organic papers
o942
Lee, Lau and Szeto C28H20O8C10H8N2 DOI: 10.1107/S1600536803012194 Acta Cryst.(2003). E59, o942±o944 Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
Diphenic acid±4,4
000-bipyridine (2/1)
Ting Wai Lee, Jasmine Po Kwan Lau and Lap Szeto*
Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 298 K
Mean(C±C) = 0.004 AÊ
Rfactor = 0.036
wRfactor = 0.054
Data-to-parameter ratio = 12.6
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
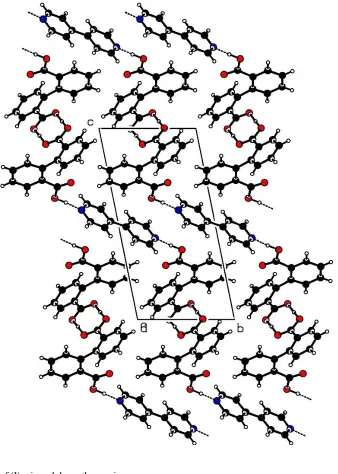
In the title co-crystal, 2C14H10O4C10H8N2, the diphenic acid molecules form a centrosymmetric dimeric pair, through strong OÐH O hydrogen bonds involving a carboxylic acid group from each molecule. In the crystal structure, the dimers are linked by CÐH O interactions to form layers parallel to the (001) plane. The adjacent layers are interlinked by bipyridine molecules, C10H8N2, through OÐH N hydrogen bonds. The bipyridine molecule lies on a centre of symmetry.
Comment
The binding properties of the carboxyl groups of benzoic acid and its derivatives are interesting themes of structural chem-istry (Lam et al., 2003). Diphenic acid (biphenyl-2,20
-di-carboxylic acid) is an extremely versatile building block for the purposes of crystal engineering. In the crystal structure of this compound, molecules form in®nite chains, via R22(8) carboxylic acid pairs (Fronczek et al., 1987). A co-crystal structure of diphenic acid, diphenic acid±acridine (1/1), has been reported (Shaameriet al., 2001). Here we report the co-crystal structure of diphenic acid±4,40-bipyridine (2/1), (I),
which comprises two acid and one base molecule.
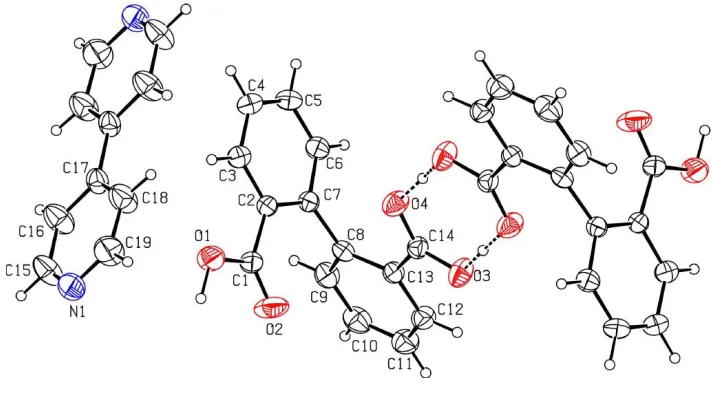
The asymmetric unit of (I) consists of one molecule of diphenic acid and a half molecule of 4,40-bipyridine. The other
diphenic acid molecule and the other half of the bipyridine molecule are generated by the inversion symmetry (2ÿx, 1ÿy, ÿz) and (1ÿx, 2ÿy, 1ÿz), respectively. In the diphenic acid, one of the carboxylic acid groups shows normal CÐO distances [O1ÐC1 = 1.321 (3) and O2ÐC1 = 1.197 (3) AÊ], while in the other these distances are nearly equal [C14ÐO3 = 1.272 (2) and C14ÐO4 = 1.256 (2) AÊ], indicating that it exists as a COOÿion. However, no proton
transfer from this carboxylic acid group to an N atom of the
bipyridine is observed. Instead, the H+ ion is located at a distance of 1.33 (4) AÊ from O3 and 1.29 (4) AÊ from O4i, indicating the formation of a centrosymmetric diphenic acid dimer through strong OÐH O hydrogen bonds (Table 2). The dimers are linked through C4ÐH4 O2iii and C6Ð H6 O3ivinteractions to form molecular layers parallel to the (001) plane. The adjacent layers are interlinked by the bi-pyridine molecules through O1ÐH1 N1ii hydrogen bonds. (Symmetry codes are given in Table 2.)
Experimental
Diphenic acid and 4,40-bipyridine were obtained from Aldrich.
Equimolar quantities of diphenic acid (27 mg) and 4,40-bipyridine
(15 mg) were dissolved in 15 ml of ethanol. The resulting solution was then set aside to crystallize at room temperature.
Crystal data 2C14H10O4C10H8N2
Mr= 640.62
Triclinic,P1
a= 6.498 (2) AÊ
b= 8.002 (7) AÊ
c= 16.208 (6) AÊ
= 99.45 (5)
= 100.93 (3)
= 98.42 (5)
V= 802.4 (8) AÊ3
Z= 1
Dx= 1.326 Mg mÿ3
MoKradiation Cell parameters from 25
re¯ections
= 13.2±16.3
= 0.09 mmÿ1
T= 298 (2) K Block, colourless 0.440.320.28 mm Data collection
AFC-7Rdiffractometer
!/2-scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.957,Tmax= 0.996 3049 measured re¯ections 2830 independent re¯ections 1448 re¯ections withI> 2(I)
Rint= 0.016
max= 25.0
h=ÿ7!7
k= 0!9
l=ÿ19!18 3 standard re¯ections
every 250 re¯ections intensity decay: 0.9% Re®nement
Re®nement onF R= 0.036
wR= 0.054
S= 1.31 2830 re¯ections 225 parameters
H atoms treated by a mixture of independent and constrained re®nement
w= 1/[()2(F
o) + 0.00016(Fo)2]
(/)max< 0.001
max= 0.30 e AÊÿ3
min=ÿ0.27 e AÊÿ3
Table 1
Selected geometric parameters (AÊ).
O1ÐC1 1.321 (3)
O2ÐC1 1.197 (3)
O3ÐC14 1.272 (2)
O4ÐC14 1.256 (2)
N1ÐC15 1.309 (3)
N1ÐC19 1.313 (3)
C1ÐC2 1.489 (3)
C7ÐC8 1.498 (3)
C13ÐC14 1.486 (3)
C15ÐC16 1.375 (4)
C16ÐC17 1.370 (3)
C17ÐC17v 1.497 (4)
C17ÐC18 1.368 (3)
C18ÐC19 1.382 (4)
Symmetry code: (v) 1ÿx;2ÿy;1ÿz.
Table 2
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
O3ÐH2 O4i 1.33 (4) 1.29 (4) 2.618 (3) 171 (3) O1ÐH1 N1ii 1.04 (3) 1.65 (3) 2.691 (4) 172 (3) C4ÐH4 O2iii 0.95 2.56 3.235 (4) 128 C6ÐH6 O3iv 0.95 2.57 3.472 (4) 158
C3ÐH3 O1 0.95 2.33 2.683 (4) 101
C12ÐH10 O3 0.95 2.39 2.730 (4) 100
Symmetry codes: (i) 2ÿx;1ÿy;ÿz; (ii) 2ÿx;1ÿy;1ÿz; (iii) x;1y;z; (iv) 1ÿx;1ÿy;ÿz.
The C-bound H atoms were placed at their geometrically calcu-lated positions, with CÐH = 0.95 AÊ and re®ned in the riding model withUiso(H) = 1.2Ueq(C). Attempts to place H2 at the calculated
position resulted in a non-planarR22(8) ring formation, with an OÐ
H O angle of 133. Hence, H1 and H2 were located in a difference
Fourier map and both positional and isotropic displacement
para-Acta Cryst.(2003). E59, o942±o944 Lee, Lau and Szeto C28H20O8C10H8N2
o943
organic papers
Figure 2
Molecular packing of (I), viewed down theaaxis. Figure 1
organic papers
o944
Lee, Lau and Szeto C28H20O8C10H8N2 Acta Cryst.(2003). E59, o942±o944meters were re®ned. H2 is involved in nearly symmetrical OÐH O hydrogen bonds with O H distances of 1.33 (4) and 1.29 (4) AÊ, respectively.
Data collection:AFC Diffractometer Control Software(Molecular Structure Corporation, 1992); cell re®nement: AFC Diffractometer Control Software; data reduction: TEXSAN (Molecular Structure Corporation, 1992); program(s) used to solve structure: SIR92 (Altomare et al., 1994); program(s) used to re®ne structure:
TEXSAN; molecular graphics:PLATON(Spek, 2003); software used to prepare material for publication:TEXSAN.
We gratefully acknowledge ®nancial support from the University of Hong Kong.
References
Altomare, A., Cascarano, G., Giacovazzo, G., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994).J. Appl. Cryst.27, 435.
Fronczek, F. R., Davis, S. T., Gehrig, L. M. B. & Gandour, R. D. (1987).Acta Cryst.C43, 1615±1618.
Lam, A. W.-H., Wong, W.-T., Gao, S., Wen, G. & Zhang, X.-X. (2003).Eur. J. Inorg. Chem.pp. 149±163.
Molecular Structure Corporation (1992).AFC Diffractometer Control Soft-ware. Molecular Structure Corporation, The Woodlands, TX, USA. Molecular Structure Corporation (1992). TEXSAN. Molecular Structure
Corporation, The Woodlands, TX, USA.
North, A. C. T., Philips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351± 359.
supporting information
sup-1
Acta Cryst. (2003). E59, o942–o944supporting information
Acta Cryst. (2003). E59, o942–o944 [doi:10.1107/S1600536803012194]
Diphenic acid
–
4,4
′
-bipyridine (2/1)
Ting Wai Lee, Jasmine Po Kwan Lau and Lap Szeto
S1. Comment
The binding properties of the carboxyl groups of benzoic acid and its derivatives are interesting themes of structural
chemistry (Lam et al., 2003). The diphenic acid (biphenyl-2,2′-dicarboxylic acid) is an extremely versatile building block
for the purposes of crystal engineering. In the crystal structure of this compound, molecules form infinite chains, via
R22(8) carboxylic acid pairs (Fronczek et al., 1987). The co-crystal structure of diphenic acid, diphenic acid-acridine (1/1)
has been reported (Shaameri et al., 2001). Herein, we report the co-crystal structure of diphenic acid-4,4′-bipyridine
(2/1), (I), which comprises two acid and one base molecule.
The asymmetric unit of (I) consists of one molecule of diphenic acid and a half molecule of 4,4′-bipyridine. The other
diphenic acid molecule and the other half of the bipyridine molecule are generated by the symmetry operations (2 − x, 1 −
y, −z) and (1 − x, 2 − y, 1 − z), respectively. In the diphenic acid, one of the carboxylic acid group shows normal C—O
distances [O1—C1 = 1.321 (3) and O2—C1 = 1.197 (3) Å], while in the other these distances are nearly equal [C14—O3
= 1.272 (2) and C14—O4 = 1.256 (2) Å] indicating that it exists as COO− ion. However, no proton transfer from this
carboxylic acid group to a N atom of the bipyridine is observed. Instead, the H+ ion is located at a distance of 1.33 (4) Å
from O3 and 1.29 (4) Å from O4i indicating the formation of centrosymmetric diphenic acid dimer through strong O—
H···O hydrogen bonds (Table 2). The dimers are linked through C4—H4···O2iii and C6—H6···O3iv interactions to form
molecular layers parallel to the (0 0 1) plane. The adjacent layers are interlinked by the bipyridine molecules through O1
—H1···N1ii hydrogen bonds. The symmetry codes are given in Table 2.
S2. Experimental
Diphenic acid and 4,4′-bipyridine were obtained from Aldrich. Equimolar quantities of diphenic acid (27 mg) and
4,4′-bi-pyridine (15 mg) were dissolved in 15 ml of ethanol. The resulting solution was then set aside to crystallize at room
temperature.
S3. Refinement
The C-bound H atoms were placed at their geometrically calculated positions, with C—H = 0.95 Å and in the riding
model with Uiso(H) = 1.2Ueq(C). Attempts to place H2 at the calculated position resulted in a non-planar R22(8) ring
formation, with an O—H···O angle of 133°. Hence, H1 and H2 were located from a difference Fourier map and both
positional and isotropic displacement parameters were refined. H2 is involved in nearly symmetrical O—H···O hydrogen
supporting information
[image:5.610.126.483.71.268.2]sup-2
Acta Cryst. (2003). E59, o942–o944Figure 1
The structure of (I), showing 50% probability displacement ellipsoids and the atom-numbering scheme for one
asymmetric unit. The other diphenic acid molecule and the other half of the bipyridine molecule are generated by the
supporting information
[image:6.610.139.477.70.544.2]sup-3
Acta Cryst. (2003). E59, o942–o944Figure 2
Molecular packing of (I), viewed down the a axis.
(I)
Crystal data
C28H20O8·C10H8N2
Mr = 640.62
Triclinic, P1 Hall symbol: -P 1 a = 6.498 (2) Å b = 8.002 (7) Å c = 16.208 (6) Å α = 99.45 (5)° β = 100.93 (3)°
γ = 98.42 (5)° V = 802.4 (8) Å3
Z = 1
F(000) = 334.00 Dx = 1.326 Mg m−3
Mo Kα radiation, λ = 0.7107 Å Cell parameters from 25 reflections θ = 13.2–16.3°
supporting information
sup-4
Acta Cryst. (2003). E59, o942–o944T = 298 K Block, colourless
0.44 × 0.32 × 0.28 mm
Data collection
AFC7R
diffractometer
Radiation source: X-ray tube Graphite monochromator ω/2–θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.957, Tmax = 0.996 3049 measured reflections
2830 independent reflections 1448 reflections with I > 2σ(I) Rint = 0.016
θmax = 25.0°, θmin = 2.2°
h = −7→7 k = 0→9 l = −19→18
3 standard reflections every 250 reflections intensity decay: −0.9%
Refinement
Refinement on F
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.036
wR(F2) = 0.054
S = 1.31 2830 reflections 225 parameters 0 restraints
H atoms treated by a mixture of independent and constrained refinement
Weighting scheme based on measured s.u.'s w = 1/[(σ)2(F
o) + 0.00016(Fo)2] (Δ/σ)max < 0.001
Δρmax = 0.30 e Å−3 Δρmin = −0.27 e Å−3
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 1.1226 (3) 0.6029 (2) 0.3652 (1) 0.0703 (6)
O2 0.8605 (3) 0.4039 (2) 0.2836 (1) 0.0895 (7)
O3 0.8094 (3) 0.3251 (2) −0.0025 (1) 0.0666 (6)
O4 0.8892 (3) 0.5837 (2) 0.0816 (1) 0.0651 (6)
N1 0.7048 (4) 0.6498 (3) 0.5733 (1) 0.0713 (8)
C1 0.9452 (4) 0.5522 (3) 0.3054 (1) 0.0495 (7)
C2 0.8630 (3) 0.6970 (3) 0.2711 (1) 0.0431 (6)
C3 0.9730 (4) 0.8641 (3) 0.3042 (1) 0.0557 (7)
C4 0.8981 (4) 1.0036 (3) 0.2778 (2) 0.0650 (8)
C5 0.7092 (5) 0.9766 (3) 0.2186 (2) 0.0663 (9)
C6 0.5989 (4) 0.8119 (3) 0.1844 (1) 0.0566 (7)
C7 0.6748 (3) 0.6686 (3) 0.2078 (1) 0.0430 (6)
C8 0.5456 (3) 0.4946 (3) 0.1649 (1) 0.0449 (6)
C9 0.3618 (4) 0.4401 (3) 0.1923 (1) 0.0579 (8)
C10 0.2305 (4) 0.2831 (4) 0.1550 (2) 0.0671 (9)
C11 0.2781 (4) 0.1768 (3) 0.0886 (2) 0.0658 (8)
C12 0.4556 (4) 0.2291 (3) 0.0586 (1) 0.0558 (7)
C13 0.5910 (3) 0.3871 (3) 0.0953 (1) 0.0439 (6)
C14 0.7746 (3) 0.4365 (3) 0.0564 (1) 0.0455 (7)
C15 0.5568 (5) 0.7174 (4) 0.6042 (2) 0.083 (1)
C16 0.4722 (4) 0.8523 (4) 0.5777 (2) 0.079 (1)
C17 0.5414 (4) 0.9248 (3) 0.5146 (1) 0.0539 (7)
C18 0.6927 (5) 0.8521 (4) 0.4809 (2) 0.083 (1)
supporting information
sup-5
Acta Cryst. (2003). E59, o942–o944H1 1.177 (5) 0.499 (4) 0.388 (2) 0.13 (1)
H2 0.973 (6) 0.369 (4) −0.038 (2) 0.16 (1)
H3 1.1026 0.8829 0.3459 0.0669
H4 0.9764 1.1171 0.3005 0.0780
H5 0.6544 1.0721 0.2011 0.0795
H6 0.4678 0.7955 0.1438 0.0679
H7 0.3253 0.5124 0.2379 0.0695
H8 0.1065 0.2486 0.1755 0.0806
H9 0.1890 0.0682 0.0637 0.0789
H10 0.4872 0.1564 0.0118 0.0669
H11 0.5043 0.6699 0.6479 0.1002
H12 0.3655 0.8955 0.6032 0.0953
H13 0.7454 0.8947 0.4361 0.0995
H14 0.8749 0.6697 0.4872 0.1037
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.072 (1) 0.058 (1) 0.071 (1) 0.0196 (9) −0.0139 (9) 0.0136 (9)
O2 0.092 (1) 0.041 (1) 0.115 (2) 0.0108 (10) −0.028 (1) 0.020 (1)
O3 0.064 (1) 0.066 (1) 0.063 (1) 0.0035 (9) 0.0230 (9) −0.0095 (9)
O4 0.070 (1) 0.052 (1) 0.070 (1) −0.0044 (9) 0.0312 (9) −0.0003 (9)
N1 0.074 (2) 0.074 (2) 0.067 (1) 0.028 (1) 0.001 (1) 0.021 (1)
C1 0.056 (1) 0.043 (1) 0.050 (1) 0.017 (1) 0.009 (1) 0.010 (1)
C2 0.051 (1) 0.040 (1) 0.040 (1) 0.012 (1) 0.013 (1) 0.0083 (10)
C3 0.064 (2) 0.045 (1) 0.054 (1) 0.008 (1) 0.006 (1) 0.009 (1)
C4 0.088 (2) 0.039 (1) 0.064 (2) 0.006 (1) 0.010 (1) 0.008 (1)
C5 0.093 (2) 0.043 (1) 0.068 (2) 0.023 (1) 0.012 (2) 0.020 (1)
C6 0.067 (2) 0.052 (1) 0.052 (1) 0.022 (1) 0.005 (1) 0.014 (1)
C7 0.050 (1) 0.041 (1) 0.041 (1) 0.012 (1) 0.014 (1) 0.0094 (10)
C8 0.046 (1) 0.047 (1) 0.045 (1) 0.013 (1) 0.008 (1) 0.018 (1)
C9 0.054 (2) 0.066 (2) 0.057 (1) 0.012 (1) 0.018 (1) 0.016 (1)
C10 0.053 (2) 0.077 (2) 0.073 (2) 0.000 (1) 0.014 (1) 0.030 (2)
C11 0.059 (2) 0.059 (2) 0.070 (2) −0.007 (1) 0.002 (1) 0.016 (1)
C12 0.057 (2) 0.049 (1) 0.056 (1) 0.005 (1) 0.005 (1) 0.008 (1)
C13 0.044 (1) 0.044 (1) 0.042 (1) 0.010 (1) 0.004 (1) 0.0094 (10)
C14 0.047 (1) 0.045 (1) 0.041 (1) 0.010 (1) 0.004 (1) 0.005 (1)
C15 0.088 (2) 0.107 (2) 0.074 (2) 0.037 (2) 0.020 (2) 0.050 (2)
C16 0.075 (2) 0.105 (2) 0.081 (2) 0.044 (2) 0.029 (2) 0.047 (2)
C17 0.053 (1) 0.064 (2) 0.044 (1) 0.015 (1) 0.002 (1) 0.013 (1)
C18 0.110 (2) 0.093 (2) 0.074 (2) 0.054 (2) 0.044 (2) 0.036 (2)
C19 0.106 (2) 0.089 (2) 0.084 (2) 0.051 (2) 0.035 (2) 0.025 (2)
Geometric parameters (Å, º)
O1—C1 1.321 (3) C8—C9 1.389 (3)
O1—H1 1.04 (3) C8—C13 1.408 (3)
supporting information
sup-6
Acta Cryst. (2003). E59, o942–o944O3—C14 1.272 (2) C9—H7 0.95
O3—H2 1.33 (4) C10—C11 1.370 (4)
O4—C14 1.256 (2) C10—H8 0.95
N1—C15 1.309 (3) C11—C12 1.372 (3)
N1—C19 1.313 (3) C11—H9 0.95
C1—C2 1.489 (3) C12—C13 1.399 (3)
C2—C3 1.388 (3) C12—H10 0.95
C2—C7 1.402 (3) C13—C14 1.486 (3)
C3—C4 1.379 (3) C15—C16 1.375 (4)
C3—H3 0.95 C15—H11 0.95
C4—C5 1.372 (3) C16—C17 1.370 (3)
C4—H4 0.95 C16—H12 0.95
C5—C6 1.375 (3) C17—C17i 1.497 (4)
C5—H5 0.95 C17—C18 1.368 (3)
C6—C7 1.393 (3) C18—C19 1.382 (4)
C6—H6 0.95 C18—H13 0.95
C7—C8 1.498 (3) C19—H14 0.95
C1—O1—H1 111 (1) C9—C10—C11 120.5 (2)
C14—O3—H2 117 (1) C9—C10—H8 119.7
C15—N1—C19 115.8 (2) C11—C10—H8 119.8
O1—C1—O2 121.9 (2) C10—C11—C12 119.3 (2)
O1—C1—C2 113.2 (2) C10—C11—H9 120.4
O2—C1—C2 124.9 (2) C12—C11—H9 120.4
C1—C2—C3 119.0 (2) C11—C12—C13 121.3 (2)
C1—C2—C7 121.5 (2) C11—C12—H10 119.3
C3—C2—C7 119.4 (2) C13—C12—H10 119.3
C2—C3—C4 121.4 (2) C8—C13—C12 119.6 (2)
C2—C3—H3 119.3 C8—C13—C14 122.7 (2)
C4—C3—H3 119.3 C12—C13—C14 117.7 (2)
C3—C4—C5 119.2 (2) O3—C14—O4 122.2 (2)
C3—C4—H4 120.4 O3—C14—C13 117.5 (2)
C5—C4—H4 120.4 O4—C14—C13 120.2 (2)
C4—C5—C6 120.2 (2) N1—C15—C16 124.3 (3)
C4—C5—H5 119.9 N1—C15—H11 117.8
C6—C5—H5 119.9 C16—C15—H11 117.8
C5—C6—C7 121.6 (2) C15—C16—C17 120.2 (3)
C5—C6—H6 119.2 C15—C16—H12 119.9
C7—C6—H6 119.2 C17—C16—H12 119.9
C2—C7—C6 118.0 (2) C16—C17—C17i 122.1 (3)
C2—C7—C8 124.6 (2) C16—C17—C18 115.6 (2)
C6—C7—C8 117.4 (2) C17i—C17—C18 122.3 (3)
C7—C8—C9 117.8 (2) C17—C18—C19 120.3 (2)
C7—C8—C13 124.4 (2) C17—C18—H13 119.8
C9—C8—C13 117.7 (2) C19—C18—H13 119.8
supporting information
sup-7
Acta Cryst. (2003). E59, o942–o944C8—C9—H7 119.2 N1—C19—H14 118.1
C10—C9—H7 119.2 C18—C19—H14 118.1
Symmetry code: (i) −x+1, −y+2, −z+1.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O3—H2···O4ii 1.33 (4) 1.29 (4) 2.618 (3) 171 (3)
O1—H1···N1iii 1.04 (3) 1.65 (3) 2.691 (4) 172 (3)
C4—H4···O2iv 0.95 2.56 3.235 (4) 128
C6—H6···O3v 0.95 2.57 3.472 (4) 158
C3—H3···O1 0.95 2.33 2.683 (4) 101
C12—H10···O3 0.95 2.39 2.730 (4) 100