tris-Buffered Saline, 0.1% Tween (TBS-T) for 1 hour. To bind to only the protein of interest, a specific primary antibody was diluted in milk and incubated with the membrane at 4º overnight. The primary antibody dilution used for this study was: 1:2000 for ACSL1, and 1:5000 for VDAC and calnexin. After the overnight incubation, the blot was washed with 1X TBS-T buffer 3 times for 5 minutes each. To detect the protein of interest, secondary antibody that can bind to the primary antibody was added to blot and kept at room temperature for 1 hour. The secondary antibodies used for this study were: goat anti-rabbit 1:5000 for ACSL1 and VDAC, and goat anti-mouse 1:10,000 for calnexin. The blot was washed 5 times with buffer and incubated with chemoluminescent substrate for 5 minutes. Finally, the blot was developed by placing film onto the membrane for 2 or 20 minutes and inserting the film into the developer machine.
Isolation and Purification of Organelles
To isolate and purify ER and mitochondria from heart tissue samples, multiple protocols were tested. The criterion used to determine whether a protocol was successful was absence of mitochondrial contamination of the ER. An enhanced protocol was established and used for subsequent experiments. Heart ventricles were rinsed in phosphate buffer saline (PBS) and minced in 1mL homogenization buffer. Samples were then homogenized in varying volumes, with different numbers of strokes, and speeds. After homogenization, samples were centrifuged in 15 mL conical tubes at 600 x g for 5 minutes to remove unbroken cells and nuclei. The
supernatants were transferred to a new tube, and the process repeated until no pellet was seen. Samples were then centrifuged at 10,000xg for 10 minutes to separate crude mitochondria and the supernatants, which contained cytosol and microsomes. To ensure that the intact
centrifuged at 18,300 rpm in a swinging-bucket rotor (Beckman-Coulter SW28) for 2 hours. In order to obtain purified ER, the layer in between 20 and 30% sucrose was removed with a syringe (this layer contains ER, whereas the layer in between 30 and 40% contains
mitochondria). The collected layer was then centrifuged at 40,000 revolutions per minute (rpm) for 1 hour to pellet the ER-enriched sample. The pellet was then resuspended in
homogenization buffer.
IV. RESULTS
Previous research conducted in our lab has shown that ACSL1 is located on the mitochondria and ER in liver. Owing to the greater ACSL contribution of ACSL1 to total ACSL activity and its presence on both mitochondria and ER, we decided to use this isoform in this study. Based on the role of mitochondria in degradative pathways, we hypothesized that ACSL1 located on mitochondria may direct fatty acyl-CoAs specifically towards β-oxidation15. Fatty acyl-CoAs can also be targeted for synthetic pathways, specifically triacylglycerol and phospholipid synthesis. Thus, it is possible that ACSL1 located on ER is important for shuttling fatty acids towards complex lipid synthesis. In order to explore pathway preference of fatty acids, we first infected cultured brown adipocytes and hepatocytes with ER-targeted and
mitochondria-targeted adenovirus ACSL1 constructs. Cells infected with ad-GFP or wild-type ad-ACSL1 were
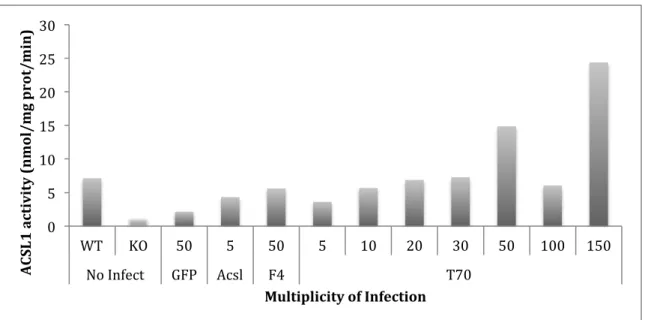
Low ACSL1 activity with a MOI of 100 may be due to an error I made in calculating the protein concentration of the sample. In the brown adipocytes, a MOI of 50 for Ad-F4-Acsl1, and a MOI of 30 for Ad-T70-Acsl1 (Figure 1) showed equal enzyme activity. One optimal dose for Ad-F4-Acsl1 and Ad-T70-Ad-F4-Acsl1 was only confirmed after analyzing results from previous experiments. Moreover, the activities of these constructs were similar to the wild-type cells. However, the ACSL1 activity in the GFP cells infected with a MOI of 50 was lower than the ACSL1 activity in wild type cells not infected with virus (Figure 1). Therefore, we concluded that GFP alters metabolism in brown adipocytes, and that brown adipocytes cannot be used in later
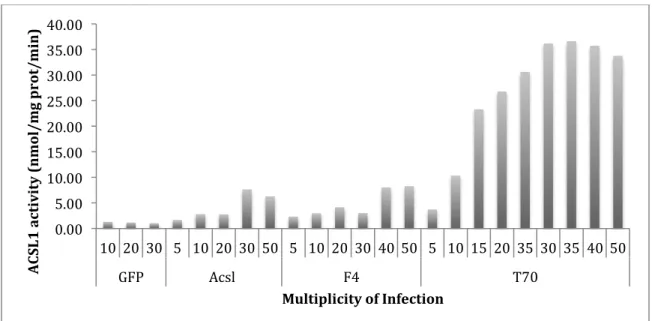
experiments. In primary hepatocytes, a MOI of 40 for Ad-F4-Acsl1, and a MOI of 10 for Ad-T70-Acsl1 (Figure 2) suggested equal enzyme activity. In addition, MOIs of 30 and 50 for Ad-Acsl showed enzyme activities that were similar to the enzyme activities for Ad-F4-Acsl1 cells infected with a MOI of 40 and Ad-T70-Acsl1 cells infected with a MOI of 10. In order to determine whether a MOI of 30 or a MOI of 50 is the optimal MOI for Ad-Acsl, additional
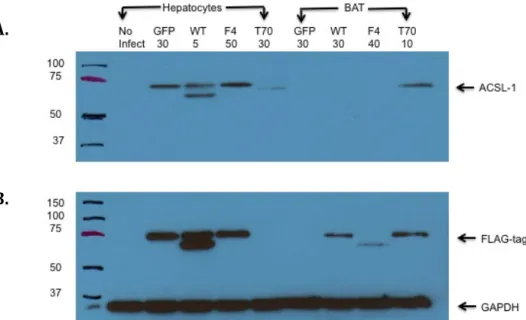
experiments must be done. Optimal MOIs were confirmed using western blot by showing equal ACSL1 expression between Ad-GFP, Ad-F4-Acsl1, and Ad-Acsl1 samples (Figure 3). The western blot for hepatocytes showed that there was equal protein expression between Ad-GFP infected with a MOI of 30, Ad-Acsl1 infected with a MOI of 5, and Ad-F4-Acsl1 infected with a MOI of 50. The optimal MOIs chosen based on the enzyme activities for hepatocytes infected with GFP and Ad-F4-Acsl1 were confirmed by protein expression (a MOI of 30 and a MOI of 50
additional experiments must be conducted in order to determine an optimal MOI for Ad-T70-Acsl1. In addition, in the western blot for hepatocytes, there are two bands at 75kDa for the expression of both ACSL1 and FLAG-tag. A possible explanation for two bands instead of one is that the proteins may have fragmented before protein loading.
Figure 1. Effect of multiplicity of infection on ACSL1 activity in virus infected brown adipocytes. Cultured pre-adipocytes from the brown adipose tissue depot were obtained from adipose-specific KO mice (Acsl1A-/-). Pre-adipocytes were differentiated into brown adipocytes in culture
media containing insulin, triiodothyronine (T3), and rosiglitazone. Brown adipocytes were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1). Equal enzyme activity between constructs was ascertained using ACSL assay. It was confirmed that an MOI of 50 for Ad-F4-Acsl1, and an MOI of 30 for Ad-T70-Acsl1 showed equal enzyme activity (n=2).
0! 5! 10! 15! 20! 25! 30!
WT! KO! 50! 5! 50! 5! 10! 20! 30! 50! 100! 150!
No!Infect! GFP! Acsl! F4! T70!!
Figure 2. Effect of multiplicity of infection on ACSL1 activity in virus infected hepatocytes. Cultured hepatocytes were obtained from liver-specific knockout (KO) (Acsl1L-/-). Hepatocytes
were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1). Equal enzyme activity between constructs was ascertained using ACSL assay. It was confirmed that an MOI of 40 for Ad-F4-Acsl1, and an MOI of 10 for Ad-T70-Acsl1 suggested equal enzyme activity (n=2).
! 0.00! 5.00! 10.00! 15.00! 20.00! 25.00! 30.00! 35.00! 40.00!
10! 20! 30! 5! 10! 20! 30! 50! 5! 10! 20! 30! 40! 50! 5! 10! 15! 20! 35! 30! 35! 40! 50!
GFP! Acsl!! F4! T70!
!
Figure 3. Effect of multiplicity of infection on ACSL1 protein expression in virus infected hepatocytes and brown adipocytes. Cultured hepatocytes and pre-adipocytes from the brown adipose tissue depot were obtained from liver-specific knockout (KO) (Acsl1L-/-) and adipose-specific KO mice (Acsl1A-/-), respectively. Hepatocytes and brown adipocytes were infected with
adenoviruses containing green fluorescent protein GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1). Equal amounts of protein were loaded for each sample (30 ug of protein) with 4X loading dye containing 10% β-mercaptoethanol. Samples were loaded with 5 µL protein marker and run at 120 V for 1.5 hours. The primary antibody dilution used for this study was 1:2000 for ACSL1. The secondary antibody dilution used was goat anti-rabbit 1:5000 for ACSL1. (A) Optimal MOI was confirmed using western blot by showing equal ACSL1
expression (at 75kDa) in hepatocytes between Ad-GFP, Ad-Acsl1, and Ad-F4-Acsl1 samples. No ACSL1 expression was seen in brown adipocytes. (B) Cells were analyzed for FLAG-tag and GAPDH. In hepatocytes, equal FLAG-tag expression was seen between GFP, Ad-Acsl1, and Ad-F4-Acsl1 samples. All samples showed equal expression of GAPDH.
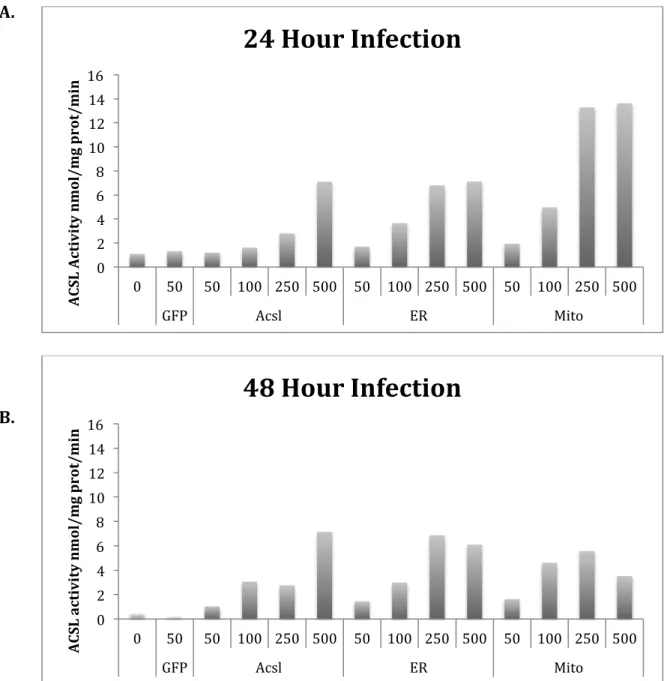
In addition to hepatocytes and brown adipocytes, cardiomyocytes were used to determine an optimal MOI. Several types of cells were used because hepatocytes, brown adipocytes, and cardiomyocytes are all highly active in energy metabolism. To determine an optimal MOI in cardiomyocytes, cells were incubated with a wide range of MOIs (50-500). Specifically, MOIs of 50, 100, 250, and 500 were initially used (Figure 4). Furthermore, from the wide range of MOIs initially used, we could narrow down which MOIs would be best, as was done in future experiments. In addition, cells were incubated for either 24 or 48 hours. In the cardiomyocytes, a higher MOI resulted in higher activity. In addition, a MOI of 50 for Ad-GFP,
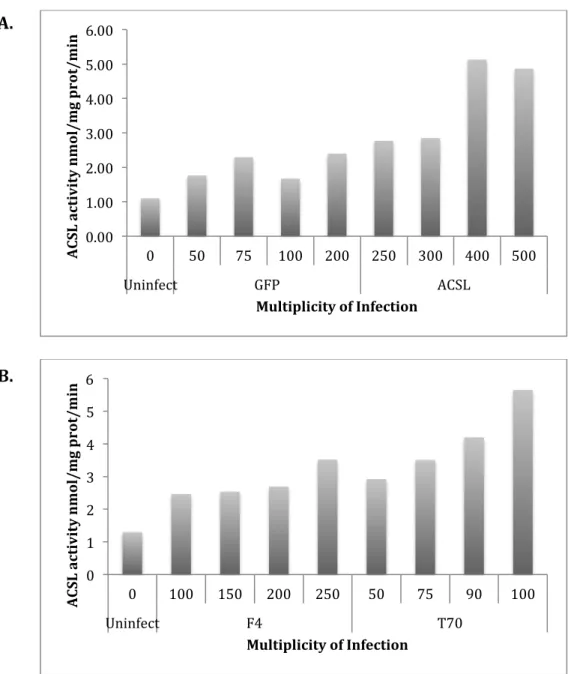
A.&
Figure 4. Effect of multiplicity of infection on ACSL1 activity in virus infected cardiomyocytes. Cardiomyocytes were obtained from a rat cardiomyoblast cell line (H9c2). The cell line was grown to confluence and then allowed to differentiate for 4 days. The growth media contained 10% fetal bovine serum, penicillin-streptomycin, and 4 mM glutamine. The differentiation media contained 1% horse media. Cardiomyocytes were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-ER-only (Ad-T70-Acsl1).
(A) Change in MOI varies ACSL activity in cardiomyocytes incubated for 24 hours (n=2). A MOI of 50 for GFP, MOIs of 250 and 500 for Ad-Acsl, MOIs of 100 and 250 for Ad-F4-Acsl1, and MOIs of 50 and 100 for Ad-T70-Acsl1 showed similar enzyme activities
(B). Change in MOI varies ACSL activity in cardiomyocytes incubated for 48 hours. A higher MOI resulted in higher activity (n=2).
0! 2! 4! 6! 8! 10! 12! 14! 16!
0! 50! 50! 100! 250! 500! 50! 100! 250! 500! 50! 100! 250! 500!
GFP! Acsl! ER! Mito!
A CS L& A ct iv it y& n m ol /m g& p ro t/ m in &
24&Hour&Infection&
0! 2! 4! 6! 8! 10! 12! 14! 16!0! 50! 50! 100! 250! 500! 50! 100! 250! 500! 50! 100! 250! 500!
GFP! Acsl! ER! Mito!
Figure 6. Effect of multiplicity of infection on health of virus infected cardiomyocytes. Cardiomyocytes were obtained from a rat cardiomyoblast cell line (H9c2). The cell line was grown to confluence and then allowed to differentiate for 4 days. The growth media contained 10% fetal bovine serum, penicillin-streptomycin, and 4 mM glutamine. The differentiation media contained 1% horse media. Cardiomyocytes were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-ER-only (Ad-T70-Acsl1). Cardiomyocytes were incubated with virus for 48 hours. MOIs of 50-500 resulted in cell death in Ad-T70-Acsl. Cells were defined as dead if they were floating. In addition, dead cells are circular, whereas healthy cells are tubular.
Figure 7. Effect of multiplicity of infection on ACSL1 activity in virus infected cardiomyocytes. Cardiomyocytes were obtained from a rat cardiomyoblast cell line (H9c2). The cell line was grown to confluence and then allowed to differentiate for 4 days. The growth media contained 10% fetal bovine serum, penicillin-streptomycin, and 4 mM glutamine. The differentiation media contained 1% horse media.
(A) Cardiomyocytes were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl). Ad-GFP cells were infected with a MOI range of 50-200, and Ad-Acsl cells were infected with a MOI range of 250-500.
(B) Cardiomyocytes were infected with adenoviruses containing ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1). Ad-F4-Acsl1 cells were infected with a MOI range of 100-250, and Ad-T70-Acsl1 cells were infected with a MOI range of 50-100. A MOI of 250 for Ad-F4-Acsl1, and a MOI of 75 for Ad-T70-Acsl1 suggested equal enzyme activity.
0.00! 1.00! 2.00! 3.00! 4.00! 5.00! 6.00!
0! 50! 75! 100! 200! 250! 300! 400! 500!
Uninfect! GFP! ACSL!
A CS L& ac ti vi ty &n m ol /m g& p ro t/ m in & Multiplicity&of&Infection& 0! 1! 2! 3! 4! 5! 6!
0! 100! 150! 200! 250! 50! 75! 90! 100!
Uninfect! F4! T70!
Figure 8. Effect of multiplicity of infection on amount of alanine aminotransferase (ALT) in the media of virus infected cardiomyocytes. Cardiomyocytes were obtained from a rat
cardiomyoblast cell line (H9c2). The cell line was grown to confluence and then allowed to differentiate for 4 days. The growth media contained 10% fetal bovine serum, penicillin-streptomycin, and 4 mM glutamine. The differentiation media contained 1% horse media.
Cardiomyocytes were infected with adenoviruses containing green fluorescent protein (Ad-GFP) (control), wild-type ACSL1 (Ad-Acsl), ACSL1 targeted to the ER-only (Ad-F4-Acsl1) or ACSL1 targeted to the mitochondria-only (Ad-T70-Acsl1). To determine the amount of ALT present in the media of cultured cells, an ALT kit containing reagent 1 (Tris buffer, L-alanine, lactate dehydrogenase) and reagent 2 (a-ketoglutarate, NADH) was obtained from Stanbio. Five parts of reagent 1 was mixed with 1 part of reagent 2, and warmed to 37°C in a water bath. Media volumes of 20 ul were added to a 90-well plate. Water was used as a blank. Using a multiple channel pipet, 200 ul of working reagent was added to each well. The plate was read at 340nm every minute for 3 minutes to determine the enzymatic rate. In Ad-T70 infected cells, high amounts (8-10 U/L) of ALT in the media were present.
It is evident that in Ad-T70 infected cells, high amounts (8-10 U/L) of ALT in the media were present (Figure 8). We can therefore conclude that for Ad-T70 infected cells, a lower MOI (50-75) should be used for infection.
0! 2! 4! 6! 8! 10! 12!
0! 50! 75! 100! 200! 250! 300! 400! 500! 100! 250! 200! 250! 50! 75! 90! 100!
Uninfect! GFP! Acsl1! F4! T70!!
U/L
&
Isolation and Purification of ER and Mitochondria
Figure 9. Effect of number of strokes used to homogenize heart on ER contamination. The positive control (as indicated by + in the western blot) was obtained from a previously purified mitochondria heart sample. The negative control (as indicated by a – in the western blot) was obtained from a previously purified heart sample that did not contain mitochondria. Heart ventricles were obtained from wild-type mice, rinsed in phosphate buffer saline (PBS) and minced in 1mL homogenization buffer. Samples were then homogenized with different numbers of strokes. The number of strokes used to homogenize the heart was either 10 or 40 strokes. Samples were centrifuged for a series of spins: 600 x g for 5 minutes, 10,000xg for 10 minutes, 10,600 rpm for 10 minutes, and 14,800 rpm for 15 minutes. A fixed-angle rotor (Beckman-Coulter Ti70.1) was used for the 10,600 rpm and 14,800 rpm spins. To purify ER, the
supernatant was layered over a sucrose gradient, which consisted of 5 mL 20%, 3 mL 30%, and 3 mL 40% sucrose with 0.4 M KCl. The sucrose gradient samples were then centrifuged at 18,300 rpm in a swinging-bucket rotor (Beckman-Coulter SW28) for 2 hours. The layer of the sucrose gradient from which samples were taken was in between 20-30% or in between 30-40%. The layer in between 20 and 30% sucrose was removed with a 25-gauge needle and syringe (this layer contains ER, whereas the layer in between 30 and 40% contains
mitochondria). The collected layer was then centrifuged at 40,000 revolutions per minute (rpm) for 1 hour to pellet the ER-enriched sample. Equal amounts of protein were loaded for each sample (30 ug of protein) with 4X loading dye containing 10% β-mercaptoethanol. Samples were loaded with 5 µL protein marker and run at 120 V for 1.5 hours. The primary antibody dilution used for this study was 1:5000 for voltage-dependent anion-selective channel protein
(VDAC). The secondary antibody dilution used was goat anti-rabbit for VDAC. The VDAC band was in between 35 kDa and 25 kDa.
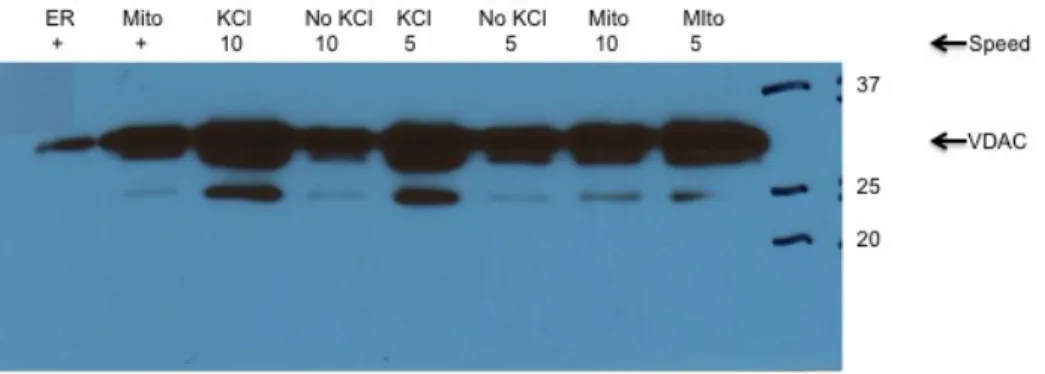
Figure 10. The speed of the homogenizer varies mitochondrial contamination of the
mitochondria.The ER positive control (as indicated by ER + in the western blot) was obtained from a previously purified ER heart sample. The mitochondria positive control (as indicated by mito + in the western blot) was obtained from a previously purified mitochondria heart sample. Heart ventricles were obtained from wild-type mice, rinsed in phosphate buffer saline (PBS) and minced in 1mL homogenization buffer. Samples were then homogenized with different speed. The speed used to homogenize the heart was either 5 or 10. Samples were centrifuged for a series of spins: 600 x g for 5 minutes, 10,000xg for 10 minutes, 10,600 rpm for 10 minutes, and 14,800 rpm for 15 minutes. A fixed-angle rotor (Beckman-Coulter Ti70.1) was used for the 10,600 rpm and 14,800 rpm spins. To purify ER, the supernatant was layered over a sucrose gradient. Two different sucrose gradients were tested and are 20, 30, 40% sucrose with 0.4 M KCl (represented as KCl in the western blot) and 1.3, 1.5, 2.0 M with no KCl (represented as no KCl in the western blot). The significance of using KCl is that it is similar to sucrose in allowing separation of mitochondria and ER. The sucrose gradient samples were then centrifuged at 18,300 rpm in a swinging-bucket rotor (Beckman-Coulter SW28) for 2 hours. The layer of the sucrose gradient from which samples were taken was in between 20-30% or in between 30-40%. The layer in between 20 and 30% sucrose was removed with a 25-gauge needle and syringe (this layer contains ER, whereas the layer in between 30 and 40% contains
mitochondria). The collected layer was then centrifuged at 40,000 revolutions per minute (rpm) for 1 hour to pellet the ER-enriched sample. Equal amounts of protein were loaded for each sample (30 ug of protein) with 4X loading dye containing 10% β-mercaptoethanol. Samples were loaded with 5 µL protein marker and run at 120 V for 1.5 hours. The primary antibody dilution used for this study was 1:5000 for voltage-dependent anion-selective channel protein
(VDAC). The secondary antibody dilution used was goat anti-rabbit for VDAC. The VDAC band was in between 35 kDa and 25 kDa.
Figure 12. Mitochondrial contamination of the ER fractions in wild type heart and liver samples. Heart ventricles obtained from wild-type mice were rinsed in PBS and minced in 2 mL
V. DISCUSSION
Currently, 1 in 3, or 34% of adult Americans are diagnosed with Metabolic Syndrome6. In addition, more people are developing type II diabetes earlier in life2. It has been suggested that there is a parallel increase in metabolic syndrome with type II diabetes4. Therefore it is
becoming even more important to diagnose the risks factors and find the underlying cause of type II diabetes. Moreover, the need to address the causes for metabolic syndrome has become increasingly important due to the changing demographic. Americans are becoming even more obese, and thus increasing their risk for metabolic syndrome. However, obesity alone cannot explain the presence of metabolic syndrome as there are individuals who are obese but do not have the syndrome. It has been proposed that in addition to obesity, insulin resistance may play a role in the development of metabolic syndrome, and type II diabetes. Although the underlying cause for insulin resistance is not known, it has been suggested that the lipid intermediate content within the cell plays a role. It is now known that increased lipid intermediate content within the cell is due to an imbalance between fatty acid delivery and intracellular fatty acid oxidation and lipid storage9. Thus, in an attempt to gain a clearer understanding of how fatty acids are shuttled to various pathways after entering the cell, this study sought to determine the amount of multiplicity of infection (MOI) necessary to attain equal ACSL1 activity between wild-type, mitochondria, and ER targeted adeno-viruses. Equal infection of cells was necessary in order to later compare fatty acid oxidation and incorporation rates.
The effect of MOI on ACSL1 activity in virus infected hepatocytes, brown adipocytes and cardiomyocytes was studied. Cells were infected with adenoviruses containing green
the activity for Ad-Acsl. However, the ACSL1 activity in the Ad-GFP cells infected with a MOI of 50 was lower than the ACSL1 activity in wild type cells not infected with virus. Therefore, we concluded that GFP alters metabolism in brown adipocytes, and that brown adipocytes cannot be used in later experiments. In primary hepatocytes, a MOI of 40 for Ad-F4-Acsl1, and a MOI of 10 for T70-Acsl1 showed equal enzyme activity. In addition, MOIs of 30 and 50 for Ad-Acsl showed enzyme activities that were similar to the enzyme activities for Ad-F4-Ad-Acsl1 cells infected with a MOI of 40 and Ad-T70-Acsl1 cells infected with a MOI of 10. In order to
determine whether a MOI of 30 or a MOI of 50 is the optimal MOI for Ad-Acsl, additional experiments must be done. However, due to GFP changing metabolism in brown adipocytes, we concluded that brown adipocytes should not be used to assess fatty acid oxidation and incorporation rates, and that further experiments using brown adipocytes would not be conducted. Furthermore, optimal MOIs were confirmed using western blot by showing equal ACSL1 expression between Ad-GFP, Ad-F4-Acsl1, and Ad-Acsl1 samples. The western blot for hepatocytes showed that there was equal protein expression between Ad-GFP infected with a MOI of 30, Ad-Acsl1 infected with a MOI of 5, and Ad-F4-Acsl1 infected with a MOI of 50. The optimal MOIs chosen based on the enzyme activities for hepatocytes infected with GFP and Ad-F4-Acsl1 were confirmed by protein expression (a MOI of 30 and a MOI of 50 respectively). However, in hepatocytes a MOI of 5 for Ad-Acsl showed lower enzyme activity when compared to the activity for Ad-F4-Acsl1 infected with a MOI of 50, but showed equal protein expression
determined that hepatocytes would not be used. Thus, determination of optimal MOIs for the various adeno-constructs in brown adipocytes and hepatocytes was not achieved.
To confirm the optimal MOI in cardiomyocytes, ACSL1 activity and health of cells were analyzed using ACSL assay, light microscopy and ALT assay. We concluded that a MOI of 50 was too low because it resulted in a low number of cells being infected but that a MOI of 500 was too high because it led to cell death. This conclusion was confirmed by the higher levels of alanine aminotransferase (ALT) in media of cells infected with a MOI of 500 (8-10 U/L) when compared to the amounts of ALT in media of cells not infected with virus (4 U/L). Furthermore, we found that cells incubated for 48 hours had higher levels of ALT in the media due to a higher observation of dead cells in the pictures taken by light microscopy. Cells were classified as dead if they were floating in the media, and looked circular and clumped under the microscope. Thus, we concluded that cells should be incubated for not more than 24 hours. We were able to further narrow down the range of optimal MOIs for each adeno-construct. We determined that a MOI range of 50-200 in Ad-GFP cells, a MOI range of 250-500 in Ad-Acsl cells, a MOI range of 100-250 in Ad-F4 cells and a MOI range of 50-100 in Ad-T70 cells showed similar ACSL1 activity. Finally, we concluded that a MOI of 250 for Ad-F4-Acsl1, and a MOI of 75 for Ad-T70-Acsl1 showed equal enzyme activity. Therefore, this project was successful in determining the optimal MOI in Ad-F4-Acsl1 and Ad-T70-Acsl1 cardiomyocytes, but not in Ad-GFP and Ad-Acsl cells.
strokes used to homogenize the heart: 10 strokes versus 40 strokes at a speed of 6. We found that 40 strokes resulted in less mitochondria contamination in the ER fraction. We then tested variation in the speed of the homogenizer: speed of 5 versus speed of 10 while using 15 up and down strokes for all samples. We concluded that a speed of 10 had less mitochondria
contamination in ER fractions. Finally, we tested variation in the amount of buffer used for homogenization: 2 mL versus 8 mL. We concluded that using 2 mL to homogenize samples resulted in less mitochondria contamination. From the series of experiments, we concluded that homogenizing samples with 40 strokes at a speed of 10 with 2 mL buffer had the least
mitochondria contamination in ER fractions. Although there was improvement (less
mitochondria contamination of ER) between each protocol, overall optimization of the protocol was not successful. Mitochondria were still present in the ER fractions. Thus, we were not able to use the modified protocol for isolation and purification of mitochondria and ER in further experiments.
Future steps will be taken to look at fatty acid oxidation and incorporation into various downstream metabolites in heart tissue using the optimal MOIs determined from this project.
VI. REFERENCES
1. Centers for Disease Control and Prevention, (2011). National diabetes fact sheet. Retrieved from website: http://www.cdc.gov/diabetes/pubs/factsheet11.htm
2. Mokdad, A., & Marks, J. (2001). The continuing increase of diabetes in the u.s. Diabetes Care, 24(2), 412. Retrieved from http://care.diabetesjournals.org/content/24/2/412.1.full? 3. American Diabetes Association, (n.d.). Diagnosing diabetes and learning about prediabetes. Retrieved from website: http://www.diabetes.org/diabetes-basics/diagnosis/
4. Sarafidis, P., & Nilsson, P. (2006). The metabolic syndrome: a glance at its history. Hypertension, 24(4), 621-6. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16531786 5. Grundy, S. (2008). Metabolic syndrome pandemic. Arteriosclerosis, Thrombosis, and Vascular Biology, Retrieved from http://atvb.ahajournals.org/content/28/4/629.full
6. Johnson, L., & Weinstock, R. (2006). The metabolic syndrome: Concepts and controversy. 81(12), 1615-1620. Retrieved from http://www.mayoclinicproceedings.org/article/S0025-6196(11)60947-6/fulltext
7. Krebs, M., & Roden, M. (2001). Free fatty acids inhibit the glucose-stimulated increase of intramuscular glucose-6-phosphate concentration in humans. Clinical Endocrinology and Metabolism, 86(5), Retrieved from http://jcem.endojournals.org/content/86/5/2153.full.pdf
8. Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase c, and ikappab-alpha. Diabetes. 2002;51:2005-2011
9. Corcoran, M., & Fielding, R. (2007). Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Clinical Nutrition, 85(3), 662-677. Retrieved from
http://ajcn.nutrition.org/content/85/3/662.long
10. Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, Coleman RA. Adipose acyl-coa synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell metabolism. 2010;12:53-64
11. Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Molecular and cellular biology. 2011;31:1252-1262
12. Li LO, Ellis JM, Paich HA, Wang S, Gong N, Altshuller G, Thresher RJ, Koves TR, Watkins SM, Muoio DM, Cline GW, Shulman GI, Coleman RA. Liver-specific loss of long chain acyl-coa synthetase-1 decreases triacylglycerol synthesis and beta-oxidation and alters phospholipid fatty acid composition. The Journal of biological chemistry. 2009;284:27816-27826
14. Li LO, Klett EL, Coleman RA. Acyl-coa synthesis, lipid metabolism and lipotoxicity. Biochimica et biophysica acta. 2010;1801:246-251
15. Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl-coa synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited
independently. The Journal of biological chemistry. 2001;276:24674-24679
16. 3. Kahn, B., & Flier, J. (2000). Obesity and insulin resistance. The Journal of Clinical Investigation, 106(4), 473-481.
17. Lopaschuk, G. (2013). Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes Care, 62(3), 711-720. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/23139350
18. Coleman, R. A., Li, L., Mashek, D., An, J., Doughman, S., & Newgard, C. (2006).
Overexpression of Rat Long Chain Acyl-CoA Synthetase 1 Alters Fatty Acid Metabolism in Rat Primary Hepatocytes. Journal of Biological Chemistry, 281, 37246-37255.
19. Yu, C., Chen, Y., Cline, G., Zhang, D., & Shulman, G. (2002). Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-associated
Phosphatidylinositol 3-Kinase Activity in Muscle. Journal of Biological Chemistry, 277, 50230-50236.
20. Lopaschuk, G. (2013). Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes Care, 62(3), 711-720. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/23139350
21. Froesch, E. R., Futo, E., & J, Z. (1992). Triiodothyronine (T3) stimulates insulin-like growth factor (IGF)-1 and IGF binding protein (IGFBP)-2 production by rat osteoblasts in vitro.
European Journal of Endocrinology, 126, 467-473.