Charge transfer induced osmylation of aromatic compounds
Full text
Figure
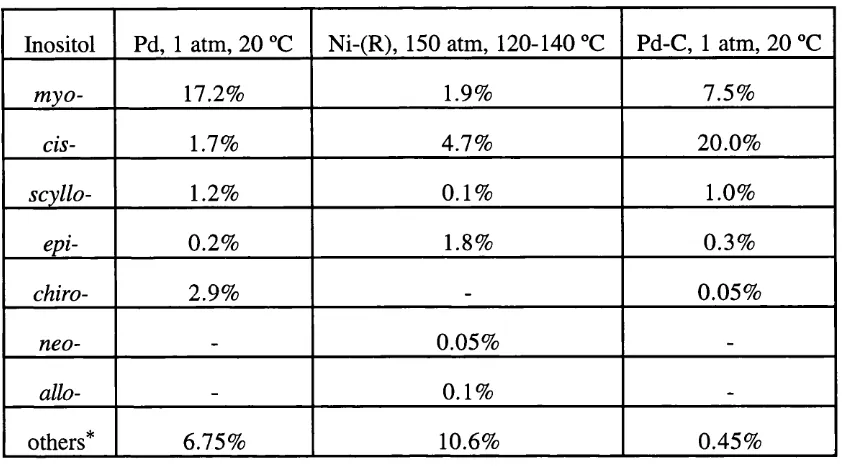
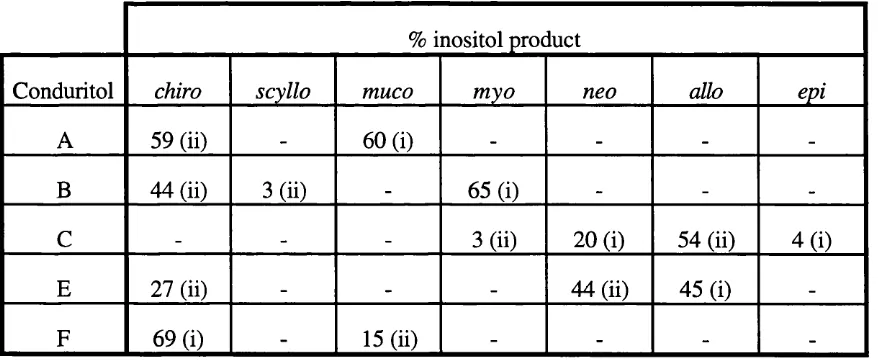

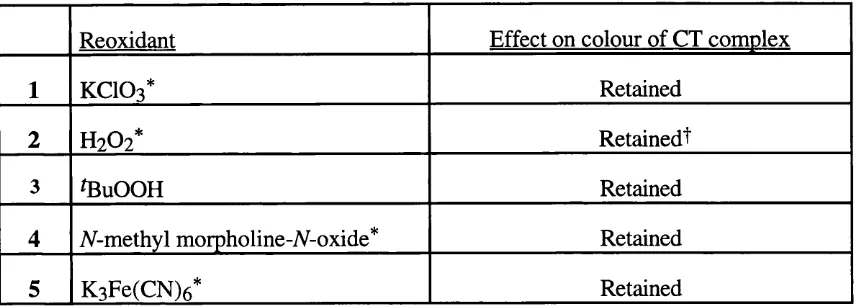

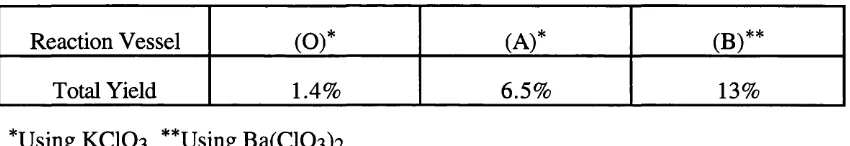



Related documents
7 Using a two standard portals technique: anterolateral portal (viewing portal) and anteromedial portal (working portal), to cut the posterior portion of the discoid medial
The range of basic shrinkage in the bulk soil was limited by delayed drying of deep soil layers, and maximum water loss in the structural shrinkage phase was 40 % of total water loss
We compared vertical water content profiles calculated from these parameters with profiles measured by x-ray attenua- tion.. A layered material model based on x-ray data was able
From the review in the next section, we know that the number of pairing computations of all existing ID-based ring signature from bilinear pairings grows linearly with the group
Dilworth DC Lens Design: Automatic and Quasi-Autonomous Computational Methods and 585. Techniques
The difficulty in controlling dengue infection stems from three root causes, ie, the presence of four different serotypes of virus, each with the independent ability to cause
To validate the set of 89 genes that were differentially expressed between distinct groups of pre- invasive lesions and are potentially involved in the early stage of DCIS
The tocopherol content of cells, fatty acids transferred and fatty acids released were all observed to increase with the addition of increasing amounts of lipoprotein lipase added
