University of Pennsylvania
ScholarlyCommons
Publicly Accessible Penn Dissertations
1-1-2015
Catalytic and Thermodynamic Studies of
Supported Core-Shell Catalysts
Chen Chen
University of Pennsylvania, chenc052@gmail.com
Follow this and additional works at:http://repository.upenn.edu/edissertations
Part of theChemical Engineering Commons
This paper is posted at ScholarlyCommons.http://repository.upenn.edu/edissertations/1646 For more information, please contactlibraryrepository@pobox.upenn.edu.
Recommended Citation
Chen, Chen, "Catalytic and Thermodynamic Studies of Supported Core-Shell Catalysts" (2015).Publicly Accessible Penn Dissertations. 1646.
Catalytic and Thermodynamic Studies of Supported Core-Shell Catalysts
Abstract
Interactions between metal catalysts and oxide supports have been known to be important in modifying the catalyst properties for many years, and catalysts with core-shell nanostructures are promising for optimizing these metal-oxide interactions. In this dissertation, core-shell nanoparticles that consist of a metal core and a metal-oxide shell were synthesized and deposited onto an alumina support. These core-shell catalysts exhibit unique catalytic and thermodynamic properties, and were investigated with different core-shell compositions as part of this thesis.
The first part of this dissertation focuses on a Pd@CeO2/Si-Al2O3 catalyst that has been developed and examined for methane-oxidation previously. To better understand this material, I investigated the catalytic, adsorption, and redox properties as they are related to the methane-steam-reforming. I also looked at the effect of calcination temperature on the catalytic properties since the catalysts were strongly influenced by the calcination temperatures, in a manner that is very different from that observed with conventional Pd/CeO2 catalysts. It was found that calcination to higher temperatures improved the performance of the Pd@CeO2 catalyst by modifying the redox properties of the ceria shell.
In the second part of this dissertation, the synthesis and investigation of core-shell catalysts was extended to other precious-metal cores and metal-oxide shells. To determine the effect of shell material, a Pd@ZrO2/Si-Al2O3 catalyst was investigated. The ZrO2 in contact with Pd was found to be reducible and to enhance the methane-oxidation.
A Au@TiO2/Si-Al2O3 catalyst was also synthesized and examined for CO oxidation. It was found that the strong interaction between Au and TiO2 not only enhanced the oxidation activity of Au but also effectively prevented Au sintering up to 873 K.
Additionally, catalysts with Pd or Pt cores and ZnO shells were prepared. The formation of Pt-Zn alloy was suggested by in-situ TEM and coulometric titration results and by catalytic properties for methanol-steam-reforming.
Finally, metal-oxide interactions were compared for Pd@CeO2 and Pt@CeO2. A very strong interaction between Pd and CeO2 helped to stabilize the core-shell structure at higher calcination temperatures and affected the CO accessibility of the core for catalyst calcined at lower temperatures, but these were not observed with Pt.
Degree Type Dissertation
Degree Name
Doctor of Philosophy (PhD)
Graduate Group
Chemical and Biomolecular Engineering
First Advisor Raymond J. Gorte
Keywords
Catalyst, Metal-oxide interaction, Nanoparticles
Subject Categories Chemical Engineering
CATALYTIC AND THERMODYNAMIC STUDIES OF SUPPORTED CORE-SHELL CATALYSTS
Chen Chen
A DISSERTATION
in
Chemical and Biomolecular Engineering
Presented to the Faculties of the University of Pennsylvania
In Partial Fulfillment of the Requirements for the
Degree of Doctor of Philosophy
2015
Supervisor of Dissertation
_______________________
Raymond J. Gorte, Professor of Chemical and Biomolecular Engineering
Graduate Group Chairperson
_______________________
Raymond J. Gorte, Professor of Chemical and Biomolecular Engineering
Dissertation Committee
John M. Vohs, Professor of Chemical and Biomolecular Engineering
Talid R. Sinno, Professor of Chemical and Biomolecular Engineering
ii
ACKNOWLEDGMENT
First, I would like to wholeheartedly thank my advisor, Professor Raymond J.
Gorte, for his continuous guidance and support. He has been a wonderful mentor to me
and I really appreciate him for giving me the opportunity to be his PhD student. Catalysis
is a fascinating research field, I have gained great insights and understanding of this
subject under his guidance and I will always be grateful for the experience working with
him.
Next, I wish to express my deep gratitude to Professor Paolo Fornasiero and
Professor John M. Vohs for their valuable suggestions and advice during the past five
years. I would like to thank professors Russell Composto and Talid Sinno for taking the
time to serve on my thesis committee and providing valuable comments. I would also like
to acknowledge Professor Christopher B. Murray for letting me use his lab facilities.
I would like to extend my sincerest thanks and appreciation to all the post doctors
and students I have been working with, particularly, Dr. Matteo Cargnello. Working with
them has made my time in graduate school enjoyable.
Finally, I would like to dedicate my dissertation of my family for their
iii
ABSTRACT
CATALYTIC AND THERMODYNAMIC STUDIES OF SUPPORTED CORE-SHELL
CATALYSTS
Chen Chen
Raymond J. Gorte
Interactions between metal catalysts and oxide supports have been known to be
important in modifying the catalyst properties for many years, and catalysts with
core-shell nanostructures are promising for optimizing these metal-oxide interactions. In this
dissertation, core-shell nanoparticles that consist of a metal core and a metal-oxide shell
were synthesized and deposited onto an alumina support. These core-shell catalysts
exhibit unique catalytic and thermodynamic properties, and were investigated with
different core-shell compositions as part of this thesis.
The first part of this dissertation focuses on a Pd@CeO2/Si-Al2O3 catalyst that has
been developed and examined for methane-oxidation previously. To better understand
this material, I investigated the catalytic, adsorption, and redox properties as they are
related to the methane-steam-reforming. I also looked at the effect of calcination
temperature on the catalytic properties since the catalysts were strongly influenced by the
calcination temperatures, in a manner that is very different from that observed with
iv
improved the performance of the Pd@CeO2 catalyst by modifying the redox properties of
the ceria shell.
In the second part of this dissertation, the synthesis and investigation of core-shell
catalysts was extended to other precious-metal cores and metal-oxide shells. To
determine the effect of shell material, a Pd@ZrO2/Si-Al2O3 catalyst was investigated. The
ZrO2 in contact with Pd was found to be reducible and to enhance the methane-oxidation.
A Au@TiO2/Si-Al2O3 catalyst was also synthesized and examined for CO
oxidation. It was found that the strong interaction between Au and TiO2 not only
enhanced the oxidation activity of Au but also effectively prevented Au sintering up to
873 K.
Additionally, catalysts with Pd or Pt cores and ZnO shells were prepared. The
formation of Pt-Zn alloy was suggested by in-situ TEM and coulometric titration results
and by catalytic properties for methanol-steam-reforming.
Finally, metal-oxide interactions were compared for Pd@CeO2 and Pt@CeO2. A
very strong interaction between Pd and CeO2 helped to stabilize the core-shell structure at
higher calcination temperatures and affected the CO accessibility of the core for catalyst
v
TABLE OF CONTENTS
ACKNOWLEDGMENT ... ii
ABSTRACT ... iii
LIST OF TABLES ... ix
LIST OF FIGURES ... x
Chapter 1. Introduction ... 1
1.1 Background ... 1
1.2 Evidence for Metal-Oxide Interactions ... 2
1.2.1 Bi-Functional Catalysts ... 2
1.2.2 Strong Metal-Support Interaction (SMSI) ... 3
1.2.3 Oxygen Storage Capacitance (OSC) ... 4
1.2.4 Alloy Formation ... 5
1.3 Mechanisms of Metal-Oxide Interactions ... 5
1.3.1 Schwab Effect ... 5
1.3.2 Bonding Interaction ... 6
1.3.3 Oxygen Transfer ... 9
1.4 Approaches to Improve Metal-Oxide Interaction ... 13
1.5 Scope of the Thesis ... 17
Chapter 2. Experimental Techniques ... 19
2.1 Catalyst Synthesis ... 19
vi
2.1.2 Supported Catalysts ... 20
2.2 Equilibrium Measurements ... 21
2.2.1 Coulometric Titration... 21
2.2.2 Transient-Pulse Measurement ... 23
2.3 Other Characterization Techniques... 24
2.3.1 TEM ... 24
2.3.2 XRD ... 25
2.3.3 Chemisorption ... 25
2.3.4 FTIR ... 26
2.3.5 TPO ... 26
2.4 Catalytic Studies ... 27
2.4.1 Steady State Rates Measurements ... 27
2.4.2 Light-Off Measurements ... 28
Chapter 3. High Temperature Calcination Improves the Catalytic Properties of Alumina-Supported Pd@Ceria Prepared by Self Assembly ... 29
3.1 Introduction ... 29
3.2 Experimental Methods ... 32
3.3 Results ... 37
3.3.1 Materials ... 37
3.3.2 Reactor Measurements ... 40
3.3.3 CO Adsorption ... 44
3.3.4 Transient-Pulse Studies ... 47
3.4 Discussion ... 52
vii
Chapter 4. Methane Oxidation on Pd@ZrO2/Si-Al2O3 is Enhanced by Surface
Reduction of ZrO2 ... 56
4.1 Introduction ... 56
4.2 Experimental Methods ... 58
4.3 Results ... 62
4.4 Discussion ... 76
4.5 Summary ... 79
Chapter 5. Au@TiO2 Core-Shell Nanostructures with High Thermal Stability ... 80
5.1 Introduction ... 80
5.2 Experimental Methods ... 82
5.2.1 Synthesis of MUA-Au Nanoparticles ... 82
5.2.2 Preparation of Au@TiO2 Core-Shell Structures ... 83
5.2.3 Preparation of Conventional Au/TiO2 ... 83
5.2.4 Characterization ... 84
5.3 Results and Discussion ... 85
5.4 Summary ... 94
Chapter 6. Supported Platinum-Zinc Oxide Core-Shell Nanoparticle Catalysts for Methanol Steam Reforming ... 95
6.1 Introduction ... 95
6.2 Experimental Methods ... 98
6.2.1 Materials ... 98
6.2.2 Synthesis of the Catalysts ... 98
6.2.3 Characterization ... 101
viii
6.4 Summary ... 115
Chapter 7. A Comparison of Hierarchical Pt@CeO2/Si-Al2O3 and Pd@CeO2 /Si-Al2O3 ... 116
7.1 Introduction ... 116
7.2 Experimental Methods ... 117
7.2.1 Synthesis of the Catalysts ... 117
7.2.2 Characterization ... 118
7.3 Results and Discussion ... 120
7.3.1 Catalyst Characterization ... 120
7.3.2 WGS activity ... 122
7.4 Summary ... 130
Chapter 8. Conclusion ... 131
ix
LIST OF TABLES
Table 2.1: Reactant gas-phase composition ... 27
Table 3.1: Pd dispersions, in percent, based on CO uptakes at room temperature for different H2 reduction temperatures. ... 45
Table 4.1: Pd dispersions based on CO uptakes at room temperature. ... 66
Table 4.2: Results of O2 titration and pulse study on different Pd based samples that used
in this chaper. ... 72
Table 6.1: Metal apparent dispersions (%) based on CO uptake at room temperature for the calcined samples after different pre-treatment conditions. .... 110
Table 6.2: Methanol steam reforming activity and CO2 selectivity of the catalysts at 573
K. ... 113
x
LIST OF FIGURES
Figure 1.1: Schematic representation of the procedure to obtain dispersible Pd@CeO2
core-shell nanostructures [60]. ... 15
Figure 1.2: Schematic representation of the agglomeration of Pd@CeO2 structures when
using the pristine alumina (A) and their deposition as single units after treatment of the same support with triethoxy(octyl)silane (TEOOS) [63]... 16
Figure 2.1: Schematic diagram of coulometric titration setup [64] ... 22
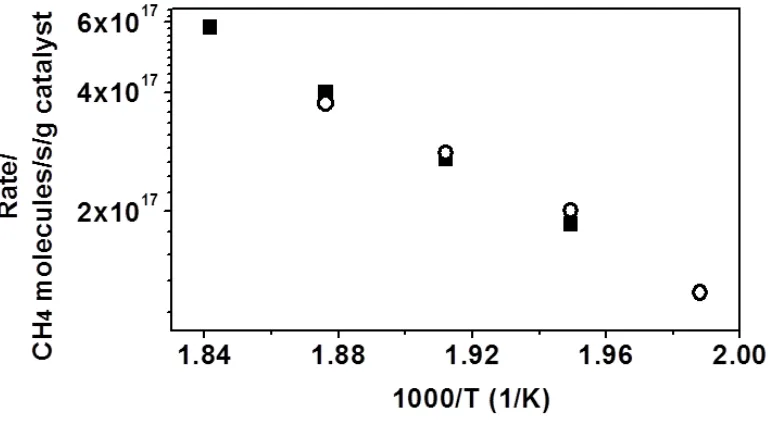
Figure 3.1: Methane-oxidation rates in 0.5% CH4 and 5% O2 over Pd@CeO2
/Si-Al2O3(1073) with total flow rate of 120ml min-1 (■), 80ml min-1 (
○
). ... 34Figure 3.2: Rates for MSR reaction over Pd@CeO2/Si-Al2O3(1073) with total flow rate
of 120ml min-1 (■), 60ml min-1 (
○
). The concentration of CH4 and H2O were held at 35torr and 70 torr respectively. ... 34
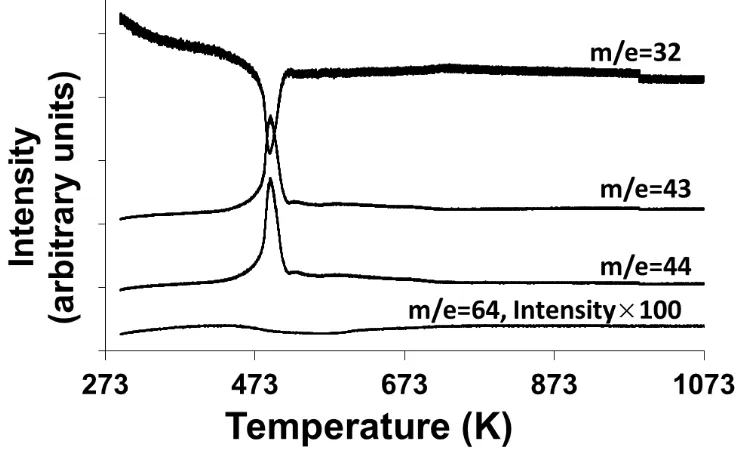
Figure 3.3: TPO results for uncalcined samples of Pd@CeO2/Si-Al2O3 in 10% O2. The
data were obtained monitoring O2 (m/e = 32), various hydrocarbons (m/e = 43), CO2 (m/e
= 44), and SO2 (m/e = 64). The data for O2, hydrocarbons, and CO2 were obtained using
0.1 g of sample, while data for SO2 were obtained using 1.0 g of catalyst for enhanced
sensitivity. ... 38
Figure 3.4: XRD powder patterns of Al2O3, Pd@CeO2/Si-Al2O3(773), and Pd@CeO2
/Si-Al2O3(1073) (Top). Bottom part: reference powder diffraction patterns of bulk Pd and
CeO2 phases labeled with Miller indices. ... 39
Figure 3.5: Methane-oxidation rates in 0.5% CH4 and 5% O2 for Pd@CeO2
/Si-Al2O3(773) (■), Pd@CeO2/Si-Al2O3(1073) (●), and Pd@CeO2/Si-Al2O3(1123) (▲). Data
were taken with 3.8 torr CH4 and 38 torr O2... 40
Figure 3.6a): CH4 conversion as a function of time during the MSR reaction at 693 K
over Pd@CeO2/Si-Al2O3(1073). Data were taken with 35 torr CH4 and 70 torr H2O. .... 42
Figure 3.6b): CH4 conversion as a function of time during the MSR reaction at 693 K
over Pd@CeO2/Si-Al2O3(773). The sample was exposed to oxidizing and reducing
xi
Figure 3.7: Rates for the MSR reaction over Pd/CeO2 (■), Pd@CeO2/Si-Al2O3(1073)
after oxidizing in O2 (
○
), and Pd@CeO2/Si-Al2O3(1073) after reducing in H2(Δ). Datawere taken with 35 torr CH4 and 70 torr H2O. ... 43
Figure 3.8: Diffuse reflectance FTIR spectra obtained in flowing He after exposure to CO at room temperature for the following samples: a) Pd@CeO2/Si-Al2O3(773) after
reduction at 423 K; b) Pd@CeO2/Si-Al2O3(773) after reduction at 673 K; c)
Pd@CeO2/Si-Al2O3(1073) after reduction at 423 K; and d) Pd@CeO2/Si-Al2O3(1073)
after reduction at 673 K. ... 46
Figure 3.9: Results from the pulse measurement performed at 673 K on Pd@CeO2
/Si-Al2O3(773). The data were obtained monitoring CO2 (m/e = 44), O2 (m/e = 32), and CO
(m/e = 28). ... 48
Figure 3.10: A plot of the amount of CO2 produced in a set of CO-O2 pulses, measured
as a function of temperature, using the Pd/Al2O3(■), Pd@CeO2/Si-Al2O3(773) (○), and
Pd@CeO2/Si-Al2O3(1073) (Δ) samples. ... 49
Figure 3.11a): A plot of selected peaks from the mass spectra of the effluent from the pulse reactor using Pd@CeO2/Si-Al2O3(773) at 673 K. The peaks that were monitored
were CO2 (m/e = 44 and 28), O2 (m/e = 32 and 16), CO (m/e = 28), and CH4 (m/e = 16).
The inlet to the reactor is shown at the top. ... 50
Figure 3.11b): A plot of selected peaks from the mass spectra of the effluent from the pulse reactor using Pd@CeO2/Si-Al2O3(1073) at 673 K. The peaks that were monitored
were CO2 (m/e = 44 and 28), O2 (m/e = 32 and 16), CO (m/e = 28), and CH4 (m/e = 16).
The inlet to the reactor is shown at the top. ... 51
Figure 4.1: TEM images of Pd@ZrO2/Si-Al2O3 catalysts. Parts A and B correspond to
the 500 °C sample, while part C to the 800 °C calcined sample. In parts D and E, a lattice profile analysis of a single particle is presented. ... 63
Figure 4.2: Powder XRD patterns of Pd/Si-Al2O3(1), and Pd@ZrO2/Si-Al2O3 that
calcined at different temperature and with different Pd:ZrO2 ratio(2-6). 500 °C 1:6 (2),
500 °C 1:9 (3), 500 °C 1:12 (4), 800 °C 1:6 (5), 800 °C 1:9 (6). Reference powder diffraction pattern of tetragonal zirconia is shown at bottom. Al2O3 phase was marked by
closed squares, ZrO2 phase was marked with open squares. ... 65
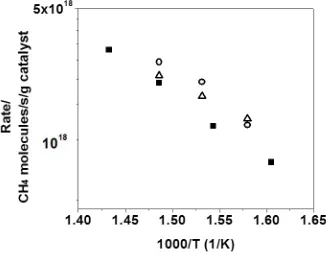
Figure 4.3: Rates for methane-oxidation reaction over 1-wt%Pd/Al2O3 calcined at 500 °C
(■),1-wt%Pd@9-wt%CeO2/Si-Al2O3 calcined at 500 °C (△),1-wt%Pd@9-wt%ZrO2
xii
and 1-wt%Pd@9-wt%ZrO2/Si-Al2O3 calcined at 800 °C (●).Data were taken with 0.5%
CH4 and 5% O2. ... 67
Figure 4.4: Effect of high temperature water poisoning over 1-wt%Pd@9-wt%ZrO2
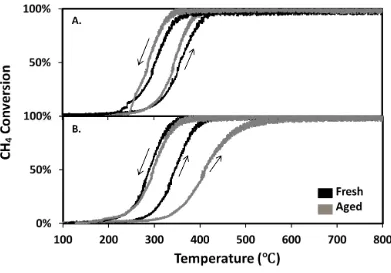
/Si-Al2O3 (A), and 1-wt%Pd@9-wt%CeO2/Si-Al2O3 (B). Both catalysts were calcined at
800 °C. The aged samples were kept under wet reaction conditions for 4 hours at 600 °C, then cooled to room temperature under He, followed by light-off test. Data were taken with 1% CH4 and 5% O2. The heating and cooling rates are 10 °C min-1. ... 68
Figure 4.5: Effect of water on methane light-off curves over 1-wt%Pd@9-wt%ZrO2
/Si-Al2O3 (A), and 1-wt%Pd@9-wt%CeO2/Si-Al2O3 (B). Both catalysts were calcined at
800 °C. Data were taken with 1% CH4, 5% O2 and 10% H2O (if present). The heating and
cooling rates are 10 °C min-1. ... 70
Figure 4.6: Redox isotherms for Pd/Al2O3 (A) with 1-wt% Pd (□) and 5-wt% Pd (◆),
1-wt% Pd/ZrO2 (B), and 1-wt% Pd@9-wt%ZrO2/Si-Al2O3 (C). ◆ symbols were obtained
starting from the oxidized state. ◇ symbols were measured while re-oxidizing the sample. All measurements were conducted at 600 ºC. ... 74
Figure 5.1: Representative TEM images of MUA-Au nanoparticles (A, B, C) and histogram of particle size distribution with fit for a Gaussian distribution (D). ... 86
Figure 5.2: Representative TEM images of as-synthesized Au@TiO2 core-shell
structures. ... 87
Figure 5.3: Representative TEM images of Au@TiO2/Si-Al2O3 (A, B) and Au/TiO2 P25
(C, D) calcined to 400 °C. Arrows point at Au particles in the two samples. ... 89
Figure 5.4: CO oxidation rates for Au@TiO2/Si-Al2O3 (■), and Au/TiO2 (◇). Both
samples were calcined at 400 °C. Data were taken with 25 torr CO and 12.5 torr O2. .. 90
Figure 5.5: Light-off curves of CO conversion against temperature for Au@TiO2
/Si-Al2O3(A), and Au/TiO2 (B) that calcined at different temperatures. Data were taken with
7.6 torr CO and 152 torr O2. ... 92
Figure 5.6: Representative TEM images of Au@TiO2/Si-Al2O3 (A, B) and Au/TiO2 P25
(C, D) catalysts calcined to 600 °C. Arrows point at Au particles in the two samples. ... 93
xiii
Figure 6.2: Representative low (A, B) and high (C-F) resolution transmission electron microscopy (TEM) images of Pt@ZnO/Si-Al2O3 sample calcined at 773 K
in air. Yellow arrows point to core-shell structures where the higher contrast is attributed to Pt cores surrounded by a lighter, ZnO shell. ... 105
Figure 6.3: TEM images taken at 20 s interval (from A to D) of two Pt@ZnO core-shell structures (A) that progressively transform into Pt-Zn alloyed particles under the electron beam irradiation. The images were taken at 200 kV. ... 106
Figure 6.4: Representative TEM image of Pd@ZnO/Si-Al2O3 sample calcined at 773 K
in air. Yellow arrows point to core-shell structures where the higher contrast is attributed to Pd cores surrounded by a lighter, ZnO shell. ... 107
Figure 6.5: X-ray diffraction (XRD) patterns of (A) Si-Al2O3, (B) Pt/Al2O3, (C) ZnO,
(D) Pt/ZnO, and (E) Pt@ZnO/Si-Al2O3. Inset in (D) show magnification of the platinum
(111) region. ... 108
Figure 6.6: X-ray diffraction (XRD) patterns of (A) Si-Al2O3, (B) Pd/Al2O3, (C) ZnO,
(D) Pd/ZnO, and (E) Pd@ZnO/Si-Al2O3 calcined at 773 K. ... 108
Figure 6.7: Redox isotherms for Pt@ZnO/Si-Al2O3 at 873 K. Closed symbols
were obtained starting from the oxidized state. Open symbols were measured while re-oxidizing the sample. ... 111
Figure 6.8: (A) Methanol steam reforming (MSR) catalytic activity and (B) CO2
selectivity over the Pt@ZnO/Si-Al2O3 core-shell (triangle symbol), and
conventional Pt/Al2O3 (circle symbol) and Pt/ZnO (diamond symbol) calcined at
773 K. The catalysts were pretreated in a reducing (5% H2/He) atmosphere at 523
K for 30 minutes. All catalysts with 1 wt% of Pt. ... 112
Figure 6.9: (A) Methanol steam reforming (MSR) catalytic activity and (B) CO2
selectivity over the Pd@ZnO/Si-Al2O3 core-shell (triangle symbol), and conventional
Pd/Al2O3 (circle symbol) and Pd/ZnO (diamond symbol) calcined at 773 K. The catalysts
were pretreated in a reducing (5% H2/He) atmosphere at 523 K for 30 minutes. All
catalysts with 1 wt% of Pd. ... 114
Figure 7.1: Representative transmission electron microscopy (TEM) images of Pt@CeO2/Si-Al2O3 samples calcined at (A) 773 K and (B) 1073 K in air. Arrows indicate
Pt particles. ... 121
/Si-xiv
Al2O3-1073K. The samples were pretreated by oxidation in 20% O2-He mixture at 573 K
for 30 min prior to rate measurements. The reactant partial pressures were fixed at 25 torr CO and 25 torr H2O. ... 123
Figure 7.3: CO conversion in the WGS reaction as a function of time at 623 K over the Pt@CeO2/Si-Al2O3-773 K (■) and Pd@CeO2/Si-Al2O3-773 K (○) catalysts. The reactant
partial pressures were fixed at 25 torr CO and 25 torr H2O. ... 124
Figure 7.4: Redox isotherms for (A) 1-wt% Pt/Al2O3 (■) and 1-wt% Pd/Al2O3 (◇), and
(B) 1-wt% Pd/CeO2 (◆). The samples were initially calcined in air at 773 K.
Measurements were conducted at 873 K, starting from the oxidized state. ... 126
Figure 7.5: Redox isotherms for 1-wt%Pt@CeO2/Si-Al2O3 (■) and 1-wt%Pd@CeO2
/Si-Al2O3 (◇)calcined at (A) 773 K and (B) 1073 K. The samples were initially calcined in
1
Chapter 1. Introduction
1.1 Background
Metal catalysts play very important roles in many industries, including petroleum
refining, and chemicals production, as well as auto catalyst emissions control and other
environmental applications. For many reactions, the catalytic activity and selectivity
depend upon metal particle size because this in turn affects the surface area and number
of catalytic active sites. To enhance the number of active sites per unit mass, most
commercial metal catalysts are dispersed in the pores of high-surface-area metal-oxide
supports in the form of nanoparticles. Although the primary function of metal-oxides that
are used as supports is to maintain metal dispersion, it has been demonstrated that contact
between an oxide support and the active metal can strongly influence the activity and
selectivity of the catalysts for some reactions. Therefore, understanding and controlling
metal-oxide interactions is crucial for developing optimal precious-metal based catalysts.
However, interactions between the metal and its oxide support are generally not well
understood and need to be further investigated with novel approaches and techniques.
The goal of my work was to understand and enhance these interactions by
maximizing the interfacial contact between the support oxide and the metal. In particular,
I prepared catalysts with core-shell structures where the active metal is encapsulated in a
layer of porous oxide. In addition to catalytic measurements, I also examined the
thermodynamic redox properties of the metal-oxide interfaces in these core-shell catalysts
2
with the metal oxide interface for the first time. A number of important
metal-oxide catalytic systems were examined in this work, including Pd-CeO2, Pd-ZrO2,
Pd-ZnO, Pt-CeO2, Pt-ZnO, and Au-TiO2. In addition to developing a better fundamental
understanding of the nature of metal-metal oxide interfaces, methods were developed for
improving catalyst stability at higher temperatures.
1.2 Evidence for Metal-Oxide Interactions
The metal-oxide interactions are complex functions of structure and composition
and will be different for different catalyst systems. While it is difficult to cover all
different types of interaction, in this section, I will choose several representative
examples to illustrate typical reactions where chemical and physical interactions between
the metal and the metal-oxide support play critical roles in determining the catalyst
properties.
1.2.1 Bi-Functional Catalysts
In some catalytic processes involving the petroleum and petrochemical industries,
the catalyst is considered to be bi-functional in that both the metal and the metal-oxide
are themselves catalytically active. One of the most important examples of this is
dehydroisomerization of methylcyclopentane over Al2O3-supported Pt in naphtha
reforming. Pt is well-known to be an efficient dehydrogenation catalyst while Al2O3 has
acid sites that are active for isomerization of olefins. For catalysts in which Pt and Al2O3
are in close contact, methylcyclopentane is first dehydrogenated on Pt to
3
cyclohexadiene. Finally, the cyclohexadiene migrates to Pt and is dehydrogenated to
benzene [1].
Although this type of metal-support interaction is certainly important, the function
of both components is well understood. Also, the support in this case does not change the
properties of the metal or affect reactions that occur on the metal. Indeed, it is known that
the metallic Pt and the alumina do not even need to be in direct contact for this reaction to
proceed. Therefore, I will not consider these types of metal-metal oxide interactions
further.
1.2.2 Strong Metal-Support Interaction (SMSI)
The concept of ‘Strong Metal-Support Interaction’ (SMSI) was first introduced in
1978 to refer to the drastic changes in the adsorption properties of group VIII metals that
were observed when deposited on a titanium oxide support after high temperature
reduction [2]. One of the characteristic properties of group VIII metals is that their
surface atoms can chemisorb H2 or CO molecules at ambient temperature and this
property is commonly used to measure the metal dispersion. However, it was found that
titania-supported metals exhibited strongly suppressed adsorption following
high-temperature reduction. It was suggested that a strong metal-support interaction between
the metal and the reduced titania modified the adsorption properties of the metal. Later, it
was confirmed by surface science investigations that reduced titania can migrate onto
metal particles and then form an overlayer over the metal surface [3], which blocked
4
are not changed by the reduced titania, the migration of a reduced oxide onto the metal
particles is still referred to as SMSI.
1.2.3 Oxygen Storage Capacitance (OSC)
The simultaneous removal of three primary exhaust pollutants (CO, unburned
hydrocarbons and NOx) remains a major challenge in emission control for automobile
engines. This is because conversion of CO and HC to CO2 and H2O requires oxidizing
conditions and conversion of NOx to N2 requires a reducing condition. This objective can
be achieved by using a so-called ‘three-way’ catalyst (TWC) within a
‘close-to-stoichiometric’ air/fuel ratio window. A typical TWC consists of a high surface area
support, such as Al2O3, catalytically active Group VIII metals that are impregnated into
the support, and an oxygen-storage component, CeO2. The facile reducibility of CeO2
makes it an efficient oxygen capacitor that is able to store oxygen under oxidizing
conditions and release oxygen under reducing conditions. Unlike the situation for
bi-functional catalysts, ceria reduction cannot occur in the automotive exhaust environment
without contact with the catalytic metal [4, 5].
However, there is still debate about how reduction of the ceria occurs, whether
oxygen is transferred from ceria to the metal or the reductant is transferred from the metal
to the ceria. There is some indication that oxygen is transferred to the metal in some cases
but it is not known if the transfer occurs only at peripheral sites or whether oxygen can be
5
1.2.4 Alloy Formation
Another possible metal-support interaction involves the formation of alloys. Some
oxides may be reduced to their metallic form and then react with the active metal to form
an alloy that has superior catalytic properties compared with the elementary metals. For
example, PtZn alloys can be formed from Pt/ZnO catalyst upon reduction. The alloy
catalysts have been shown to be more selective for hydrogenation of α, β-unsaturated
aldehydes to the corresponding saturated alcohols [6]. In another example, CO2
selectivity for the methanol-steam-reforming reaction was found to be greatly improved
when Pd or Pt is supported on ZnO, Ga2O3 and In2O3 due to the formation of Pd–Zn, Pd–
Ga, Pd–In, Pt–Zn, Pt–Ga and Pt–In alloys [7].
1.3 Mechanisms of Metal-Oxide Interactions
As pointed out earlier, the nature of metal-metal oxide interactions are not well
understood in many cases. Various mechanisms have been proposed to explain how the
oxide affects the metal [8-10]. Here, I will briefly discuss some of the more important
cases that have been proposed for understanding support effects. Some of these will be
relevant for explaining the work I have carried out in this thesis.
1.3.1 Schwab Effect
One of the first models developed for understanding how support oxides and
metals interact was proposed by Schwab and Koller [11]. They suggested that, when a
catalytic metal is supported on a semiconducting oxide, electrons could transfer between
6
was invoked initially when the SMSI phenomenon was first observed with
titania-supported catalysts. It was suggested at that time that titania became semiconducting
under reducing conditions and that it could then donate electrons to the supported metal.
Because electron densities of metals are orders of magnitude larger than electron
densities in semiconducting oxides, such long-range electron-transfer mechanisms are
difficult to explain. Also, electron-transfer mechanisms would imply that support effects
should depend on metal loading, which was not observed with SMSI in titania-supported
metals. Therefore, electron-transfer mechanisms have grown out-of-favor as a way for
explaining support effects.
1.3.2 Bonding Interactions
The effects observed with titania-supported metals are presently thought to arise
from bonding between the metal and reduced titania. The attraction between the metal
and reduced titania is analogous to wetting phenomena observed at liquid-solid interfaces
and results in migration of reduced titania onto the metal particles [3, 12]. When the
metal surface is completely covered, adsorption suppression is caused by simple site
blocking.
In less extreme cases, bonding interactions between a metal and its oxide support
could also induce changes in geometry of the metal particles. For example, it was shown
that the sublimation energy of Au on SiO2 is abnormally low (50 kcal mol-1) at 0.2
monolayers (mL), but increases rapidly with increasing Au coverage to the bulk value (90
kcal mol-1) at 5mL. The low value of sublimation energy at low Au coverage is
7
atoms are not attracted to silica; at the edge of small clusters, the Au atoms are bonded to
fewer Au neighbors and hence are weakly held to the surface. By contrast, the
sublimation energy remains at 50 kcal mol-1 with increasing Au coverage up to 2mL for
the case of Au/TiO2. This has been attributed to a stronger interaction between Au and
TiO2, which in turn leads to greater wetting of TiO2 by Au compared with the situation
for Au/SiO2. These bonding interactions result in higher Au dispersions and improved the
thermal stabilities of Au with TiO2.[13].
There is evidence that bonding interactions between an active metal and its oxide
support can also modify the thermodynamic redox properties of supported metal particles.
In a thermodynamic investigation of supported Co catalysts for Fischer-Tropsch synthesis
(FTS) [14], it was found that redox properties of Co that had been deposited onto a ZrO2
support can be affected significantly by the metal-support interface, while redox
properties of Co supported on mesoporous silica were indistinguishable from that found
for bulk Co. It was reported that this observation is critical for understanding FTS
reaction over supported Co catalyst because metallic Co is the active phase for
low-temperature FTS reaction. Although bulk thermodynamic calculations indicate that Co
should remain metallic under FTS conditions, redox isotherms obtained by coulometric
titration of Co/ZrO2 catalysts suggested that interactions between Co and ZrO2 can cause
particles to be oxidized under FTS conditions. It was further suggested that this effect is
due to bonding between highly dispersed Co and oxygen from the zirconia support [15].
It is important to notice that interfacial effects could be very different with metal
8
supported nanoparticles differ from bulk materials and this could change bonding
interactions with the oxide support. One case where this appears to occur is the situation
when Au is supported on titania. Au/TiO2 catalysts prepared by deposition-precipitation
were reported to be capable of catalyzing CO oxidation at temperatures as low as 90 K
[16], while neither unsupported Au nor TiO2 alone is active for this reaction. The activity
of Au nanoparticles is not dependent simply on Au particle size; the composition of the
support is also critically important, with rates for water-gas shift reaction over Au/TiO2
that are 20 times higher than rates over the same Au particles supported on Al2O3 [17].
In the Au and TiO2 example, there is a question whether the bonding interactions
are important over the entire interface between the metal and the oxide or just at edge
sites where the metal and oxide are both exposed to the gas phase. Although many
authors have proposed that only the peripheral sites are affected [18-23], it has also been
proposed that the active sites on Au particles are those that are separated from the titania
by one monolayer of Au [24]. In this case, the entire surface of a “flat” particle, two
monolayers thick, would be active. With either model, the active sites are in the
proximity of the TiO2 support and the enhanced catalytic activity has been ascribed to the
strong bonding interaction between Au and TiO2 support.
The manner by which titania bonding affects Au is uncertain. One recent study of
Au supported on highly reduced TiOx film has suggested that strong bonding between Au
and reduced Ti atoms leads to electron-rich Au and it is this form of Au that exhibits
exceptionally high activity for CO oxidation [25]. Theoretical studies also show that
9
electronic properties of Au clusters through charge transfer from TiO2 to Au [26, 27],
which in turn affects the catalytic properties of Au.
It should be emphasized that, while there are numerous studies reporting evidence
for electronic interactions between metal nanoparticles and the oxide support, the
situation is complicated by the fact that the properties of metal nanoparticles are already
intrinsically different from that of bulk materials. Separating effects of particles size and
metal-support interactions is challenging.
1.3.3 Oxygen Transfer
For reducible oxides, such as ceria, the role of the support also includes
transferring oxygen to and from the supported metal. The ability of ceria to donate
oxygen is crucial for three-way catalysis [28, 29], water-gas shift (WGS) catalysis
[30-33], hydrocarbon-reforming catalysis [34], and hydrocarbon oxidation catalysis [35, 36].
For example, reaction rates for the WGS reaction on ceria supported Pd has been shown
to be orders of magnitude higher than on non-reducible Al2O3 supported Pd [33].
Oxygen transfer appears to be also important for methane combustion over
supported-Pd catalysts [37, 38]. It is widely acknowledged that PdO is more active than
Pd for this reaction. However, the PdO phase tends to decompose to metallic Pd at high
temperatures (over 1073 K) resulting in decreased activity. The re-oxidation only occurs
until the temperature decreases to below 873 K [39]. It has been reported that PdO phase
can be stabilized when it is supported on reducible oxides such as TiO2 and CeO2, with a
10
The key reason for the special properties of ceria-based catalysts is the facile
redox shift between Ce4+ and Ce3+. Under reducing conditions, oxygen atoms can be
removed from ceria and result in a non-stoichiometric composition of partially reduced
CeO2-x. Each released O atom leaves behind an oxygen vacancy and creates two Ce3+
cations by transferring two electrons to two Ce4+ cations. Under oxidizing conditions, a
reverse process takes place.
It has been found that oxygen from the ceria support can be transferred to active
metal and then used to oxidize species that adsorbed on the metal. Reduced ceria can be
oxidized by molecular O2 or by O from another reactant such as water. This proposed
mechanism has been used successfully to explain the enhanced activity observed with
several reactions which can be carried out on these catalysts, including CO oxidation [40],
water-gas shift [30], and steam-reforming [34]. Considering WGS reaction over Pd/ceria,
the mechanism can be written as follows:
2 2 3
2CeO +PdPdO+Ce O (1.1)
2
CO+PdOPd+CO (1.2)
2 3 2 2 2
Ce O +H OH +2CeO (1.3)
Interestingly, this proposed mechanism appears to contradict the thermodynamic
redox properties of ceria. Thermodynamics indicates that CeO1.98 is the equilibrium
stoichiometry at 873 K and P(O2)~10-26 atm [41]. Therefore, ceria should not be able to
provide oxygen to the supported metal in most reactions, i.e. the reaction conditions are
too oxidizing and reduction of ceria should not occur. The likely explanation for these
11
Usually, ceria that is used in catalysis is in nanoparticle form [42-45] or is promoted by
other materials, such as zirconia [46, 47].
It is possible to rationalize why ‘catalytic’ forms of ceria might be more reducible.
With regards to pure ceria, Ce+4 is the most stable state in stoichiometric CeO2 because
the symmetry of the fluorite crystal places the Ce atoms in such a geometry that bonding
with the oxygen matches the direction of the 4f orbitals, which is critical for stabilizing
Ce+4. When ceria is nanostructured, the crystalline structure becomes highly defective
and Ce+4 are expected to be destabilized relative to Ce+3. The presence of additives (for
example, Ti+4) can also create distortions in the fluorite structures. In both cases, the
perfect fluorite structure of CeO2 can be interrupted, causing a relatively high stability of
Ce+3 that result in an increased reducibility. In the case of zirconia-doped ceria, it has
been suggested that Zr+4 can pair with Ce+3 to form Zr2Ce2O7 locally within the
mixed-oxide structure. Again, formation of such compounds would then stabilize Ce+3. Finally,
small cerium crystallites are expected to have a high surface energy. If this free energy of
the surface is large enough, the reducibility of ceria as a whole will be affected.
It should be noted that some have suggested certain crystal surfaces of ceria are
more easily reduced than others. For example, theoretical calculations have argued that
the (100) surface is preferentially reduced in ceria relative to the (111) surface [48]. Some
support for this idea has come from an experimental study which found that nano-rods
synthesized to preferentially expose (111) planes were easier to reduce than normal
12
the surface could only be reduced with great difficulty [50] (e.g. by sputtering with Ar+).
Therefore, crystal surface dependence on reducibility has yet to be proven.
While the reducibility of ceria can promote many reactions, questions have been
raised regarding the optimal level of reducibility. For example, a study by Bakhmutsky et
al. [51]has shown that making ceria too reducible does not always have a positive effect
on catalytic activity. In their work, addition of Pr to ceria in a Pd/ceria catalyst leads to a
mixed oxide that can give over-reduction of ceria, which in turn gives a much lower
activity for the WGS reaction. In this case, oxygen in the praseodymium-ceria mixed
oxide was so weakly bound that oxygen could not be restored by oxidation in steam.
Therefore, there appears to be an optimum reducibility for ceria that will lead to the most
active catalysts.
Zirconia is another support material that has attracted considerable interest in a
variety of catalyst systems. Even though ZrO2 is normally considered an irreducible
oxide, and should therefore not promote reaction in the same way that CeO2 does, there
are some indications that interaction between the active metal and ZrO2 support can
affect the catalytic activity for CO hydrogenation [52], methanol decomposition [53], and
methane-steam-reforming [54]. This may be associated with the fact that oxygen mobility
of ZrO2 supports is relatively high at moderate and high temperatures, so that oxygen can
be exchanged between the active metal and the support. For example, it has been
demonstrated that on a conventional Pd/ZrO2 catalyst, oxygen from a ZrO2 support may
contribute to methane-oxidation reaction, and the involvement of ZrO2 increases with
13
In metal-oxide catalytic systems, interactions between phases are crucial for
reactions. Whether the mechanism involves bonding interactions, oxygen transfer, or
both simultaneously, there must be direct contact between the metal and the oxide
support. Therefore, any potential advantages associated with these interactions require
maximizing the interface between the metal and the oxide. Some methods that have been
developed to maximize interfacial contact will be discussed in the next section.
1.4 Approaches to Improve Metal-Oxide Interaction
For traditional supported metal catalysts which consist of small metal particles
deposited onto high-surface-area oxide support, the metal-oxide interaction can be
enhanced by simply increasing the metal dispersion. Significant effort in the field of
catalysis has gone into synthesizing particles with sizes down to the nanometer scale in
order to enhance the specific surface area, the interaction with the support and,
consequently, the catalytic properties. However, such enhancement is limited by the
‘single interfacial perimeter’ that forms between the metal particles and the oxide support.
With the rise of nanotechnology, ‘multiple points of contact’ between the metal
and the oxide becomes possible; and this provides three dimensional control of the
reaction zone and greatly modifies the properties of the catalysts. For example, Pietron et
al. [56] formed isolated Au nanoparticles in Au-TiO2 composite aerogels by adding
alkanethiolate-monolayer-protected Au clusters to a titania sol before gelation. In the
resulting aerogel nanoarchitecture, each Au particle was in contact with multiple TiO2
14
with much larger Au particle sizes. Indeed, the particle size in their study was so large
that previous reports would have predicted the Au would be inactive.
The ‘3-D’ design can also be used to address another common problem in
catalysis: thermal stability of highly dispersed active metal. Nanoparticles tend to rapidly
sinter into larger clusters due to their high surface energy, resulting in a loss of activity. It
would be desirable to use new approaches to prevent such sintering by encapsulating the
dispersed metal nanoparticles with additional materials. Co-precipitation and
microemulsion are two of the most commonly used techniques to make encapsulated
nanostructures. In the former method, the preformed metal particles (or their precursors)
are precipitated together with the metal oxide precursor. The latter one involves using
micro-emulsions as a nano-reactor; metal particles are produced inside micelles, which
are in turn coated with the oxide precursor to form the encapsulated metal. However,
these methods cannot provide great control over the final structures. Our group used both
of these methods to prepare Pd in ceria nanostructures for water-gas shift reaction and
found that ceria layers made by co-precipitation method were insufficient to prevent
sintering of the metal, while with the microemulsion method, the ceria layer underwent
condensation that blocked the active sites of the metal [57].
Novel nanotechnologies have allowed great improvements in the synthesis of
catalysts with well-controlled size, shape, and surface properties. The deliberate tailoring
of the nanostructure can lead to unique catalytic properties, and provide the level of
control that is required to maximize the metal-support interface. The controlled structure
15
shown in the work of Yeung et al. [58], who prepared Pt@CeO2 core-shell structures by a
modified microemulsion procedure with controllable ceria thickness. The Pt core was
shown to interact with CeO2 shell in an optimum geometry to form a unique interface,
which exhibited high activity for the water-gas shift reaction, but was completely inactive
for the undesirable side reaction, methanation. In another example, Zhang et al. [59]
prepared Au nanoparticles supported on TiO2/SiO2 core-shell composites, which were in
turn encapsulated in mesoporous silica. The small TiO2 particles anchored on SiO2 beads
led to a better dispersion of Au and the encapsulation improved sinter-resistance during
calcination at 773 K.
Recently, our group used self-assembly method to achieve even more precise
control over core-shell structures, allowing finer tuning of the oxide shell and
maximizing the metal–oxide interactions [60-62]. A schematic of the method is shown in
Figure 1.1. Pd@CeO2 was prepared by reacting preformed 11-mercaptoundecanoic acid
(MUA)-protected Pd nanoparticles with cerium (IV) alkoxide, followed by a controlled
hydrolysis in the presence of dodecanoic acid.
Figure 1.1: Schematic representation of the procedure to obtain dispersible Pd@CeO2
16
After forming the dispersible Pd@CeO2 nanoparticles in solution, they could be
deposited onto Al2O3 supports by impregnation. However, Pd@CeO2 particles prepared
in this manner are slightly hydrophobic and tend to present as agglomerates rather than as
isolated particles adhering to the hydrophilic Al2O3 support. In an alternative method, the
dispersed Pd@CeO2 can be adsorbed onto a silane modified Al2O3. In this case, the
alumina surface is made hydrophobic by reaction with triethoxy(octyl)silane (TEOOS)
(Figure 1.2). After adsorption of the core-shell particles, the solid residue can be
recovered, crushed, and calcined.
In previous work, it was demonstrated that these core-shell catalysts exhibit very
exciting properties [63]. First, this material shows outstanding thermal stability, with the
Pd cores remaining isolated even after heating the catalyst to 1123 K. More importantly,
the methane-oxidation activity for the Pd@CeO2/Si-Al2O3 catalyst was exceptional,
exhibiting rates that were at least 50 times higher than that of a normal Pd/CeO2 catalyst,
well beyond what a simple optimization of interfacial site concentrations would provide.
Figure 1.2: Schematic representation of the agglomeration of Pd@CeO2 structures
17
While it has been inferred that this exceptional activity may result from improved
metal-oxide interaction between the Pd core and the ceria shell, many questions remain. A
further investigation on metal-oxide interaction and a better understanding of the
properties of this core-shell material are needed.
1.5 Scope of the Thesis
My thesis work fell into two categories. The first part focuses on Pd@CeO2
/Si-Al2O3 catalysts. The goal was to gain a better understanding of the properties of this
material that make it such good catalyst. The second category involved extending the
synthesis to other metal@oxide systems and into investigations of metal-oxide
interactions with different core-shell compositions.
The thesis is divided into 8 chapters. Chapter 2 provides descriptions of the
sample preparations, characterization techniques and experimental methods used in this
work. In Chapter 3, I describe an investigation of Pd@CeO2/Si-Al2O3 catalysts for the
methane-steam-reforming reaction. In this work, I looked at the effect of calcination
temperature for this catalyst and this reaction. The oxidation and reduction properties of
Pd@CeO2/Si-Al2O3 calcined at different temperatures were explored using pulse
measurements and related to the catalytic rates.
Chapter 4 focuses on the interactions between Pd and ZrO2 in Pd@ZrO2/Si-Al2O3
catalysts, and investigates how they relate to the observed high activity this material has
for the methane-oxidation reaction. This work provides for the first time a quantitative
18
addition, because steam has been reported to strongly inhibit catalytic reactions and cause
severe deactivation over Pd-based catalysts, the stability of Pd@ZrO2/Si-Al2O3 for
methane-oxidation in the presence of steam was also investigated.
Chapter 5 reports the synthesis and characterization of Au@TiO2/Si-Al2O3
core-shell catalysts. Two of the major issues with Au-based catalysts are poor thermal stability
due to the very low surface energy compared to other transitional-metal based catalysts
and a strong particle-size dependence on activity. The interaction between Au and TiO2
in the core-shell catalysts helped stabilize the materials against sintering, maintaining a
high activity.
In Chapter 6, the synthesis and characterization of Pt@ZnO/Si-Al2O3 and
Pd@ZnO/Si-Al2O3 core-shell catalysts are described. In order to investigate whether
Pt-Zn alloys can be formed upon mild reduction due to the intimate contact between the core
and the shell, in-situ TEM and coulometric titration experiments were conducted. The
applications of these catalysts in methanol-steam-reforming reactions were also discussed
and compared to that of the conventional supported catalysts.
Chapter 7 compares the catalytic and adsorption properties of Pd@CeO2/Si-Al2O3
with Pt@CeO2/Si-Al2O3 catalysts. The interactions between the ceria shell and the
different metal cores were inferred from their thermodynamic isotherms and used to
explain different stabilities that were observed on these two catalysts.
19
Chapter 2. Experimental Techniques
This chapter will describe the methods used to synthesize the catalysts, as well as
the experimental principles and applications of the various techniques used to
characterize the catalysts.
2.1 Catalyst Synthesis
All catalysts used in this thesis were synthesized in our laboratory. The catalysts
can be categorized into two types: core-shell catalysts supported on hydrophobic alumina
(referred as metal@oxide/Si-Al2O3); and metal-oxide, supported, precious-metal catalysts
(referred as metal/oxide). In this section, general synthesis methods will be discussed,
while the details of the preparation procedure, such as ingredients and calcination
temperatures for particular metal-metal oxide combinations, will be described in
corresponding chapters.
2.1.1 Core-Shell Catalysts
The general method for synthesis of the dispersible core-shell nanostructures
includes 3 steps [61, 62]: 1) preparation of 11-mercaptoundecanoic acid (MUA) protected
metal nanoparticles, 2) the self-assembly of a metal alkoxide on the MUA protected
metal particles by reaction of metal alkoxide with the acid functionality of the MUA
ligand, and 3) the controlled hydrolysis of the remaining alkoxide functionality in the
presence of protective ligands (dodecanoic acid) to obtain metal@oxide nanoparticles
20
The metal@oxide nanoparticles, dispersed in THF, were then adsorbed from
solution onto an Al2O3 support that had been modified by reaction with Triethoxy Octyl
Silane (TEOOS) [63]. This modification of the support is required to make the Al2O3
hydrophobic so that core-shell nanoparticles could adsorb onto the surface as isolated
units. The Al2O3 itself was purchased from Alfa Aesar as γ-Al2O3 and then stabilized by
calcining to 1173 K for 24 h. After removing the catalysts from solution by centrifugation
and drying, the resulting powders were calcined to various temperatures with a heating
ramp of 3 K min-1.
2.1.2 Supported Catalysts
For comparison purpose, two types of metal/oxide catalysts were prepared in this
thesis. Conventional types of supported catalysts were prepared by impregnation of
metal-oxide support with aqueous solutions of metal ammonium nitrate precursors. The
amount of precursor and support were carefully chosen to obtain the desired weight
loadings. The slurry was then dried and calcined in air to decompose the precursors and
obtain a catalyst in the oxidized state.
Nanoparticles deposited on metal-oxide support were also prepared from the
MUA-protected metal nanoparticles without oxide shells. In this case, oxide supports
were first dispersed in THF, and then an appropriate amount of MUA-protected metal
nanoparticles, also dispersed in THF, were added to the mixture dropwise. After stirring
overnight to allow complete adsorption, the solid residue was recovered by centrifugation,
21
2.2 Equilibrium Measurements 2.2.1 Coulometric Titration
The redox properties of the catalysts were measured using coulometric titration,
which is an electrochemical technique that provides the equilibrium oxygen
stoichiometry of the oxide sample as a function of P(O2) that above the sample. For
instance, for the Pd-PdO equilibrium:
( ) 2( ) ( )
2Pds O g 2PdOs (2.1)
The equilibrium constant can be calculated using:
2 -1 2 2 2 α
K = =P(O )
α ×P(O ) PdO eq
Pd
(2.2)
where the activity of solids is equal to 1. The differential Gibbs Free Energy of the
oxidation reaction can then be calculated using the following equation:
2
RT ln(K )eq RTln[P(O )]
G
(2.3)
It is important to recognize that the experimental P(O2) values are so low that
they are not actual partial pressures of O2 over most of the range reported in this thesis,
but rather oxygen fugacity established by an equilibrium with another reaction:
2 2 2
2H +O 2H O (2.4)
22
2 2
2
P(H O) log[P(O )] 2 log(K ) 2log[ ]
P(H ) eq
(2.5)
The coulometric titration setup used in this thesis is shown in Figure 2.1 [64]. The
setup consists of a YSZ (yttria-stabilized zirconia) tube with Ag electrodes painted on
both inside and outside. Pt wires were attached to the inner and outer electrode using Ag
paste. The inner Pt wire was then spot-welded to an Ultra-Torr Swagelok fitting which
had been fitted over one of the ends of the YSZ tube.
Figure 2.1: Schematic diagram of coulometric titration setup [64].
During measurements, approximately 1g of sample was placed in an alumina
crucible that was then inserted into the coulometric titration cell. The entire apparatus
was heated to 873 K; and a mixture of 5% O2, 11% H2O, and 84% Ar was allowed to
23
measurements were started from the oxidized state of the samples. After stopping the
flow, the ends of the YSZ tube were sealed using glass stoppers. To perform the actual
measurements, oxygen was electrochemically pumped out of the YSZ tube by applying a
potential across the electrodes with a Gamry Instruments potentiostat. The amount of
oxygen removed was determined by integrating the current as a function of time. After
removing the desired amount of oxygen, the system was allowed to come to equilibrium
with the electrodes at open-circuit. The criterion that used for establishing equilibrium
was that the open-circuit potential across the electrodes changed by less than 3 mV day-1,
which typically took 4 to 10 days. Finally, the equilibrium P(O2) was calculated from the
Nernst equation (equation 2.6) and the open-circuit voltage (OCV).
2 2
P(O )
OCV
ln(
)
4
P(O )
in out
RT
F
(2.6)As an additional check that equilibrium was achieved and that there were no
system leaks, most isotherms were measured again starting with the reduced sample and
pumping oxygen back into the electrochemical cell.
2.2.2 Transient-Pulse Measurement
The oxidation and reduction of some of the samples were also examined by the
transient-pulse experiments. By separating interactions between the catalyst and different
reactive molecules sequentially, pulse measurements could be used to investigate the
redox properties and deactivation mechanisms on the core-shell catalysts. These
24
spectrometer to analyze the concentrations of the effluent gases [65, 66].
Computer-controlled solenoid valves allowed step changes in the gas composition. In all
measurements, helium was the major component of the gas phase, with a total flow rate
of 20 mL min-1, while the concentrations of the reactive component (either CO, O2, H2, or
H2O) were chosen to be 10% of the total gas stream. Water was introduced to the reactor
by saturation of a He carrier gas flowing through deionized water and condensed with an
ice-trap before the products were analyzed. 1.0 g of catalyst was used for these
experiments. Prior to loading the samples in the reactor, each sample was pressed into
thin wafers that were then broken into smaller pieces. Integration of the partial pressures
of the reactor effluent as a function of time allows determination of the amounts of
oxygen that could be added or removed from the catalyst by the various reactive
components at different temperatures. No attempt was made to analyze the shapes of the
pulses because coupling between desorption, re-adsorption, reaction, and diffusion does
not allow for a unique determination of rate processes in transient experiments of this
type [67].
2.3 Other Characterization Techniques 2.3.1 TEM
Transmission Electron Microscopy (TEM) characterization was performed on a
Jeol JEM 2100 operating at 200 kV. Samples were prepared either by drop-casting the
particles directly from THF solution onto 300 mesh carbon-coated Cu grids (Electron
Microscopy Sciences) or by dispersing the powders into isopropanol and then
25
2.3.2 XRD
Powder X-ray Diffraction (XRD) was used to determine the structures and the
chemical phase composition of the core-shell catalysts. The XRD patterns were recorded
on a Rigaku Smartlab diffractometer equipped with a Cu Kα source (λ=1.5405 Å). The
sample powders were finely dispersed in 2-propanol by sonication and then drop-cast
onto glass slides. The intensities of the diffracted beam were measured while sampling
different diffraction angles. The crystallite particle size (d) could be estimated using the
Scherrer equation:
κλ d=
B(2θ)cosθ (2.7)
where κ is a shape factor usually equal to 1, λ is the X-ray wavelength, B is the peak
widthat half the maximum intensity, and θ is the diffraction angle.
2.3.3 Chemisorption
The apparent metal dispersions were measured using CO chemisorption at room
temperature in a home-built adsorption apparatus [68]. Two different protocols, with
different reducing temperatures, were used to pretreat the core-shell catalysts. The
calcined samples were first placed in the adsorption apparatus, oxidized at 673 K in 200
Torr O2 for two minutes, evacuated, and then re-oxidized. After repeating the oxidation
step three times, the samples were evacuated and then reduced by exposure to 200 Torr
H2 at either 423 K or 673 K for 5 minutes, followed by a second evacuation. The
26
observed. CO chemisorption was performed at room temperature by adding small pulses
of CO to the samples until a pressure rise in the sample cell was detected [69].
2.3.4 FTIR
Fourier Transform Infrared Spectroscopy (FTIR) was used to analyze the
adsorption properties on the catalyst surface. The spectra were recorded using Mattson
Galaxy 2020 FTIR spectrometer. The spectrometer was equipped with a Spectra-Tech
Collector II diffuse-reflectance accessory to allow measurements on powdered samples
with control over temperature and atmosphere.
2.3.5 TPO
In order to determine the temperature required for removing the organic
protecting agents from core-shell nanoparticles, Temperature Programmed Oxidation
(TPO) measurements were performed in the same system used in the transient-pulse
measurements on samples dried at 338 K. Experiments were conducted with either 0.1 g
or 1.0 g of sample to determine when most of the carbon leave the sample and to look for
the possibility of sulfur contamination from the 11-mercaptoundecanoic acid (MUA)
ligands used in preparing the metal nanoparticles. The gas-phase composition during
TPO was 20% O2 in He, with a total flow rate of 25 mL min-1; and the heating rate was 3
27
2.4 Catalytic Studies
2.4.1 Steady-State Rates Measurements
Several reactions were used in this thesis to characterize catalytic properties of the
core-shell catalysts with different metal-metal oxide compositions. The steady-state rates
were measured using 100 mg of sample in a ¼-in, quartz, flow reactor with an on-line gas
chromatograph (SRI8610C) equipped with a Hayesep Q column and a TCD detector. The
reactor pressure was always atmospheric. The partial pressure of each reactant was
controlled by adjusting the relative flow rate of each component, with a total flow rate of
120 ml min-1, corresponding to Gas Hourly Space Velocity of 72,000 mL g-1 h-1. Detailed
reactant gas-phase composition for each reaction was summarized in Table 2.1. For those
experiments in which H2O was added, H2O was introduced by bubbling He through a
H2O saturator and the content of H2O was controlled by the temperature of the saturator.
For all measurements where rates are reported, the conversions of reactant were kept well
below 15%, so that differential conditions could be assumed.
Table 2.1: Reactant gas-phase composition.
Reaction Catalyst Reactant composition (balanced in He)
Methane oxidation Pd@CeO2, Pd@ZrO2 3.8 torr (0.5%) CH4, 38 torr (5%) O2
Methane steam reforming Pd@CeO2 35 torr (4.6%) CH4, 70 torr (9.2%) H2O
CO oxidation Au@TiO2 25 torr (3.3%) CO, 12.5 torr(1.65%) O2
Water-gas shift Pd@CeO2, Pt@CeO2 25 torr (3.3%) CO, 25 torr(3.3%) H2O
28
2.4.2 Light-Off Measurements
The light-off measurements were performed in the same system used in the
transient-pulse measurements using 100 to 400 mg of sample. For CO oxidation reaction,
the composition of the reactant mixture was chosen to be 1% CO and 20% O2 in He and
the total flow rate was maintained at 60 ml min-1. For methane oxidation, the composition
of the reactant mixture was chosen to be 1% CH4 and 5% O2 in He and the total flow rate
was maintained at 120 ml min-1. In both cases, the conversions of reactant were measured
29
Chapter 3. High Temperature Calcination Improves the Catalytic
Properties of Alumina-Supported Pd@Ceria Prepared by Self Assembly
3.1 Introduction
High activity and thermal stability are required for catalysts used in
high-temperature reactions, such as oxidation. The most active catalysts for
methane-oxidation are based on Pd [38, 70], which is susceptible to sintering at high temperatures;
but both activity and thermal stability can be improved through the proper choice of
oxide support [71-77]. Ceria can affect rates on supported Pd catalysts because its redox
properties can provide an additional channel for oxygen on the Pd. Further improvement
of these catalysts requires the optimization of the Pd-support interactions and one
approach for accomplishing this is through the development of core-shell catalysts in
which a porous oxide “shell” surrounds the metal “core”.
Various methods have been reported for preparing catalysts with a core-shell
structure [78]; and, not surprisingly, the different preparation methods can result in
materials with vastly different structures and catalytic properties. For example, materials
having an “egg-yolk” structure, in which the metal core is in a void volume made from a
larger oxide shell [79], will have very different properties from materials prepared using
microemulsions, in which the metal core will be in physical contact and completely
surrounded by an oxide shell that is typically thicker [80]. Additionally, many of the