MiR 9 and miR 200 regulate PDGFRβ mediated endothelial differentiation of tumor cells in triple negative breast cancer
Full text
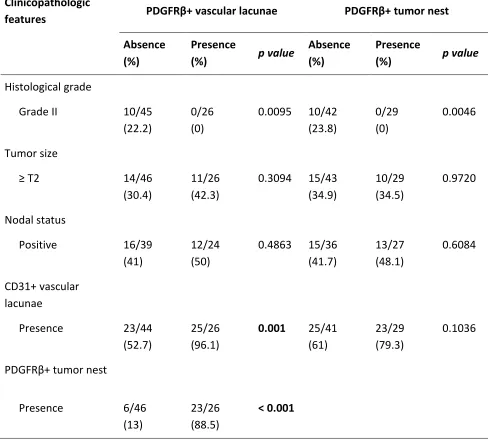
Figure
Related documents
The various parameters considered for study are Nusselt number, heat transfer coefficient and heat transfer rate for a constant Prandtl number (0.7) and Grashof
Figure 3, 4, and 5 shows the effect of tie thickness on the maximum total settlement, differential settlement, and bending moment generated for collapsible soil
An interesting research problem in this context is the STS- class: if secure, it would allow efficient encryption schemes based on M ultivariate Q uadratic polynomials.. Still,
As a “disease with no cure” and recognized as a threat for the Borana community, individual susceptibility to the risk of HIV infection is related to men’s mobility, “other
' '4 These suggested levels of professional responsibility created con- flicts with the Code of Professional Responsibility (Code) 5 and elic- ited strong favorable and
Watugala, Sumudu transform: a new integral transform to solve differential equations and
The urban ecological characteristics of the first two surface material categories can still have a positive effect on the urban climate even at a high degree of soil sealing..
Theanine levels were also determined in the different partitions of the tea shoot as was done in the above study but now considering the effect of withering time varying from three
