molecules
ISSN 1420-3049 www.mdpi.com/journal/molecules ArticleNovel Fluorinated Phosphorus–Sulfur Heteroatom Compounds:
Synthesis and Characterization of Ferrocenyl- and
Aryl-Phosphonofluorodithioic Salts, Adducts, and Esters
Guoxiong Hua, Junyi Du, Brian A. Surgenor, Alexandra M. Z. Slawin and J. Derek Woollins *
EaStCHEM School of Chemistry, University of St. Andrews, St Andrews, Fife KY16 9ST, UK; E-Mails: gh15@st-and.ac.uk (G.H.); jd83@st-and.ac.uk (J.D.); basurgenor@gmail.com (B.A.S.); amzs@st-and.ac.uk (A.M.Z.S.)
* Author to whom correspondence should be addressed; E-Mail: jdw3@st-andrews.ac.uk; Tel./Fax: +44-1334-463-861.
Academic Editor: Jean Jacques Vanden Eynde
Received: 07 June 2015 / Accepted: 30 June 2015 / Published: 3 July 2015
Abstract: A series of novel ferrocenyl- and aryl-phosphonofluorodithioic salts, adducts, and esters has been prepared. The reaction of 2,4-diferrocenyl-1,3,2,4-diathiadiphosphetane 2,4-disulfide {[FcP(μ-S)S]2, FcLR} with dry KF or tetrabutylammonium fluoride (TBAF) led to
the corresponding potassium and tetrabutylammonium salts of ferrocenyldithiofluorophosphinic acids. Treating potassium ferrocenyldithiofluorophosphinic acid with an equimolar amount of tetraphenylphosphonium chloride readily yielded the corresponding organic adducts, and with mono- and di-halogenated alkanes generated a series of the corresponding esters of ferrocenylphosphonofluoridodithioates. Similarly, using 1,3-epithionaphtho[1,8-cd][1,2,6] oxadiphosphinine 1,3-disulfide or Belleau’s Reagent in place of FcLR resulted in the corresponding novel salts, adducts, and ester derivatives. All new compounds have been characterized by means of multi-NMR (1H, 13C, 31P, 19F) spectroscopy and accurate mass
measurement in conjunction with single crystal X-ray crystallography of four structures.
1. Introduction
Organophosphorus-fluorine heteroatom compounds (OPFHACs) bearing a P–F bond are of interest due to their diverse chemical or biological activities, such as selective phosphorylating agents in synthesis, and efficient inhibition of several classes of enzyme [1–12]. This new class of thiophosphates bearing one or more anion FPS− units was first prepared from the reaction of alkali metal fluorides and P4S10 by
Roesky’s group [13,14]. Since then, the synthesis of thiophosphoryl halides S=PF3 and S=PFCl2, and
their derivatives S=PF2NH2, S=PFClNH2, S=PF2N=PF2X (X = Br, NH2 or OH), S=PF2N=PCl3, and
S=PF2N=PF2N=C=NSiMe3 was reported successively [15–19]. There are few examples of the synthesis
of simple phosphonofluorodithioates ROP(S)(S−)F containing a fluorine atom attached directly to the
phosphorus atom [15,20,21]. The nucleoside phosphonofluoridodithioate monoesters were also prepared via oxidation of nucleoside phosphonodithioate with I2 in pyridine in the presence of TMSCl, followed
by addition of triethylamine trihydrofluoride (TAF) [22,23]. Similar analogues were obtained from a one-pot sequential reaction of 1,3,2-dithiaphospholane P(III) derivatives, which were converted readily into the corresponding P(V) compounds by addition of elemental sulfur and finally into phosphonofluoridodithioates by further treating with TBAF [24]. The importance of phosphoro-fluorine compounds in pure and applied chemistry invigorated our interest in synthesizing new phosphorodithioates bearing the P–F group. Recently, we have reported the synthesis of a series of phenylphosphonofluorodiselenoic salts, adducts, and esters [25]. Herein, we extend this procedure for the synthesis of potassium and tetrabutylammonium salts of ferrocenyl- and aryl-phosphonofluoridodithioates, and the related organic adducts and esters. To the best of our knowledge, this is the first reported synthesis and characterization of ferrocenyl-phosphonofluorodithioates [FcPS2F]− and their structural
analogues, providing a valuable addition to the library of phosphodithioate compounds. 2. Results and Discussion
The preparation, spectroscopic characterization, and crystal structures of a ferrocene analogue of Lawesson Reagent, 2,4-diferrocenyl-1,3,2,4-diathiadiphosphetane 2,4-disulfide {[FcP(μ-S)S]2, FcLR}
has been reported by our group [26,27]. FcLR reacted with two equivalents of fresh dry potassium fluoride in dry acetonitrile at 80 °C under N2 atmosphere for 1 h, giving rise to potassium
ferrocenylphosphonofluoridodithioate 1 in 97% yield; or with two equivalents of tetrabutylammonium fluoride (TBAF) in tetrahydrofuran at room temperature for 1 h providing tetrabutylammonium ferrocenylphosphonofluoridodithioate 2 in 99% yield (Scheme 1). Both reactions were fast and very straightforward and must be performed in a moisture and oxygen-free atmosphere. Treatment of FcLR with dry KCl or KBr in acetonitrile or with HCl and HBr in the presence of triethylamine in dry acetonitrile or tetrahydrofuran at room temperature did not result in the similar chloride and bromide products to 1 and 2, indicating that the Cl− and Br− anions are much less reactive nucleophiles than
Scheme 1. Salts 1 and 2 derived from FcLR and KI or TBAF.
Using an analogous process to Yilmaz and coworkers [28], salt 1 was obtained as a yellow solid and is insoluble in organic solvents but soluble in oxygen-free water and slowly decomposed when stored at room temperature. Organic salt 2 was prepared as golden sticky oil, is soluble in organic solvents, and shows good air stability at room temperature. Both compounds show the anticipated molecular ion peaks [M − K]− or [M – N(n-Bu)
4]− and satisfactory accurate mass measurement. The 31P-NMR spectra
exhibits doublets at δp = 132.1 ppm in compound 1 and 127.3 ppm in compound 2, respectively,
attributable to the presence of the P–F single bond, and the values are significantly bigger than that in their selenium counterpart PSe2 ions [25]. In the 19F-NMR spectra, doublets are observed with 1J(P,F)
coupling constants of 1008 Hz for both 1 and 2 within the known literature values [14,25,28,29]. It should be noted that the 1H-NMR spectrum for compound 1 is poor quality (Figure S1); this may be
explained by the inherent “shielding” effect of salt ions in the solution leading to sample conductivity [30] or, more likely, the presence of traces of oxidized paramagnetic ferrocenium species. However, the
1H-NMR spectrum still clearly shows that only ferrocenyl ring protons are present.
We presumed that compounds 1 and 2 are, like phenylphosphonofluorodiselenoic salts, strong nucleophiles [25] and therefore are able to serve as useful precursors for the synthesis of a wide variety of functionalized heteroatom systems and ligands. Compounds 1 and 2 should have the same reactivity toward organic substituents; therefore we chose compound 1 as a target staring material to explore their reactivity. Treating 1 with an equal molar amount of tetraphenylphosphonium chloride in degassed water at room temperature led to the formation of tetraphenylphosphonium ferrocenylphosphonofluoridodithioate 3 in 91% yield, though it should be noted that we have not established quantitative exchange of the cations in this or subsequent reactions. Reacting 1 with half equimolar amount of p-xylylene dibromide in dry tetrahydrofuran in room temperature generated 1,4-phenylenebis(methylene) bis(ferrocenylphosphonofluoridodithioate) 4 in 87% yield. Similarly, compound 1 was allowed to react with an equivalent of mono-halogenated alkanes, giving a series of esters of ferrocenylphosphonofluoridodithioates 5–9 in 64%–86% yields, respectively (Scheme 2). It is worth noting that products 5 and 8, in which the phenyl groups bear the strong electron-withdrawing group NO2 and C≡N group, were obtained in rather lower yields (64% and 66%, respectively); thus,
Scheme 2. Synthesis of heteroatom derivatives 3–9 from salt 1.
Compounds 3–9 are air- and moisture-stable oils, pastes, or solids and are soluble in common organic solvents such as dichloromethane, chloroform, acetone, and tetrahydrofuran. All new compounds show the anticipated molecular ion peaks [M]+, and were confirmed by satisfactory accurate mass
measurements. Not surprisingly, the 31P- and 19F-NMR spectra of 3 show similar patterns to 1 and 2
with identical 1J(P,F) coupling constants apart from another singlet signal in the 31P-NMR spectrum at
δP = 23.2 ppm, assigned to the PPh4 cation ion. The 31P-NMR spectra of 4–9 display signals ranging
from δP = 116.0 to 118.0 ppm with 31P–19F coupling constants differing from J(P,F) = 1098–1101 Hz.
In the 19F-NMR spectra of compounds 4–9, two equal signals in the range δF = −42.6–−38.3 ppm being
considerately bigger than that in their selenium counterpart esters (δF = −58.5–−55.8 ppm) [25] are
observed with the matching 31P–19F coupling constants.
By using the same procedure, we have carried out the synthesis of potassium 3-fluoronaphtho[1,8-cd] [1,2,6]oxadiphosphinine-1-thiolate 1,3-disulfide 11 and tetrabutylammonium, 3-fluoronaphtho[1,8-cd] [1,2,6]oxadiphosphinine-1-thiolate 1,3-disulfide salt 12 as shown in Scheme 3. 1,3-Epithionaphtho[1,8-cd] [1,2,6]oxadiphosphinine 1,3-disulfide 10 was obtained from the half-oxidization of the know compound 1,3-epithionaphtho[1,8-cd][1,2,6]thiadiphosphinine 1,3-disulfide [31]. Treating 10 with two equivalents of fresh dry potassium fluoride at 80 °C in dry acetonitrile under N2 atmosphere for 1 h led
Scheme 3. Synthesis of salts 11 and 12.
Once again, presuming that both compounds 11 and 12 possessing the same ArP2OS3F− ion should
have similar reactivity, we selected salt 11 as a target starting material to explore their reactivity toward organic substituents. Complete conversion of 11 to the corresponding organic tetraphenylphosphonium salt 13 was carried on straightforwardly. Similarly, the reactions of 1,3,4-triphenyl-1H-1,2,4-triazol-4-ium tetrafluoroborate and 1,3-dimesityl-4,5-dihydro-1H-imidazol-3-ium chloride with 1 under identical conditions afforded the corresponding organic salts 14 and 15 in excellent yields (Scheme 4). Alkylation product 18 was obtained in 46% yield when 11 or 12 was dissolved in medium dichloromethane and the solution was stirred at room temperature for 48 h. It is believed that the mechanism for this reaction involved the intermediate 16, a product of the alkylation of two molecules of 11 or 12 with one molecule of dichloromethane; afterward, the intermediate, 16, broke and cyclized to give newly formed six-membered P2S2CO heterocyclic compound 18 by loss of a molecule of
1,3-difluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine 1,3-disulfide 17. Unfortunately, we are not able to grasp and identify compound 17 due possibly to its instability on the chromatography column or ready decomposition when exposed to air. Attempts to study the mechanism of the reaction by 31P-NMR were unsuccessful because
complex mixtures were observed.
Salt 11 was obtained as a pale yellow solid and is poorly soluble in organic solvents but soluble in oxygen-free water. All of 11–15 are very stable under an inert atmosphere of nitrogen, but they appear to undergo slow decomposition when exposed to the air after days at room temperature. Organic salts 12–15 were synthesized as a sticky paste, foam, or solid, are soluble in organic solvents, and also show good air stability at room temperature. Heterocycle 18 was isolated as yellow foam and was very stable in air and moisture. Surprisingly, in contrast to its starting material 10, compound 18 is soluble in normal organic solvents such as dichloromethane, chloroform, acetone, and so on. All salts show the anticipated molecular cation and anion ion peaks and satisfactory accurate mass measurement. Meanwhile, compound 18 displays the anticipated molecular ion peak and satisfactory accurate mass measurement. The 31P-NMR spectra of salts 11–15 exhibit a similar pattern of two unequal phosphorus
signals ranging from δP = 106.4–107.1 ppm for the non-fluorinated phosphorus and 78.1–78.6 ppm for
the fluorinated phosphorus atom with the corresponding 1J(P,F) and 2J(P,P) coupling constants. The 1J(P,F) coupling constants differing from 1099 to 1106 Hz are considerately lower than that in the
analogous tetrabutylammonium, 3-fluoronaphtho[1,8-cd][1,2,6]thiadiphosphinine-1-thiolate 1,3-disulfide salt (1144 Hz) [32], and in the selenium counterpart PhPSe2F ions (1141–1146 Hz) [26]. The most
significant difference is that the much bigger 2J(P,P) coupling constant of 54.0 Hz was found in 11–15,
comparing to its reported analogy (12.6 Hz) [33], indicating the noticeably differing effects of the P–S–P and P–O–P bridges. The singlet due to the PPh4 cation in 13 is observed at δP = 23.8 ppm. In the 19F-NMR spectra, doublets at δF = −42.6–−28.4 ppm are observed along with the matching 1J(P,F)
coupling constants, falling within the literature values [14,25,28,29]. Detailed analysis revealed that the small 3J(P,F) = 3.2 or 3.3 Hz was observed in compounds 12, 13, and 15. For 18, the 31P-NMR
spectrum shows a singlet at δP = 106.0 ppm with a 2J(P,P) coupling constant of 54.0 ppm. The 1H-NMR spectrum displays signals from the aryl and CH2 protons present within compound.
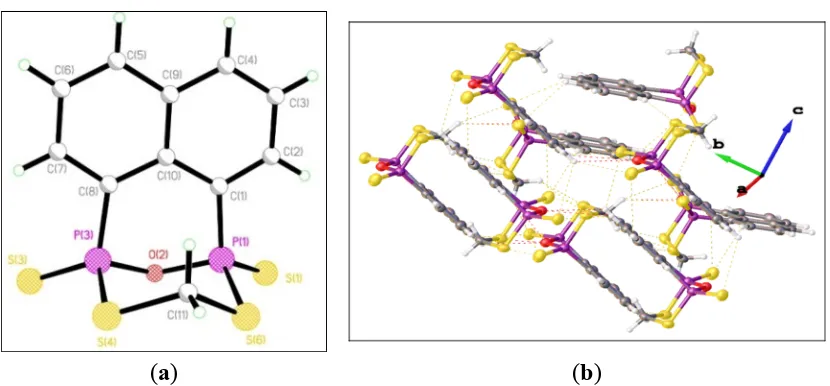
Crystals of 3, 13, 14, and 18 suitable for X-ray analysis were obtained by diffusion of hexane into the dichloromethane solution at room temperature. Crystal data and structure refinement are summarized in Table 1. All structures have a single molecule of the compound in the asymmetric unit, aside from compound 14, in which the asymmetric unit comprises two independent molecules. The X-ray structural analysis of 3, 13, and 14 as shown in Figures 1–3 reveals they crystallize as cation and anion ion-separated species. The X-ray structure of 3 shows that the ferrocenylphosphonofluoridodithioate anionic part contains a distorted tetrahedral phosphorus atom with P–S bond distances of 1.963(3) and 1.953(3) Å, which are shorter than in the reported ferrocenylphosphonodithioate [Fc(RO)PS2]−
complexes [1.973(2)–2.046(2) Å] [33,34]; however, the values are still intermediate between the P–S single bond [ca. 2.10 Å] [35] and the P=S double bond [ca. 1.91 Å] [36], indicating the delocalization of the negative charge over the FPS2− fragment. The P–F bond length [1.615(4) Å] is statistically
indistinguishable from that in PSe2F− [1.604(3)–1.610(5) Å] [25]. The three-dimensional network
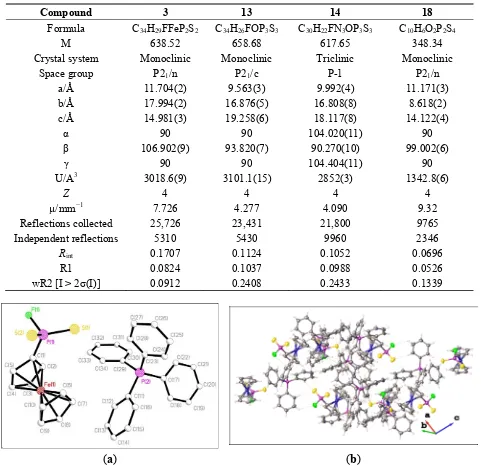
Table 1. Details of the X-ray data collections and refinements for 3, 13, 14, and 18.
Compound 3 13 14 18 Formula C34H29FFeP2S2 C34H26FOP3S3 C30H22FN3OP3S3 C10H6O2P2S4
M 638.52 658.68 617.65 348.34
Crystal system Monoclinic Monoclinic Triclinic Monoclinic
Space group P21/n P21/c P-1 P21/n
a/Å 11.704(2) 9.563(3) 9.992(4) 11.171(3)
b/Å 17.994(2) 16.876(5) 16.808(8) 8.618(2)
c/Å 14.981(3) 19.258(6) 18.117(8) 14.122(4)
α 90 90 104.020(11) 90
β 106.902(9) 93.820(7) 90.270(10) 99.002(6)
γ 90 90 104.404(11) 90
U/A3 3018.6(9) 3101.1(15) 2852(3) 1342.8(6)
Z 4 4 4 4
µ/mm−1 7.726 4.277 4.090 9.32
Reflections collected 25,726 23,431 21,800 9765
Independent reflections 5310 5430 9960 2346
Rint 0.1707 0.1124 0.1052 0.0696
R1 0.0824 0.1037 0.0988 0.0526
wR2 [I > 2σ(I)] 0.0912 0.2408 0.2433 0.1339
(a) (b)
Figure 1. (a) Single crystal structure of 3 (Hydrogen atoms omitted for clarity). Selected bond lengths (Å) and angles (°) (esds in parentheses): S(1)–P(1) 1.963(3), S(2)–P(1) 1.953(3), P(1)–F(1), 1.615(4), P(1)–C(1) 1.801(6), P(1)···P(2) 7.096; S(1)–P(1)–F(1) 105.70(18), S(2)–P(1)–F(1) 106.35(19), F(1)–P(1)–C(1) 98.6(2), S(1)–P(1)–S(2) 121.66(11), S(1)–P(1)–C(1) 110.7(2), S(2)–P(1)–C(1) 111.0(2); (b) Packing diagram of 3.
[image:7.595.60.540.97.566.2]of steric interactions of the Ph4P+ cation ion with the anionic fragments FPS2− and C10H6P2S3OF−. The
three-dimensional network in Figure 2b shows the weak intramolecular and intermolecular C–H···S interactions (yellow dashed line), intermolecular C–H···F interactions (blue dashed line), intermolecular C–H···O interactions (pink dashed line), and π–π stacking interactions responsible for the stabilization of the crossed-layers supramolecular assembly.
(a) (b)
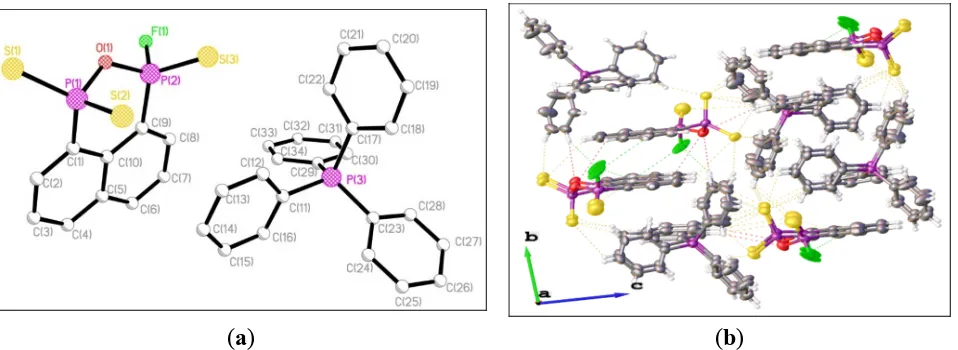
Figure 2. (a) Single crystal structure of 13 (hydrogen atoms omitted for clarity). Selected bond lengths (Å) and angles (°) (esds in parentheses): S(1)–P(1) 1.957(3), S(2)–P(1) 1.957(3), S(3)–P(2) 1.883(4), P(1)–O(1) 1.689(6), P(1)–C(1) 1.813(8), P(2)–F(1) 1.593(8), P(2)–O(1) 1.573(6), P(2)–C(9) 1.794(8); P(1)–O(1)–P(2) 125.5(4), S(1)–P(1)–S(2) 121.42(13), S(1)–P(1)–O(1) 105.0(2), S(1)–P(1)–C(1) 111.9(3), S(2)–P(1)–O(1) 107.9(2), S(2)–P(1)–C(1) 108.6(3), O(1)–P(1)–C(1) 99.8(3), S(3)–P(2)–F(1) 100.7(4), S(3)–P(2)–O(1) 119.5(3), S(3)–P(2)–C(9) 120.0(3), F(1)–P(2)–O(1) 102.4(4), F(1)–P(2)–C(9) 107.3(4), O(1)–P(2)–C(9) 104.9(4); (b) Packing diagram of 13.
The single crystal structure of compound 14 has the same anionic part as that in the structure of 13. However, in contrast to 13, the C10H6P2 part of the anion is planar with the oxygen atom being
significantly distorted and lying 0.495 [0.490] Å above the mean plane. The C3P2O ring is buckled
with the C3P2 and P2O planes, inclined with respect to each other by 46.3° in 13 and 42.0° [44.1°] in
14. It is worth noting that the opening of the P2SO ring results in substantial lengthening of the
transanular P···P distances {2.900 Å in 13 and 2.923 Å [2.927 Å] in 14 vs. 2.73 Å} [35,37]. The internal P–O–P angle in 13 [125.5(4)°] is significantly smaller than the corresponding values in 14 {128.0(4) [128.9(4)]°}, indicating the steric effect of the different cationic parts. However, the P–O–P angles are different from each other {125.5(4)° in 13 and 128.0(4) [128.9(4)]° in 14}; these values are much wider than in similar P–S–P structures [100.21(4)–102.69(4)°] [31,35,37]. Surprisingly, the two P–F bond distances in 14 [1.563(8) Å] are slightly shorter than in 13 [1.593(8) Å], 2 [1.615(4) Å], and PSe2F− [1.604(3)–1.610(5) Å] [25]. All P–S bond lengths are not substantially different from those
[1.950(4)–1.974(4) Å] in 2, 13, and 14, falling between the P–S single bond lengths [2.3307(8) Å] and the P=S double bond lengths [1.8939(12) Å] [35,38]. The P=S distances in 13 [1.883(4) Å] and 14 {1.891(4) [1.819(5)] Å} are slightly shorter than that in PO3S systems [1.888(8)–1.8987(16) Å] [38],
[image:8.595.61.540.172.347.2]{1.689(6) or 1.680(6) [1.671(6)] Å} is significantly longer than the other {1.573(6) or 1.577(8) [1.569(8)] Å}. The aggregation in Figure 3b is dominated by the weak intramolecular and intermolecular C–H···S interactions (yellow dashed line), intermolecular C–H···F interactions (blue dashed line), intermolecular C–H···O interactions (pink dashed line), which involve the H atoms from the aryl rings and the triazole rings, to construct the crossed-layers supramolecular assembly without the presence of the π–π stacking interactions.
(a) (b)
Figure 3. (a) Single crystal structure of 14. Selected bond lengths (Å) and angles (°) (esds in parentheses) (dimensions for second independent molecule in square parentheses): S(1)–P(1) 1.950(4) [1.955(5)], S(2)–P(1) 1.957(4) [1.974(4)], S(3)–P(2) 1.891(4) [1.819(5)], P(1)–O(1) 1.680(6) [1.671(6)], P(1)–C(1) 1.816(10) [1.817(9)], P(2)–F(1) 1.563(8) [1.715(8)], P(2)–O(1) 1.577(8) [1.569(8)], P(2)–C(9) 1.776(10) [1.772(9)]; S(1)–P(1)–S(2) 117.45(17) [117.88(17)], S(1)–P(1)–O(1) 105.7(3) [105.6(3)], S(1)–P(1)–C(1) 112.7(4) [110.9(4)], S(2)–P(1)–O(1) 108.7(3) [110.3(3)], S(2)–P(1)–C(1) 111.7(4) [111.6(3)], O(1)–P(1)–C(1) 98.6(4) [98.6(4)], S(3)–P(2)–F(1) 103.8(4) [103.8(4)], S(3)–P(2)–O(1) 114.3(3) [113.5(3)], S(3)–P(2)–C(9) 118.7(4) [113.0(4)], F(1)–P(2)–O(1) 106.7(5) [109.1(5)], F(1)–P(2)–C(9) 106.3(6) [111.7(5)], O(1)–P(2)–C(9) 106.2(5) [105.8(5)], P(1)–O(1)–P(2) 128.0(4) [128.9(4)]; (b) Packing diagram of 14.
The X-ray structure of 18 as shown in Figure 4 reveals a very different geometry, compared to the structures of 2, 13 and 14. The naphthalene part of the molecule and two phosphorus atoms lies very close to a mean plane [maximum deviation 0.071 Å for P(3)]. The C3P2O is non-planar with a dihedral
angle of 46.73° about P···P axis. The small newly formed six-membered P2S2CO ring is notable
distorted from planar: atom O(2) lies 0.631 Å below and atom C(11) lies 0.915 Å above the P(1)–P(3)–S(4)–S(6) mean plane with two S(1) and S(3) atoms below the mean plane. The newly formed eight-membered C3P2S2C ring is hinged with the C3P2 and P2S2 mean planes inclined by 74.2°
with respect to each other. The transanular P···P distance is 2.916 Å. The P(1)–S(1) [1.9020(13) Å] and P(3)–S(3) [1.9097(13) Å] and P(1)–S(6) [2.0762(14) Å] and P(3)–S(4) [2.0730(14) Å] distances are comparable to those in other known C3P2S ring derivatives [31,36,37,39]. The supramolecular
[image:9.595.65.544.192.341.2](a) (b)
Figure 4. (a) Singlecrystal structure of 18. Selected bond lengths (Å) and angles (°) (esds in parentheses): P(1)–S(1) 1.9020(13), P(3)–S(3) 1.9097(13), P(1)–S(6) 2.0762(14), P(3)–S(4) 2.0730(14), P(1)–O(2) 1.629(2), P(3)–O(2) 1.628(2), S(4)–C(11) 1.830(4), S(6)–C(11) 1.828(4); S(1)–P(1)–S(6) 111.30(6), S(1)–P(1)–C(1) 117.56(11), S(1)–P(1)–O(2) 113.27(9), S(6)–P(1)–C(1) 107.31(12), S(6)–P(1)–O(2) 103.96(9), P(1)–S(6)–C(11) 99.25(12), P(3)–S(4)–C(11) 100.33(12), O(2)–P(1)–C(1) 102.22(13), S(3)–P(3)–S(4) 110.09(6), S(3)–P(3)–O(2) 112.10(9), S(3)–P(3)–C(8) 118.41(11), S(4)–P(3)–O(2) 105.51(9), S(4)–P(3)–C(8) 107.80(12), O(2)–P(3)–C(8) 101.96(13), P(1)–O(2)–P(3) 127.10(14); (b) Packing structure of 18.
Using the Yilmaz methodology [28], we also carried out a similar reaction with Belleau’s Reagent. The reaction of Belleau’s Reagent with two molar equivalents of dry potassium fluoride at 80 °C gave potassium (4-phenoxyphenyl)phosphonofluoridodithioate 19 in 97% yield as a pale yellow paste; or with two equivalents of tetrabutylammonium fluoride (TBAF) at room temperature in tetrahydrofuran for 1 h it led to tetrabutylammonium (4-phenoxyphenyl)phosphonofluoridodithioate 20 in 99% yield as a slightly greenish yellow oil (Scheme 5). Furthermore, reacting 19 with equivalent of tetraphenylphosphonium chloride [28] at room temperature in degassed water/acetone medium furnished tetraphenylphosphonium (4-phenoxyphenyl)phosphonofluoridodithioate 21 in 86% yield as a white foam. All three reactions took place immediately and must be performed in a moisture- and oxygen-free atmosphere. Similar to salts 1 and 11, compound 19 is poorly soluble in organic solvents but soluble in oxygen-free water and is air and moisture stable. Compounds 20 and 21 are insoluble in oxygen-free water but soluble in normal organic solvents. The 31P-NMR spectra of salts 19–21
reveal similar pattern to salts 1–3. Doublets at δP = 130.2, 126.3, and 126.4 ppm, respectively, were
found with the 1J(P,F) coupling constants differing from 1016 to 1028 Hz in 19–21. The 19F-NMR
spectra of 19–21 displays doublets at δF = −26.3, −20.9, and −35.3 ppm, quite similar to literature
[image:10.595.91.506.71.264.2]P P S S S S MeCN, r.t. [nBu
4N]F
KF MeCN, 80°C
19
PPh4Cl
H2O, r.t., 2 h
P F S S K P F S
S NnBu 4 20 O O O P F S
S PPh4
O
21
O
Belleau's Reagent
Scheme 5. Synthesis of derivatives 19–21 from Belleau’s Reagent.
3. Experimental Section
3.1. General
Unless otherwise stated, all reactions were carried out under on oxygen-free nitrogen atmosphere using pre-dried solvents and standard Schlenk techniques; subsequent chromatographic and work-up procedures were performed in air. NMR spectra were recorded on Bruker Avance-400 (1H at 400 MHz, 13C at 100.6 MHz, 31P at 162.0 MHz, and 19F at 376.5 MHz, Blue Lion Biotech, Carnation, WA, USA)
and JEOL GSX-270 (77Se at 51.5 MHz referenced to external Me
2Se, JEOL USA, Inc. Peabody, MA,
USA) at 25 °C (unless stated otherwise). IR spectra were recorded as KBr pellets in the range of 4000–250 cm−1 on a Perkin-Elmer 2000 FTIR/Raman spectrometer(Perkin Elmer, Beaconsfield, UK).
Crystals of 3, 13, 14, and 18 suitable for X-ray analysis were obtained by diffusion of hexane into the dichloromethane solution at room temperature. X-ray crystal structures were determined at −148(1) °C on a Rigaku ACTOR-SM, Saturn 724 CCD area detector [the St Andrews Automated Robotic Diffractometer (STANDARD), Rigaku, Houston, TX, USA] [40] with SHINE optic using Mo Ka radiation (k = 0.71073 Å). The data were corrected for Lorentz, polarization, and absorption. The data was collected and processed using CrystalClear (Rigaku) [41]. The structures were solved by direct methods [42] and expanded using Fourier techniques [43]. Hydrogen atoms were refined using the riding model. All calculations were performed using the CrystalStructure [44] and SHELXL 97 [45]. CCDC contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; Fax (+44)-1223-336-033; E-Mail: deposit@ccdc.cam.ac.uk. CCDC No. 1061138-1061141.
3.2. Synthesis
3.2.1. Synthesis of Potassium Ferrocenylphosphonofluoridodithioate (1)
the title compound 1 as yellow solid (1.305 g, 97%). M.p. 147–149 °C. Selected IR (KBr, cm−1):
1638 (w), 1411 (m), 1259 (m), 1176 (m), 1106 (m), 1019(s), 812(m), 760(s), 697(vs), 598(s), 502 (s), 435 (m). 1H-NMR (D2O, δ), 4.65–4.48 (m, 4H, ferrocenyl-H), 4.36 (s, 5H, ferrocenyl-H) ppm. 13C-NMR
(D2O, δ), 71.7 (d, J(P,C) = 13.1 Hz, ferrocenyl-C), 71.5 (d, J(P,C) = 20.1 Hz, ferrocenyl-C), 71.1 (d
and weak, J(P,C) = 57.9 Hz, ferrocenyl-C), 70.6 (s, ferrocenyl-C) ppm. 31P-NMR (D2O, δ), 132.1 (d,
J(P,F) = 1016 Hz) pm. 19F-NMR (282.34 MHz, D
2O, δ), −18.7 (d, J(F,P) = 1016 Hz) ppm. MS (CI−,
m/z), 299 [C10H9FFePS2]−. Accurate mass measurement (CI−MS): 298.9208 [C10H9FFePS2]−, calculated
mass for [C10H9FFePS2]−: 298.9222.
3.2.2. Synthesis of Tetrabutylammonium Ferrocenylphosphonofluoridodithioate (2)
A suspension of FcLR (3.36 g, 6.0 mmol) and tetrabutylammonium fluoride (12 mL of 1 M solution in THF, 12.0 mmol) in THF (50 mL) was heated at 80 °C for 1 h. Upon cooling to room temperature the reaction mixture was filtered to remove unreacted solid and the filtrate was evaporated in vacuo to give the title compound 2 as golden sticky oil (6.470 g, 99%). Selected IR (KBr, cm−1):
1638 (m), 1467 (s), 1382 (m), 1172 (m), 1106 (m), 1023 (s), 884 (m), 822 (m), 738 (m), 698 (vs), 586 (m), 489 (s). 1H-NMR (CD2Cl2, δ), 4.60–4.55 (m, 2H, ferrocenyl-H), 4.31–4.27 (m, 2H, ferrocenyl-H), 4.22 (s,
5H, ferrocenyl-H), 3.25–3.18 (m, 8H, CH2), 1.67–1.58 (m, 8H, CH2), 1.42–1.38 (m, 8H, CH2), 0.97 (t,
J(H,H) = 6.6 Hz, 12H, CH3) ppm. 13C-NMR (CD2Cl2, δ), 72.5 (d, J(P,C) = 67.6 Hz, ferrocenyl-C),
71.5 (d, J(P,C) = 15.5 Hz, ferrocenyl-C), 70.6 (d, J(P,C) = 17.7 Hz, ferrocenyl-C), 70.2 (s, ferrocenyl-C), 59.0, 24.1, 19.9, 13.6 ppm. 31P-NMR (CD2Cl2, δ), 127.3 (d, J(P,F) = 1009 Hz) pm. 19F-NMR (CD2Cl2,
δ), −14.0 (d, J(F,P) = 1008 Hz) ppm. MS (CI+, m/z), 242 [C
16H36N]+. Accurate mass measurement
(CI+MS): 242.2840 [C16H36N]+, calculated mass for [C16H36N]+: 242.2846; MS (CI−, m/z), 299
[C10H9FFePS2]−. Accurate mass measurement (CI−MS): 298.9260 [C10H9FFePS2]−, calculated mass for
[C10H9FFePS2]−: 298.9269.
3.2.3. Synthesis of Tetraphenylphosphonium Ferrocenylphosphonofluoridodithioate (3)
Potassium ferrocenylphosphonofluoridodithioate (0.339 g, 1.0 mmol) was dissolved in degassed water (10 mL). To this solution was added dropwise an equivalent of tetraphenylphosphonium chloride (0.374 g, 1.0 mmol) in water (10 mL). The precipitation was almost immediately formed and the mixture was allowed to continue stirring for 1 h. The salt was harvested by filtration and was dried over 110 °C in vacuo to give the title compound 3 as pale yellow solid (0.581 g, 91%). M.p. 175–176 °C. Selected IR (KBr, cm−1): 1585 (m), 1483 (m), 1437 (s), 1168 (m), 1107 (s), 1021 (m), 997 (m), 817 (m),
758 (m), 721 (s), 690 (vs), 585 (m), 529 (s). 1H-NMR (CD2Cl2, δ), 7.97–7.91 (m, 4H, Ar-H), 7.81–7.71
(m, 8H, Ar-H), 7.67–7.63 (m, 8H, Ar-H), 4.60–4.57 (m, 2H, ferrocenyl-H), 4.24 (s, 5H, ferrocenyl-H), 4.22–4.20 (m, 2H, ferrocenyl-H) ppm. 13C-NMR (CD2Cl2, δ), 136.6 (d, J(P,C) = 3.0 Hz, Ar-C), 135.3 (d,
J(P,C) = 10.1 Hz, Ar-C), 131.5 (d, J(P,C) = 12.7 Hz, Ar-C), 118.3 (d, J(P,C) = 89.8 Hz, Ar-C), 72.3 (d, J(P,C) = 15.2 Hz, ferrocenyl-C), 71.3 (d, J(P,C) = 56.4 Hz, ferrocenyl-C), 70.9 (s, ferrocenyl-C), 70.4 (d, J(P,C) = 13.1 Hz, ferrocenyl-C) ppm. 31P-NMR (Acetone-d
6, δ), 126.8 (d, J(P,F) = 1010 Hz),
23.2 (s) pm. 19F-NMR (Acetone-d6, δ), −14.5 (d, J(F,P) = 1008 Hz) ppm. MS (CI−, m/z), 299
[C10H9FFePS2]−: 298.9222. MS (CI+, m/z), 339 [C24H20P]+. Accurate mass measurement (CIMS):
339.1299 [C24H20P]+, calculated mass for [C24H20P]+: 339.1297.
3.2.4. Synthesis of 1,4-Phenylenebis(methylene) Bis(ferrocenylphosphonofluoridodithioate) (4)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.108 g, 2.0 mmol) in THF (30 mL) was added to p-xylene dibromide (0.264 g, 1.0 mmol). The mixture was stirred at room temperature for 24 h. After filtration to remove the unreacted solid and drying under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 4 as greenish yellow oil (0.608 g, 87%). Selected IR (KBr, cm−1): 1492 (w),
1454 (w), 1412 (m), 1186 (s), 1107 (m), 1027 (s), 1002 (m), 820 (s), 764 (m), 700 (s), 607 (m), 548 (s), 488 (s). 1H-NMR (CD
2Cl2, δ), 7.36–7.27 (m, 4H, Ar-H), 4.70–4.60 (m, 8H, ferrocenyl-H), 4.57 (s,
10H, ferrocenyl-H), 4.37–4.31 (m, 4H, CH2) ppm. 13C-NMR (CD2Cl2, δ), 131.2 (Ar-C), 131.0 (Ar-C),
129.4 (Ar-C), 128.7 (Ar-C), 75.0 (d, J(P,C) = 75.1 Hz, ferrocenyl-C), 72.6 (d, J(P,C) = 13.5 Hz, ferrocenyl-C), 72.2 (d, J(P,C) = 19.3 Hz, ferrocenyl-C), 70.8 (s, ferrocenyl-C), 31.0 ppm. 31P-NMR
(CD2Cl2, δ), 115.5 (d, J(P,F) = 1103 Hz) pm. 19F-NMR (CD2Cl2, δ), −39.6 (d, J(F,P) = 1104 Hz) ppm. MS
(EI+, m/z), 702 [C28H26F2Fe2P2S4]+. Accurate mass measurement (EI+MS): 701.9057 [C28H26F2Fe2P2S4]+,
calculated mass for [C28H26F2Fe2P2S4]+: 701.9059.
3.2.5. Synthesis of 4-Nitrobenzyl Ferrocenylphosphonofluoridodithioate (5)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.541 g, 1.0 mmol) in THF (30 mL) was added to 4-nitrobenzyl bromide (0.216 g, 1.0 mmol). The mixture was stirred at room temperature for 24 h. After filtration to remove the unreacted solid and drying under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 5 as pale yellow paste (0.286 g, 64%). Selected IR (KBr, cm−1): 1604 (m), 1521 (s),
1347 (s), 1186 (m), 1108 (m), 1029 (m), 821 (m), 801 (m), 699 (s), 548 (m), 493 (m). 1H-NMR
(CD2Cl2, δ), 8.17 (d, J(H,H) = 7.5 Hz, 2H, Ar-H), 7.56 (d, J(H,H) = 7.5 Hz, 2H, Ar-H), 4.64–4.34 (m,
6H, CH2 + ferrocenyl-H), 4.33 (s, 5H, ferrocenyl-H) ppm. 13C-NMR (CD2Cl2, δ), 144.9 (Ar-C), 130.0
(Ar-C), 129.4 (Ar-C), 123.8 (Ar-C), 72.7 (d, J(P,C) = 13.6 Hz, ferrocenyl-C), 72.2 (d, J(P,C) = 19.7 Hz, ferrocenyl-C), 71.1 (d, J(P,C) = 14.5 Hz, ferrocenyl-C), 70.8 (s, ferrocenyl-C), 36.7 (SCH2) ppm. 31P-NMR
(CD2Cl2, δ), 116.0 (d, J(P,F) = 1103 Hz) ppm. 19F-NMR (CD2Cl2, δ), −38.8 (d, J(F,P) = 1104 Hz)
ppm. MS (CI+, m/z), 453 [C17H15FFeNO2PS2NH4]+. Accurate mass measurement (CI+MS): 451.0002
[C17H15F54FeNO2PS2NH4]+, calculated mass for [C17H15F54FeNO2PS2NH4]+: 451.0000.
3.2.6. Synthesis of 4-Bromobenzyl Ferrocenylphosphonofluoridodithioate (6)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.567 g, 1.05 mmol) in THF (30 mL) was added to 4-bromobenzyl bromide (0.260 g, 1.05 mmol). The mixture was stirred at room temperature for 24 h. After filtered to remove unreacted solid and dried under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 6 as reddish yellow paste (0.400 g, 86%). Selected IR (KBr, cm−1): 1468 (m),
1H-NMR (CD2Cl2, δ), 7.57 (d, 2H, J(H,H) = 8.0 Hz, Ar-H), 7.26 (d, 2H, J(H,H) = 8.0Hz, Ar-H), 4.63–4.36
(m, 6H, CH2 + ferrocenyl-H), 4.34 (s, 5H, ferrocenyl-H) ppm. 13C-NMR (CD2Cl2, δ), 133.1 (Ar-C),
131.4 (Ar-C), 129.6 (Ar-C), 127.9 (Ar-C), 72.3 (d, J(P,C) = 13.4 Hz, ferrocenyl-C), 72.1 (d, J(P,C) = 19.7 Hz, ferrocenyl-C), 71.3 (d, J(P,C) = 15.6 Hz, ferrocenyl-C), 70.8 (s, ferrocenyl-C), 38.1 (SCH2) ppm. 31P-NMR (CD2Cl2, δ), 116.4 (d, J(P,F) = 1103 Hz) ppm. 19F-NMR (CD2Cl2, δ), −38.3
(d, J(F,P) = 1105 Hz) ppm. MS (CI+, m/z), 469 [C
17H15BrFFePS2H]]+. Accurate mass measurement
(CI+MS): 466.8988 [C17H15BrF54FePS2H], calculated mass for [C17H15BrF54FePS2H]+: 466.8989.
3.2.7. Synthesis of Methylyl Ferrocenylphosphonofluoridodithioate (7)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.541 g, 1.0 mmol) in THF (30 mL) was added to iodomethane (0.142 g, 1.0 mmol). The mixture was stirred at room temperature for 24 h. After filtration to remove the unreacted solid and drying under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 7 as red oil (0.270 g, 86%). Selected IR (KBr, cm−1): 1413 (m), 1312 (m), 1186 (s), 1107 (m),
1028 (s), 1003 (w), 820 (vs), 699 (vs), 551 (s), 492 (s). 1H-NMR (CD
2Cl2, δ), 4.67–4.39 (m, 4H,
ferrocenyl-H), 4.36 (s, 5H, ferrocenyl-H), 2.44 (dd, J(P,H) = 16.5 Hz, J(F,H) = 1.7 Hz, 3H, SCH3) ppm. 13C-NMR (CD
2Cl2, δ), 72.5 (d, J(P,C) = 12.6 Hz, ferrocenyl-C), 72.2 (d, J(P,C) = 19.3 Hz, ferrocenyl-C),
70.8 (s, ferrocenyl-C), 14.6 (d, J(P,C) = 4.2 Hz, SCH3) ppm. 31P-NMR (CD2Cl2, δ), 118.0 (d,
J(P,F) = 1099 Hz) pm. 19F-NMR (CD
2Cl2, δ), −42.6 (d, J(F,P) = 1102 Hz) ppm. MS (CI+, m/z), 315
[M + H]+. Accurate mass measurement (CI+MS): 312.9569 [M + H]+, calculated mass for
[C11H12F54FePS2H]+: 312.9571.
3.2.8. Synthesis of 4-Cyanobenzyl Ferrocenylphosphonofluoridodithioate (8)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.541 g, 1.0 mmol) in THF (30 mL) was added to 4-cyanobenzyl bromide (0.198 g, 1.0 mmol). The mixture was stirred at room temperature for 24 h. After filtration to remove the unreacted solid and driying under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 8 as pale golden paste (0.290 g, 66%). Selected IR (KBr, cm−1): 2229 (s), 1484 (m),
1433 (m), 1411 (m), 1276 (m), 1186 (s), 1028 (m), 1002 (m), 905 (m), 823 (s), 804 (s), 701 (vs), 601 (m), 545 (s), 492 (s). 1H-NMR (CD
2Cl2, δ), 7.69–7.34 (m, 4H, Ar-H), 4.60–4.35 (m, 4H, ferrocenyl-H), 4.25
(s, 5H, ferrocenyl-H), 4.28–4.22 (m, 2H, CH2) ppm. 13C-NMR (CD2Cl2, δ), 133.6 (Ar-C), 132.9 (Ar-C),
132.6 (Ar-C), 132.1 (Ar-C), 131.4 (Ar-C), 129.7 (Ar-C), 72.6 (d, J(P,C) = 13.6 Hz, ferrocenyl-C), 72.3 (d, J(P,C) = 19.6 Hz, ferrocenyl-C), 71.1 (d, J(P,C) = 15.4 Hz, ferrocenyl-C), 70.8 (s, ferrocenyl-C), 36.6 (SCH2) ppm. 31P-NMR (CD2Cl2, δ), 116.1 (d, J(P,F) = 1101 Hz), 116.0 (d, J(P,F) = 1101 Hz) pm. 19F-NMR (CD2Cl2, δ), −39.1 (d, J(F,P) = 1103 Hz) ppm. MS (CI+, m/z), 433 [C18H15FFeNPS2NH4]+.
Accurate mass measurement (CI+MS): 431.0101 [C
18H15F54FeNPS2NH4]+, calculated mass for
3.2.9. Synthesis of Benzyl Ferrocenylphosphonofluoridodithioate (9)
A mixture of tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.541 g, 1.0 mmol) in THF (30 mL) was added to benzyl bromide (0.170 g, 1.0 mmol). The mixture was stirred at room temperature for 24 h. After filtration to remove the unreacted solid and drying under reduced pressure, the residue was purified by column chromatography on silica gel using dichloromethane as eluent to give the title compound 9 as reddish-yellow oil (0.312 g, 80%). Selected IR (KBr, cm−1): 1493 (m), 1454 (m),
1412 (m), 1186 (s), 1107 (m), 1027 (s), 1002 (m), 820 (vs), 698 (vs), 548(s), 492 (s). 1H-NMR
(CD2Cl2, δ), 7.34–7.21 (m, 5H, Ar-H), 4.64–4.35 (m, 4H, ferrocenyl-H), 4.32 (s, 5H, ferrocenyl-H),
4.24–4.21 (m, 2H, CH2) ppm. 13C-NMR (CD2Cl2, δ), 137.0 (Ar-C), 129.1 (Ar-C), 128.8 (Ar-C), 127.8
(Ar-C), 72.9 (dd, J(P,C) = 11.6 Hz, ferrocenyl-C), 72.5 (d, J(P,C) = 13.6 Hz, ferrocenyl-C), 72.4 (d, J(P,C) = 19.7 Hz, ferrocenyl-C), 71.2 (d, J(P,C) = 15.4 Hz, ferrocenyl-C), 70.8 (s, ferrocenyl-C), 37.5 (SCH2) ppm. 31P-NMR (CD2Cl2, δ), 116.3 (d, J(P,F) = 1101 Hz) pm. 19F-NMR (CD2Cl2, δ), −39.1 (d,
J(F,P) = 1103 Hz) ppm. MS (CI+, m/z), 391 [C17H16FFePS2H]+. Accurate mass measurement (CI+MS):
388.9880 [C17H16F54FePS2H]+, calculated mass for [C17H16F54FePS2H]+: 388.9884.
3.2.10. Synthesis of Potassium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (11)
A mixture of 1,3-epithionaphtho[1,8-cd][1,2,6]oxadiphosphinine 1,3-disulfide (0.600 g, 2.0 mmol) and freshly dried and finely ground KF (0.232 g, 4.0 mmol) in dry acetonitrile (50 mL) was stirred at ambient temperature. After mixing for 10 min, the mixture was heated at 80 °C for 2 h. Upon cooling to room temperature the reaction mixture was filtered and the filtrate was evaporated in vacuo to give the title compound 11 as pale yellow solid (0.700 g, 98%). M.p. 170–171 °C. Selected IR (KBr, cm−1):
1494 (m), 1219 (m), 1159 (m), 1029 (m), 926 (vs), 847 (s), 821 (s), 762 (s), 682 (vs), 650 (vs), 584 (s), 561 (s), 438 (m). 1H-NMR (CD3OD, δ), 8.53 (dd, J(H,H) = 8.2 Hz, J(P,H) = 1.4 Hz, 1H, Ar-H), 8.48–8.44
(m, 1H, Ar-H), 8.21 (d, J(H,H) = 8.2 Hz, 1H, Ar-H), 8.04–8.01 (m, 1H, Ar-H), 7.70–7.61 (m, 2H, Ar-H) ppm. 13C-NMR (CD3OD, δ), 140.0 (d, J(P,C) = 95.5 Hz, Ar-C), 134.9 (s, Ar-C), 134.1 (d,
J(P,C) = 14.5 Hz, Ar-C), 131.9 (d, J(P,C) = 15.6 Hz, Ar-C), 130.9 (s, Ar-C), 126.4 (s, Ar-C), 126.1 (s, Ar-C), 125.1 (s, Ar-C), 124.9 (s, Ar-C), 117.0 (s, Ar-C) ppm. 31P-NMR (CD3OD, δ), 107.1 (d, 2J(P,P) = 54.0 Hz), 78.1 (dd, J(P,F) = 1099 Hz, 2J(P,P) = 54.0 Hz) pm. 19F-NMR (CD
3OD, δ), −42.6
(d, J(F,P) = 1104 Hz) ppm. MS (CI−, m/z), 319 [C10H7FOP2S3]−. Accurate mass measurement
(CI−MS): 318.9030 [C
10H7FOP2S3]−, calculated mass for [C10H7FOP2S3]−: 318.9040.
3.2.11. Synthesis of Tetrabutylammonium 3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (12)
772 (m), 695 (s), 642 (m), 587 (s), 552 (m). 1H-NMR (CD2Cl2, δ), 8.52–7.42 (m, 6H, Ar-H), 3.13 (t,
J(H,H) = 8.0 Hz, 8H, CH2), 1.55–1.53 (m, 8H, CH2), 1.33 (q, J(H,H) = 8.0 Hz, 8H, CH2), 0.92 (t,
J(H,H) = 8.0 Hz, 12H, CH3) ppm. 13C-NMR (CD2Cl2, δ), 142.0 (d, J(P,C) = 95.8 Hz, Ar-C), 134.8 (s,
Ar-C), 134.1 (d, J(P,C) = 15.6 Hz, Ar-C), 131.7 (d, J(P,C) = 14.5 Hz, Ar-C), 130.4 (s, Ar-C), 128.9 (s, Ar-C), 126.7 (s, Ar-C), 126.4 (s, Ar-C), 125.2 (s, Ar-C), 124.9 (s, Ar-C), 58.7 (CH2), 23.9 (CH2), 19.7
(CH2), 13.5 (CH3) ppm. 31P-NMR (CD2Cl2, δ), 106.9 (d, 2J(P,P) = 54.0 Hz), 78.6 (dd, J(P,F) = 1099 Hz, 2J(P,P) = 54.0 Hz) pm. 19F-NMR (CD2Cl2, δ), −28.4 (dd, 1J(F,P) = 1104 Hz, 3J(F,P) = 3.2 Hz) ppm. MS
(CI+, m/z), 242 [C
16H36N]+. Accurate mass measurement (CI+MS): 242.2841 [C16H36N]+, calculated
mass for [C16H36N]+: 242.2842; MS (CI−, m/z), 319 [C10H7FOP2S3]−. Accurate mass measurement
(CI−MS): 318.9032 [C
10H7FOP2S3]−, calculated mass for [C10H7FOP2S3]−: 318.9040.
3.2.12. Synthesis of Tetraphenylphosphonium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (13)
Potassium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (0.358 g. 1.0 mmol) was dissolved in degassed water (10 mL). To this solution was added dropwise an equivalent amount of tetraphenylphosphonium chloride (0.374 g, 1.0 mmol) in water (10 mL). The precipitate almost immediately formed and the mixture was allowed to continue stirring for 1 h. The salt was harvested by filtration and was dried over 100 °C in vacuo to give the title compound 13 as off-white solid (0.446 g, 68%). M.p. 163–165 °C. Selected IR (KBr, cm−1): 1585 (m), 1481 (m), 1436 (s), 1185 (m),
1163 (m), 1107 (vs), 922 (s), 826 (s), 753 (s), 72 3(s), 689 (vs), 647 (s), 583 (s), 526 (vs). 1H-NMR
(CD2Cl2, δ), 8.52–8.31 (m, 2H, Ar-H), 8.09 (d, J(H,H) = 8.3 Hz, 1H, Ar-H), 7.91–7.85 (m, 4H, Ar-H),
7.75–7.71 (m, 7H, Ar-H), 7.64–7.56 (m, 12H, Ar-H) ppm. 13C-NMR (CD2Cl2, δ), 141.9 (d, J(P,C) =
96.0 Hz, Ar-C), 135.7 (d, J(P,C) = 3.2 Hz, Ar-C), 134.6 (s, Ar-C), 134.5 (s, Ar-C), 134.4 (Ar-C), 134.0 (Ar-C), 133.9 (Ar-C), 131.7 (Ar-C), 131.6 (Ar-C), 130.6 (d, J(P,C) = 12.5 Hz, Ar-C), 130.1 (Ar-C), 126.5 (d, J(P,C) = 10.8 Hz, Ar-C), 124.9 (d, J(P,C) = 12.5 Hz, Ar-C), 117.9 (d, Ar-C), 117.1 (d, Ar-C) ppm.
31P-NMR (CD2Cl2, δ), 106.9 (d, 2J(P,P) = 54.0 Hz), 78.2 (dd, J(P,F) = 1099 Hz, 2J(P,P) = 54.0 Hz),
23.8 (s) pm. 19F-NMR (CD
2Cl2, δ), −28.7 (dd, 1J(F,P) = 1100 Hz, 3J(F,P) = 3.2 Hz) ppm. MS (CI+,
m/z), 339 [C24H20P]+. Accurate mass measurement (CI+MS): 339.1296 [C24H20P]+, calculated mass for
[C24H20P]+: 339.1297. MS (CI−, m/z), 319 [C10H7FOP2S3]−. Accurate mass measurement (CI−MS):
318.9031 [C10H7FOP2S3]−, calculated mass for [C10H7FOP2S3]−: 318.9040.
3.2.13. Synthesis of 1,3,4-Triphenyl-1H-1,2,4-triazol-4-ium3-fluoronaphtho[1,8-cd][1,2,6] oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (14)
841 (m), 762 (s), 687 (vs), 644 (s), 584 (s). 1H-NMR (CD2Cl2, δ), 11.20 (s, 1H, triazol-H), 8.37–8.07
(m, 3H, Ar-H), 7.66–7.39 (m, 18H, Ar-H) ppm. 13C-NMR (CD
2Cl2, δ), 154.0 (triazol-C), 142.0 (triazol-C),
135.0 (Ar-C), 134.8 (Ar-C), 134.5 (Ar-C), 134.2 (Ar-C), 132.7 (Ar-C), 131.7 (Ar-C), 131.2 (Ar-C), 130.9 (Ar-C), 130.5 (Ar-C), 130.2 (Ar-C), 129.8 (Ar-C), 129.7 (Ar-C), 129.5 (Ar-C), 129.3 (Ar-C), 128.7 (Ar-C), 128.5 (Ar-C), 128.2 (Ar-C), 126.6 (Ar-C), 126.3 (Ar-C), 125.3 (Ar-C), 125.0 (Ar-C), 124.4 (Ar-C), 122.0 (Ar-C), 121.3 (Ar-C) ppm. 31P-NMR (CD
2Cl2, δ), 106.5 (d, 2J(P,P) = 54.0 Hz),
78.6 (dd, J(P,F) = 1106 Hz, 2J(P,P) = 54.0 Hz) pm. 19F-NMR (CD2Cl2, δ), −28.5 (d, J(F,P) = 1106 Hz)
ppm. MS (CI+, m/z), 298 [C
20H16N3]+. Accurate mass measurement (CI+MS): 298.1335 [C20H16N3]+,
calculated mass for [C20H16N3]+: 298.1339; MS (CI−, m/z), 319 [C10H7FOP2S3]−. Accurate mass
measurement (CI−MS): 318.9031 [C
10H7FOP2S3]−, calculated mass for [C10H7FOP2S3]−: 318.9040.
3.2.14. Synthesis of 1,3-Dimesityl-4,5-dihydro-1H-imidazol-3-ium3-fluoronaphtho[1,8-cd][1,2,6] oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (15)
Potassium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (0.108 g, 0.30 mmol) was dissolved in degassed water (10 mL). To this solution was added dropwise an equivalent amount of 1,3-dimesityl-4,5-dihydro-1H-imidazol-3-ium chloride (0.102 g, 0.30 mmol) in water (10 mL). The precipitation was almost immediately formed and the mixture was allowed to continue stirring for 2 h. The precipitate was harvested by filtration and dissolved in dichloromethane (30 mL). The solution was dried over MgSO4 and dried in vacuo to give the title compound 15 as
white foam (0.175 g, 93%). Selected IR (KBr, cm−1): 1628 (vs), 1481 (m), 1378 (m), 1261 (s), 1217 (m),
1162 (m), 942 (s), 904 (m), 849 (m), 82 5(m), 769 (m), 691 (s), 643 (s), 585 (s). 1H-NMR (CD
2Cl2, δ),
8.61 (s, 2H, Ar-H), 8.32–7.34 (m, 6H, Ar-H), 7.00 (s, 2H, Ar-H), 4.49 (s, 1H, imidazol-H), 3.66 (t, 2H, CH2), 2.36 (s, 12H, CH3), 2.32 (s, 6H, CH3), 1.80 (t, 3H, CH2) ppm. 13C-NMR (CD2Cl2, δ), 159.7
(imidazol-C ), 140.9 (Ar-C), 135.1 (Ar-C), 134.6 (Ar-C), 34.2 (Ar-C), 134.0 (Ar-C), 131.7 (Ar-C), 131.5 (Ar-C), 130.3 (Ar-C), 130.1 (Ar-C), 129.6 (Ar-C), 126.5 (Ar-C), 126.3 (Ar-C), 125.1 (Ar-C), 124.9 (Ar-C), 53.1 (imidazol-C), 51.8 (imidazol-C), 20.9 (CH3), 17.9 (CH3) ppm. 31P-NMR (CD2Cl2, δ),
106.4 (d, 2J(P,P) = 54.0 Hz), 78.2 (dd, J(P,F) = 1101 Hz, 2J(P,P) = 54.0 Hz) pm. 19F-NMR (CD
2Cl2, δ),
−28.7 (dd, 1J(F,P) = 1100 Hz, 3J(P,F) = 3.3 Hz) ppm. MS (CI+, m/z), 307 [C21H27N2]+. Accurate mass
measurement (CI+MS): 307.2170 [C
21H27N2]+, calculated mass for [C21H27N2]+: 307.2169; MS (CI−,
m/z), 319 [C10H7FOP2S3]−. Accurate mass measurement (CI−MS): 318.9031 [C10H7FOP2S3]−, calculated
mass for [C10H7FOP2S3]−: 318.9040.
3.2.15. Synthesis of 1,5-Epoxynaphtho[1,8-cd][1,7,2,6]dithiadiphosphocine 1,5-disulfide (18)
A mixture of potassium3-fluoronaphtho[1,8-cd][1,2,6]oxadiphosphinine-1(3H)-thiolate 1,3-disulfide (0.101 g, 0.30 mmol) or tetrabutylammonium ferrocenylphosphonofluoridodithioate (0.162 g, 0.30 mmol) and dry dichloromethane (10 mL) was stirred at room temperature for 48 h. After removing the unreacted solid by filtration the remaining filtrate was concentrated to 2 mL and was purified by column (eluted by dichloromethane) to give the title compound 18 as yellow foam (0.048 g, 46%) for both starting materials 11 and 12. M.p. 78–80 °C. Selected IR (KBr, cm−1): 1584 (m), 1483 (m),
7.63–7.55 (m, 2H, Ar-H) ppm. 13C-NMR (CD2Cl2, δ), 141.7 (d, J(P,C) = 3.0 Hz, Ar-C), 138.6 (d,
J(P,C) = 10.8 Hz, Ar-C), 134.3 (d, J(P,C) = 13.0 Hz, Ar-C), 130.5 (d, J(P,C) = 117.9 Hz, Ar-C), 129.9 (Ar-C), 124.6 (Ar-C) ppm. 31P-NMR (CD2Cl2, δ), 106.9 (d, 2J(P,P) = 54.0 Hz) pm. MS (EI+, m/z), 348
[C10H6O2P2S4]+. Accurate mass measurement (EI+MS): 347.8719 [C10H6O2P2S4]+, calculated mass for
[C10H6O2P2S4]+: 347.8727.
3.2.16. Synthesis of Potassium (4-phenoxyphenyl)phosphonofluoridodithioate (19)
A mixture of 2,4-bis(4-phenoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (Belleau’s Reagent) (1.056 g, 2.0 mmol) and freshly dried and finely ground KF (0.232 g, 4.0 mmol) in dry acetonitrile (50 mL) was stirred at ambient temperature. After mixing for 10 min, the mixture was heated at 80 °C for 2 h. Upon cooling to room temperature the reaction mixture was filtered and the filtrate was evaporated in vacuo to give the title compound 19 as pale yellow paste (1.250 g, 97%). Selected IR (KBr, cm−1):
1596 (s), 1571 (m), 1501 (s), 1406 (m), 1296 (s), 1253 (s), 1181 (m), 1112 (s), 1022 (s), 973 (m), 829 (m), 688 (s), 556 (s), 385 (m). 1H-NMR (CD3OD, δ), 8.07–7.96 (m, 1H, Ar-H), 7.94–7.86 (m, 2H, Ar-H),
7.80–7.70 (m, 2H, Ar-H), 6.84–6.70 (m, 2H, Ar-H) ppm. 13C-NMR (CD
3OD, δ), 163.6 (Ar-C), 163.1
(Ar-C), 132.2 (d, J(P,C) = 112.1 Hz, Ar-C), 131.3 (d, J(P,C) = 13.5 Hz, Ar-C), 118.2 (Ar-C), 116.9 (s, Ar-C), 112.6 (Ar-C), 112.3 (s, Ar-C) ppm. 31P-NMR (CD
3OD, δ), 130.2 (d, J(P,F) = 1028 Hz) pm. 19F-NMR (CD3OD, δ), −26.3 (d, J(F,P) = 1030 Hz) ppm. MS (CI−, m/z), 283 [C12H9FOPS2]−. Accurate
mass measurement (CI−MS): 283.2954 [C
12H9FOPS2]−, calculated mass for [C12H9FOPS2]−: 283.2951.
3.2.17. Synthesis of Tetrabutylammonium (4-phenoxyphenyl)phosphonofluoridodithioate (20)
To a suspension of 2,4-bis(4-phenoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (Belleau’s Reagent) (0.528 g, 1.0 mmol) in THF (30 mL) was added dropwise tetrabutylammonium fluoride (2 mL of 1 M solution in THF, 2.0 mmol) at room temperature. The off-white suspension disappeared immediately and a colorless solution formed. The mixture was stirred for one more hour for completion. After filtering through a Celit lawyer, the filtrate was evaporated in vacuum to give the title compound 20 as slightly greenish yellow oil (0.520 g, 99%). Selected IR (KBr, cm−1): 1595 (s), 1499 (s), 1426 (m),
1292 (m), 1252 (s), 1177 (m), 1108 (s), 1027 (m), 831 (m), 800 (m), 738 (m), 691 (s), 554 (m).
1H-NMR (CD2Cl2, δ), 8.08–7.91 (m, 5H, Ar-H), 6.87–6.81 (m, 4H, Ar-H), 3.23–3.04 (m, 8H, NCH2),
1.63–1.51 (m, 8H, CH2), 1.37 (m, 8H, CH2), 0.98 (t, J(H,H) = 7.3 Hz, 12H, CH3) ppm. 13C-NMR
(CD2Cl2, δ), 162.2, 160.9, 136.5 (d, J(P,C) = 112.0 Hz, Ar-C), 131.6 (d, J(P,C) = 13.6 Hz, Ar-C),
131.4 (s, Ar-C), 113.0 (s, Ar-C), 112.8 (s, Ar-C), 112.5 (s, Ar-C), 58.8 (CH2), 24.0 (CH2), 19.7 (CH2),
13.4 (CH3) ppm. 31P-NMR (CD2Cl2, δ), 126.3 (d, J(P,F) = 1016 Hz) pm. 19F-NMR (CD2Cl2, δ),
−20.9 (d, J(F,P) = 1017 Hz) ppm. MS (CI+, m/z), 242 [C
16H36N]+. Accurate mass measurement
(CI+MS): 242.2841 [C16H36N]+, calculated mass for [C16H36N]+: 242.2842; MS (CI−, m/z), 283
[C12H9FOPS2]−. Accurate mass measurement (CI−MS): 283.2953 [C12H9FOPS2]−, calculated mass for
3.2.18. Synthesis of Tetraphenylphosphonium (4-phenoxyphenyl)phosphonofluoridodithioate (21)
Potassium (4-phenoxyphenyl)phosphonofluoridodithioate (0.322 g. 1.0 mmol) was dissolved in degassed water/acetone (1:1, 30 mL). To this solution was added dropwise an equivalent amount of tetraphenylphosphonium chloride (0.374 g, 1.0 mmol) in water (10 mL). The mixture was allowed to continue stirring at room temperature for 3 h. After removing the organic solvent, the mixture was extracted with dichloromethane (2 × 30 mL). The organic lawyer was washed with water and dried over MgSO4 to give the title compound 21 as white form (0.534 g, 86%) after removing the solvent
and drying in vacuo. Selected IR (KBr, cm−1): 1594 (m), 1497 (m), 1438 (m), 1250 (m), 1179 (m),
1108 (s), 1024 (m), 830 (m), 800 (m), 723 (s), 688 (s), 554 (m), 526 (s). 1H-NMR (CD2Cl2, δ), 8.04–7.87
(m, 6H, Ar-H), 7.77–7.73 (m, 10H, Ar-H), 7.65–7.57 (m, 10H, Ar-H), 6.79–6.76 (m, 3H, Ar-H) ppm.
13C-NMR (CD2Cl2, δ), 160.7, 135.8 (d, J(P,C) = 3.1 Hz, Ar-C), 134.6 (Ar-C), 134.5 (Ar-C), 132.7 (Ar-C),
132.6 (Ar-C), 131.3 (d, J(P,C) = 14.5 Hz, Ar-C), 130.7 (d, J(P,C) = 13.5 Hz, Ar-C), 119.3 (Ar-C), 116.9 (Ar-C), 112.6 (Ar-C), 112.3 (Ar-C) ppm. 31P-NMR (CD2Cl2, δ), 126.4 (d, J(P,F) = 1019 Hz),
23.8 (s) pm. 19F-NMR (CD
2Cl2, δ), −35.3 (d, J(F,P) = 1018 Hz) ppm. MS (CI+, m/z), 339 [C24H20P]+.
Accurate mass measurement (CI+MS): 339.1297 [C24H20P]+, calculated mass for [C24H20P]+: 339.1297;
MS (CI−, m/z), 283 [C
12H9FOPS2]−. Accurate mass measurement (CI−MS): 283.2952 [C12H9FOPS2]−,
calculated mass for [C12H9FOPS2]−: 283.2951.
4. Conclusions
In summary, the reaction of the ferrocene analogue of Lawesson reagent, 2,4-diferrocenyl-1,3,2,4-diathiadiphosphetane 2,4-disulfide {[FcP(μ-S)S]2, FcLR} with dry KF or tetrabutylammonium fluoride,
led to the corresponding potassium and tetrabutylammonium salts of phenyldithiofluorophosphinic acids in excellent yields. Potassium phenyldithiofluorophosphinic acid is readily converted into the corresponding organic adduct by treating it with an equimolar amount of tetraphenylphosphonium chloride, and treating potassium phenyldiselenofluorophosphinic acid with mono- and di-halogenated alkanes gave rise of a series of the corresponding esters of phenylphosphonofluoridodithioates in high yields. Furthermore, using 1,3-epithionaphtho[1,8-cd][1,2,6]oxadiphosphinine 1,3-disulfide or Belleau’s reagent in its place resulted in the formation of a series of novel salts, adducts, and ester derivatives.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/07/12175/s1.
Acknowledgments
Author Contributions
G.H. and J.D.W. conceived and designed; G.H. and B.A.S performed the experiments; J.D. and A.M.Z.S. finished the X-ray structure measurement; J.D.W. provided critical intellectual input in this study; All authors participated in the preparation of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. References
1. Wilson, B.W.; Walker, C.R. Regulation of newly synthesized acetyleholinesterase in muscle cultures treated with diisopropylfluorophosphate. Proc. Natl. Acad. Sci. USA 1974, 71, 3194–3198. 2. Bartlett, P.A.; Lamdem, L.A. Inhibition of chymotrypsin by phosphonate and phosphonamidate
peptide analogs. Bioorg. Chem. 1986, 14, 356–377.
3. Engel, R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977, 77, 349–367. 4. Camps, F.; Coll, J.; Fabrias, G.; Guerrero, A. Synthesis of dientic fluorinated analogs of insect sex
pheromones. Tetrahedron 1984, 40, 2871–2878.
5. Sikder, A.K.; Ghosh, A.K.; Jaiswal, D.K. Quaternary salts of 3,3′-bis-pyridinium monooximes: Synthesis and biological activity. J. Pharm. Sci. 1993, 82, 258–261.
6. Marjit, D.N.; Sharma, U.S. Hydrolysis of diisopropyl phosphorofluoridate catalysed by copper(II)-diamine complexes. Indian J. Chem. 1989, 28A, 958–960.
7. Sikder, A.K.; Pandey, K.S.; Jaiswal, D.K.; Dube, S.N.; Kumar, D.; Hussain, K.; Bhattacharya, R.; Das Gupta, S. The 3.3′-bispyridinium monooximes as antidotes agaist organophosphorous intoxication. J. Pharm. Pharmacol. 1992, 44, 1038–1040.
8. Anderson, C.; Freeman, J.; Lucas, L.H.; Farley, M.; Dalhoumi, H.; Widlanski, T.S. Estrone sulfatase: Probing structural requirements for substrate and inhibitor. Biochemsitry 1997, 36, 2586–2594. 9. Ashani, Y.; Leader, H.; Rothschild, N.; Dosoretz, C. Combined effect of organophosphorus
hydrolase and oxime on the reactivation rate of diethylphosphoryl-acetylcholinesterase conjugates. Biochem. Pharmacol. 1998, 55, 159–168.
10. Bollmark, M.; Stawinski, J. H-phosphonates-chemistry and applications. Nucleosides Nucleotides 1998, 17, 663–680.
11. Eyer, P. The role of oximes in the management of organophosphorus pesticide poisoning. Toxicol. Rev. 2003, 22, 165–190.
12. Kim, T.H.; Oh, K.A.; Park, N.J.; Park, N.S.; Kim, Y.J.; Yum, E.K.; Jung, Y.S. Reactivation study of pyridinium oximes for acetylcholinesterases inhibited by paraoxon or DFP. J. Appl. Biomed. 2006, 4, 67–72.
13. Roesky, H.W.; Tebbe, F.N.; Muetterties, E.L. New phosphorus-sulfur chemistry. J. Am. Chem. Soc. 1967, 89, 1272–1274.
14. Roesky, H.W.; Tebbe, F.N.; Muetteries, E.L. Thiophosphate chemistry: Anion set X2PS2−, (XPS2)2S2−,
15. Roesky, H.W. Phosphorus compounds. XIX. Preparation and reactions of thiophosphoramidic dihalides and alkyldithiophosphonic acid fluorides. Chem. Ber. 1968, 101, 3679–3687.
16. Roesky, H.W.; Beyer, H. Phosphorus compounds. XXX. Substitution reactions in thiophosphinyl halide compounds. Chem. Ber. 1969, 102, 2588–2594.
17. Roesky, H.W.; Grimm, L.F. Phosphorus compounds. XXVIII. Preparation and characterization of thiophosphoryl compounds containing a phosphorus-nitrogen double bond. Chem. Ber. 1969, 102, 2319–2329.
18. Roesky, H.W.; Grimm, L.F. Phosphorus compounds. 50. Reactions with N-halophosphoranyliden ethiophosphorylhalidde amides. Chem. Ber. 1970, 103, 1664–1673.
19. Roesky, H.W.; Grimm, L.F. N,N′-Sulfonylbis(sulffur difluoride diimide). Angew. Chem. Int. Ed. 1970, 9, 244–245.
20. Harris, R.K.; Woplin, J.R.; Murruy, M.; Schmutzler, R. Preparation and nuclear magnetic resonance spectra of symmetrical spin systems containing phosphorus: Bis(fluorophosphinothioyl) sulfphides. J. Chem. Soc. Dalton Trans. 1972, 1590–1596, doi:10.1039/DT9720001590.
21. Fluck, E.; Schimdt, R.; Haubold, W. Reaktionen des pyridinium-fluordithiophosphorsaure-betains. Phosphorus Sulfur Silicon Relat. Elem. 1978, 5, 141–146.
22. Stawinski, J.; Bollmark, M. A facile access to nucleoside phosphorofluoridate, nucleoside phosphorofluoridothioate, and nucleoside phosphorofluoridethioate monoesters. Tetrahedron Lett. 1996, 37, 5739–5741.
23. Dabkowski, W.; Michalska, M.; Tworowska, I. Highly efficient synthesis of phosphorodithioates derived from 3′-thiothymidine by anhydro-ring opening of 2,3′-anhydro5′-tritylthylidine with O,O-disubstituted phosphorodithioic acids. Chem. Commun. 1998, 427–428, doi:10.1039/A706211G. 24. Tworowska, I.; Dabkowski, W. New efficient synthesis of phosphonofluorodithioates ROP(S)(S−)F
and their structural analogues. Chem. Commun. 1998, 2611–2612, doi:10.1039/A807029F.
25. Hua, G.; Du, J.; Slawin, A.M.Z.; Woollins, J.D. Novel fluorinated phosphorus-selenium heteroatom compounds: Phenylphosphonofluorodislenoic salts, adducts and esters. Inorg. Chem. 2013, 52, 8214–8217.
26. St. J. Foreman, M.R.; Slawin, A.M.Z.; Woollins, J.D. 2,4-Diferrocenyl-1,3-dithiadiphosphetane 2,4-disulfide; structure and reactions with catechols and [PtCl2(PR3)2](R = Et or Bun). J. Chem.
Soc. Dalton Trans. 1996, 3653–3657, doi:10.1039/DT9960003653.
27. Parveen, S.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and characterisation of four- and six-membered P-Se heterocycles. Dalton Trans. 2006, 2586–2590, doi:10.1039/B517093A.
28. Sağlam, E.G.; Çelik, Ö.; Yılmaz, H.; Acar, N. Synthesis and spectroscopic characterization of novel aryl-dithiofluorophosphonate derivatives and X-ray studies of [(4-CH3OC6H4)(F)P(S)S−]
[PH4P+]. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 1339–1346.
29. Roesky, H.W. Über die darstellung von alkyldithiofluorophosphaten. Z. Naturforschung 1967, 22, 716–718.
31. Slawin, A.M.Z.; Williams, D.J.; Wood, P.T.; Woollins, J.D. The preparation and X-ray structure of naphthalenedithiadiphosphetanedisulphide. J. Chem. Soc. Chem. Commun. 1987, 1741–1741, doi:10.1039/C39870001741.
32. Gray, I.P.; Slawin, A.M.Z.; Woollins, J.D. Synthesis and structure of [Fc(EtO)PS2]-complexes.
Z. Anorg. Allg. Chem. 2004, 630, 1851–1857.
33. Fei, Z.; Slawin, A.M.Z.; Woollins, J.D. Nucleophilic reactions of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide. Polyhedron 2001, 20, 2577–2581.
34. Liang, C.X.; Allen, L.C.; Sulfur does not form double bonds in phosphorothioate anions. J. Am. Chem. Soc. 1987, 109, 6449–6453.
35. Wong, C.Y.; McDonald, R.; Cavell, R.G. Hexacoordinate phosphorus. 7. Synthesis and characterization of neutral phosphorus(V) compounds containing divalent tridentate diphenol imine, azo, and thio ligants. Inorg. Chem. 1996, 35, 325–334.
36. Hernandez, J.; Goycoolea, F.M.; Rivera, D.Z.; Onofre, J.J.; Martinez, K.; Lizardi, J.; Reyes, M.S.; Gordillo, B.; Contreras, C.V.; Barradas, O.G.; et al. Substituent effects on the 31P-NMR chemical
shifts of arylphosphorothionates. Tetrahedron 2006, 62, 2520–2528.
37. Kilian, P.; Marek, J.; Marek, R.; Tousek, J.; Humpa, O.; Slawin, A.M.Z.; Touzin, J.; Novosad, J.; Woollins, J.D. New P–S–N containing ring systems. Reaction of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide with methylbis(trimethylsily)amine. Dalton Trans. 1999, 2231–2236, doi:10.1039/A901698H.
38. Kilian, P.; Marek, J.; Marek, R.; Touin, J.; Novosad, J.; Woollins, J.D. New P–S–N containing ring systems. Reaction of 2,4-(naphthalene-1,8-diyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide and its 4-methoxynaphthalene derivative with hexamethyldisilazane. Dalton Trans. 1998, 1175–1180, doi:10.1039/A708348C.
39. Eleftheriou, M.E.; Novosad, J.; Williams, D.J.; Woollins, D.J. Reaction of 1,3-epithionaphtho [1,8-cd][1,2λ5,6λ5]thiadiphosphinine-1,3-dithione; the preparation and X-ray structure of
Np(S)(SMe)SP(S)(OMe), the first C3P2S ring. J. Chem. Soc. Chem. Commun. 1991, 116–117,
doi:10.1039/C39910000116.
40. Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated chemical crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802.
41. Flugrath, J.W.P. The finer things in X-ray diffraction data collection. Acta Crystallogr. 1999, D55, 1718–1725.
42. Altomare, A.; Burla, M.; Camalli, M.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999, 32, 115–119.
43. Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; Israel, R.; Smits, J.M.M. The DIRDIF-99 Program System; Technical Report of the Crystallography Laboratory, University of Nijmegen: Nijmegen, The Netherlands, 1999.
45. Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. 2008, A64, 112–122.
Sample Availability: Samples of all compounds are not available except compounds 1 and 2 from the authors.
![Figure 3. ([110.9(4)], S(2)–P(1)–O(1) 108.7(3) [110.3(3)], S(2)–P(1)–C(1) 111.7(4) [111.6(3)], a) Single crystal structure of 14](https://thumb-us.123doks.com/thumbv2/123dok_us/8928873.391823/9.595.65.544.192.341/figure-s-p-o-s-single-crystal-structure.webp)