organic papers
o1818
Genivaldo Julio Perpe´tuoet al. C3H7N6+CHO2 doi:10.1107/S1600536805015151 Acta Cryst.(2005). E61, o1818–o1820 Acta Crystallographica Section E
Structure Reports
Online
ISSN 1600-5368
Melaminium formate
Genivaldo Julio Perpe´tuo,a Marcos Antoˆnio Ribeiroaand Jan Janczakb*
aDepartamento de Fisica, Instituto de Cieˆncias
Exatas e Biolo´gicas, Universidade Federal de Ouro Preto, CEP 35.400-000 – Ouro Preto, MG, Brazil, andbInstitute of Low Temperature and
Structure Research, Polish Academy of Sciences, PO Box 1410, 50-950 Wrocław, Poland
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 295 K
Mean(O–C) = 0.001 A˚ Rfactor = 0.036 wRfactor = 0.096
Data-to-parameter ratio = 16.9
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
Crystals of a new melaminium salt, 2,4,6-triamino-1,3,5-triazin-1-ium formate, C3H7N6
+ CHO2
, consist of singly protonated melaminium residues and formate anions. The components are linked by hydrogen bonds into a three-dimensional framework structure. The melaminium residues are interconnected by two pairs of N—H N hydrogen bonds into chains in the form of stacks, with a distance of 3.34 (1) A˚ between the triazine rings, clearly indicating–interactions. The chains of melaminium residues are interconnected via
N—H O hydrogen bonds by the formate anions, forming a non-covalent superstructure.
Comment
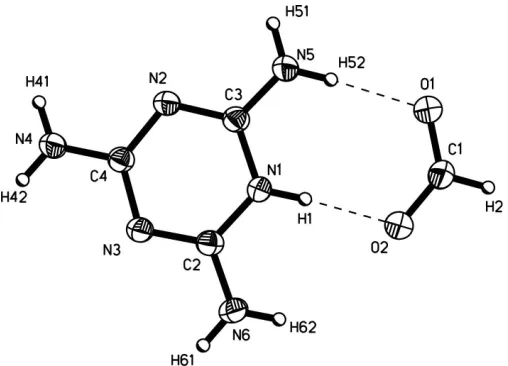
Melamine and its derivatives and inorganic or organic salts can develop well defined non-covalent supramolecular nanoarchitectures via multiple hydrogen bonds by self-assembly of components containing complementary arrays of hydrogen-bonding sites (Desiraju, 1990; MacDonald & Whitesides, 1994; Row, 1999; Krische & Lehn, 2000; Janczak & Perpe´tuo, 2001; Sherington & Taskinen, 2001). In order to expand the understanding of the solid-state physical-organic chemistry of compounds containing multiple N—H N and N—H O hydrogen-bonding systems, we present here the solid-state structure of melaminium formate, (I). The asym-metric unit of (I) consists of a melaminium cation, singly protonated at a ring N atom, and the formate anion (Fig. 1). Selected geometric parameters are given in Table 1.
The amine groups (NH2) are almost coplanar with the
six-membered aromatic triazine ring [the N atoms of the NH2
groups are displaced from the plane by 0.040 (1)–0.059 (1) A˚ ]. The triazine ring is planar, but it exhibits significant distortions from the ideal hexagonal form as reported for other mela-minium salts (Janczak & Perpe´tuo, 2002, 2003). Protonation of the ring also disturbs the C—N bonds within the ring when compared with the neutral melamine crystal structure (Vargheseet al., 1977). The C—N bonds involving the proto-nated N atom are slightly longer than the others within the
ring. Thus there is overwhelming evidence for a partially localized double bond form,i.e.the bond order of the N2—C3 and C2—N3 bonds is greater than the other C—N intra-ring bonds. Theab initiomolecular orbital calculations performed for the neutral melamine molecule revealD3hsymmetry with
C—N distances of 1.325 A˚ within the ring (Drozd & March-ewka, 2005), while corresponding calculations for the singly protonated melaminium cation predictC2vsymmetry (Janczak
& Perpe´tuo, 2004). These calculations support the conclusion concerning the partial localization of the double-bond form. Additionally, protonation of the triazine ring of melamine leads to shortening of the C—NH2 bonds in relation to the
non-protonated melamine molecule in the solid state (Vargheseet al., 1977), as well as in the gas phase (Drozd & Marchewka, 2005). These variations are supported byab initio
calculations, as reported previously (Janczak & Perpe´tuo, 2002, 2003, 2004).
A search of the Cambridge Structural Database (Version 5.26; Allen, 2002) for systems containing a singly protonated melaminium residue yields 23 structures, all of which show melaminium ring distortions quite similar to those found here. The formate anion is planar. The C—O bond lengths (Table 1) are intermediate between the single Csp2—O (1.308–1.320 A˚ ) and double Csp2 O bond lengths (1.214–1.224 A˚ ; Allenet al., 1987), indicating delocalization of the charge over both O atoms of the carboxyl group. The C—O distances and O—C— O angle in the formate ion correlate well with those obtained byab initiomolecular orbital calculations (C1—O1 = 1.232 A˚ , C1—O2 = 1.250 A˚ and O—C—O = 126.5; Frischet al., 1998). An extensive set of hydrogen bonds (Table 2) links the independent components of (I) into a continuous framework superstructure. Each melaminium residue is involved in nine hydrogen bonds. In seven of these it acts as a donor and as an acceptor in the remaining two. Two pairs of N—H N hydrogen bonds link pairs of melaminium cations, related to one another by a combination of n-glide plane and 21screw
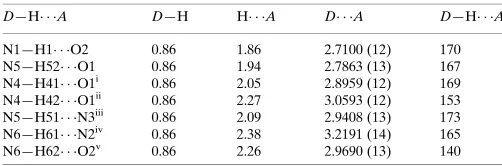
axis and, together with translation, these form stacked chains
parallel to [100] (Fig. 2). In these hydrogen-bonded chains, the alternate triazine rings are not parallel but are inclined at 42.5 (1)to each other. This chain formation is different from that found in the other singly protonated melaminium crystal structures (Allen, 2002), in which the melaninium residues form planar linear chains. In the present case, the triazine rings within each stack are separated by 3.34 (1) A˚ . This distance is slightly shorter than that for a-aromatic ring system (3.4 A˚ ; Pauling, 1960) and indicates strong–interactions between the triazine rings within the stack. Five N—H O hydrogen bonds link each melaminium cation to four formate anions (Table 2).
The formate HCOOanions interconnect the neighbouring stacks of melaminium chains into a three-dimensional non-covalent bonded superstructure. Atom O1 of the formate anion is involved as an acceptor in hydrogen bonds with three melaminium residues, while the other O atom (O2) acts as an acceptor in hydrogen bonds with two melaminium cations that are related by an inversion centre and translation.
Experimental
Melamine (99%) was dissolved in hot 10% formic acid (>98%) and the resulting solution cooled to room temperature. After several days, colourless single crystals of (I) had formed.
Crystal data
C3H7N6+CHO2
Mr= 172.16
Monoclinic,P21=n a= 3.957 (1) A˚
b= 15.798 (3) A˚
c= 11.461 (2) A˚
= 94.14 (1)
V= 714.6 (3) A˚3
Z= 4
Dx= 1.600 Mg m
3
Dm= 1.60 (1) Mg m
3
Dmmeasured by flotation in
chloroform/bromoform MoKradiation Cell parameters from 1252
reflections
= 3.1–29.5
= 0.13 mm1
T= 295 (2) K
Parallelepiped, colourless 0.360.200.12 mm
organic papers
Acta Cryst.(2005). E61, o1818–o1820 Genivaldo Julio Perpe´tuoet al. C
[image:2.610.45.299.69.252.2]3H7N6+CHO2
o1819
Figure 2 [image:2.610.315.563.73.228.2]A view of the crystal packing in (I), showing the hydrogen-bonded N— H N chains that form the – stacks (along the [100] direction). Hydrogen bonds are drawn as dashed lines.
Figure 1
Data collection
Kuma KM-4 diffractometer with CCD area-detector
!scans
Absorption correction: analytical [face-indexed;SHELXTL
(Sheldrick, 1990)]
Tmin= 0.950,Tmax= 0.984 8423 measured reflections
1840 independent reflections 1300 reflections withI> 2(I)
Rint= 0.018
max= 29.5
h=5!4
k=21!21
l=15!15
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.036
wR(F2) = 0.096
S= 1.01 1840 reflections 109 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.057P)2]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.19 e A˚
3
min=0.23 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
O1—C1 1.2448 (12) O2—C1 1.2416 (12) N1—C2 1.3550 (12) N1—C3 1.3670 (12) N2—C3 1.3283 (13) N2—C4 1.3522 (12)
N3—C2 1.3241 (13) N3—C4 1.3560 (12) N4—C4 1.3224 (13) N5—C3 1.3120 (12) N6—C2 1.3225 (13)
O2—C1—O1 128.16 (10) C2—N1—C3 118.90 (9) C3—N2—C4 115.84 (9) C2—N3—C4 116.00 (8)
[image:3.610.44.295.433.516.2]N3—C2—N1 122.01 (9) N2—C3—N1 121.75 (9) N2—C4—N3 125.42 (10)
Table 2
Hydrogen-bonding geometry (A˚ ,).
D—H A D—H H A D A D—H A
N1—H1 O2 0.86 1.86 2.7100 (12) 170 N5—H52 O1 0.86 1.94 2.7863 (13) 167 N4—H41 O1i 0.86 2.05 2.8959 (12) 169 N4—H42 O1ii
0.86 2.27 3.0593 (12) 153 N5—H51 N3iii
0.86 2.09 2.9408 (13) 173 N6—H61 N2iv 0.86 2.38 3.2191 (14) 165 N6—H62 O2v
0.86 2.26 2.9690 (13) 140
Symmetry codes: (i)1 2x;y
1 2;
1
2z; (ii)x 1 2;
1 2y;z
1 2; (iii)x
1 2;
1 2y;
1 2þz; (iv) 1
2þx; 1 2y;z
1
2; (v) 1x;1y;z.
H atoms were treated as riding, with N—H distances of 0.86 A˚ , C—H distances of 0.93 A˚ andUiso(H) = 1.2Ueqof the parent N or C
atoms.
Data collection:KM-4 CCD Software(Kuma, 2001); cell refine-ment:KM-4 CCD Software; data reduction:KM-4 CCD Software; program(s) used to solve structure: SHELXS97 (Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:SHELXTL(Sheldrick, 1990); software used to prepare material for publication:SHELXL97.
GJP thanks the FAPEMIG foundation (Brazil) for financial support.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388.
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans.2, pp. S1–19.
Desiraju, G. R. (1990). InCrystal Engineering. The Design of Organic Solids.
Amsterdam: Elsevier.
Drozd, M. & Marchewka, M. K. (2005).J. Mol. Struct. (Theochem),716, 175– 192.
Frisch, J. M., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery, J. A. Jr, Stratmann. R. E., Burant, J. C., Dapprich, S., Millam, J. M., Daniels, A. D., Kudin, K. N., Strain, M. C.et al.(1998).GAUSSIAN98. Revision A3. Gaussian Inc., Pittsburgh, PA, USA.
Janczak, J. & Perpe´tuo, G. J. (2001).Acta Cryst.C57, 873–875. Janczak, J. & Perpe´tuo, G. J. (2002).Acta Cryst.C58, o455–o459. Janczak, J. & Perpe´tuo, G. J. (2003).Acta Cryst.C59, o349–o352. Janczak, J. & Perpe´tuo, G. J. (2004).Acta Cryst.C60, o211–o214. Krische, M. J. & Lehn, J. M. (2000).Struct. Bonding,96, 3–29.
Kuma (2001).KM-4 CCD Software. Version 171. Kuma Diffraction, Wrocław, Poland.
MacDonald, J. C. & Whitesides, G. M. (1994).Chem. Rev.94, 2383–2420. Pauling, L. (1960).The Nature of the Chemical Bond, 3rd ed., p. 262. Ithaca:
Cornell University Press.
Row, T. R. (1999).Coord. Chem. Rev.183, 81–100.
Sheldrick, G. M. (1990).SHELXTL.Siemens Analytical X-ray Instruments Inc., Madison, Wiconsin, USA.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Sherington, D. C. & Taskinen, K. A. (2001).Chem. Soc. Rev.30, 83–91. Varghese, J. N., O’Connel, A. M. & Maslen, E. N. (1977).Acta Cryst.B33,
2102–2108.
organic papers
o1820
Genivaldo Julio Perpe´tuoet al. Csupporting information
sup-1
Acta Cryst. (2005). E61, o1818–o1820
supporting information
Acta Cryst. (2005). E61, o1818–o1820 [https://doi.org/10.1107/S1600536805015151]
Melaminium formate
Genivaldo Julio Perp
é
tuo, Marcos Ant
ô
nio Ribeiro and Jan Janczak
Melaminium formate
Crystal data
C3H7N6+·CHO2− Mr = 172.16
Monoclinic, P21/n
Hall symbol: -P 2yn
a = 3.957 (1) Å
b = 15.798 (3) Å
c = 11.461 (2) Å
β = 94.14 (1)°
V = 714.6 (3) Å3 Z = 4
F(000) = 360
Dx = 1.600 Mg m−3 Dm = 1.60 (1) Mg m−3 Dm measured by floatation in
chloroform/bromoform Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1252 reflections
θ = 3.1–29.5°
µ = 0.13 mm−1 T = 295 K
Parallelepiped, colourless 0.36 × 0.20 × 0.12 mm
Data collection
Kuma KM-4 with CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 1024 x 1024 with blocks 2 x 2 pixels mm-1
ω scans
Absorption correction: analytical
[face-indexed; SHELXTL (Sheldrick, 1990)]
Tmin = 0.950, Tmax = 0.984
8423 measured reflections 1840 independent reflections 1300 reflections with I > 2σ(I)
Rint = 0.018
θmax = 29.5°, θmin = 3.1° h = −5→4
k = −21→21
l = −15→15
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.036 wR(F2) = 0.096 S = 1.01 1840 reflections 109 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.057P)2]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.19 e Å−3
Δρmin = −0.23 e Å−3
Special details
supporting information
sup-2
Acta Cryst. (2005). E61, o1818–o1820
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.1135 (2) 0.50059 (5) 0.31401 (7) 0.0392 (2)
O2 0.3259 (2) 0.49984 (5) 0.13841 (7) 0.0382 (2)
C1 0.2787 (3) 0.53078 (7) 0.23547 (9) 0.0331 (3)
H2 0.3793 0.5831 0.2515 0.040*
N1 0.0197 (2) 0.34890 (5) 0.09196 (7) 0.0262 (2)
H1 0.0927 0.3994 0.1062 0.031*
N2 −0.2590 (2) 0.22705 (6) 0.15420 (7) 0.0274 (2)
N3 −0.0349 (2) 0.23285 (5) −0.03535 (7) 0.0269 (2)
N4 −0.2942 (2) 0.11420 (6) 0.02892 (7) 0.0320 (2)
H41 −0.3955 0.0866 0.0808 0.038*
H42 −0.2576 0.0906 −0.0366 0.038*
N5 −0.2131 (3) 0.34656 (6) 0.26877 (7) 0.0338 (2)
H51 −0.3216 0.3219 0.3215 0.041*
H52 −0.1434 0.3978 0.2792 0.041*
N6 0.2457 (2) 0.35239 (6) −0.08704 (8) 0.0343 (2)
H61 0.2851 0.3293 −0.1526 0.041*
H62 0.3173 0.4028 −0.0711 0.041*
C2 0.0750 (2) 0.31042 (7) −0.01060 (8) 0.0246 (2)
C3 −0.1539 (2) 0.30613 (6) 0.17235 (8) 0.0249 (2)
C4 −0.1949 (2) 0.19302 (6) 0.04982 (8) 0.0241 (2)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0535 (5) 0.0312 (5) 0.0349 (4) −0.0078 (4) 0.0168 (4) −0.0029 (3)
O2 0.0511 (5) 0.0324 (5) 0.0324 (4) −0.0079 (4) 0.0130 (4) −0.0038 (3)
C1 0.0424 (6) 0.0266 (6) 0.0304 (6) −0.0073 (5) 0.0044 (5) −0.0016 (4)
N1 0.0344 (5) 0.0198 (4) 0.0251 (4) −0.0030 (3) 0.0067 (4) −0.0008 (3)
N2 0.0345 (5) 0.0251 (5) 0.0233 (4) −0.0021 (4) 0.0076 (4) 0.0000 (3)
N3 0.0336 (5) 0.0235 (5) 0.0243 (4) −0.0004 (4) 0.0076 (4) −0.0004 (3)
N4 0.0475 (6) 0.0254 (5) 0.0241 (4) −0.0054 (4) 0.0095 (4) −0.0013 (4)
N5 0.0521 (6) 0.0242 (5) 0.0268 (5) −0.0059 (4) 0.0137 (4) −0.0020 (4)
N6 0.0467 (6) 0.0262 (5) 0.0323 (5) −0.0044 (4) 0.0176 (4) 0.0004 (4)
C2 0.0268 (5) 0.0232 (5) 0.0242 (5) 0.0035 (4) 0.0043 (4) 0.0023 (4)
C3 0.0286 (5) 0.0231 (5) 0.0232 (5) 0.0019 (4) 0.0036 (4) 0.0018 (4)
supporting information
sup-3
Acta Cryst. (2005). E61, o1818–o1820
Geometric parameters (Å, º)
O1—C1 1.2448 (12) N4—C4 1.3224 (13)
O2—C1 1.2416 (12) N4—H41 0.8600
C1—H2 0.9300 N4—H42 0.8600
N1—C2 1.3550 (12) N5—C3 1.3120 (12)
N1—C3 1.3670 (12) N5—H51 0.8600
N1—H1 0.8600 N5—H52 0.8600
N2—C3 1.3283 (13) N6—C2 1.3225 (13)
N2—C4 1.3522 (12) N6—H61 0.8600
N3—C2 1.3241 (13) N6—H62 0.8600
N3—C4 1.3560 (12)
O2—C1—O1 128.16 (10) H51—N5—H52 120.0
O2—C1—H2 115.9 C2—N6—H61 120.0
O1—C1—H2 115.9 C2—N6—H62 120.0
C2—N1—C3 118.90 (9) H61—N6—H62 120.0
C2—N1—H1 120.5 N6—C2—N3 119.84 (9)
C3—N1—H1 120.5 N6—C2—N1 118.15 (10)
C3—N2—C4 115.84 (9) N3—C2—N1 122.01 (9)
C2—N3—C4 116.00 (8) N5—C3—N2 121.12 (9)
C4—N4—H41 120.0 N5—C3—N1 117.13 (9)
C4—N4—H42 120.0 N2—C3—N1 121.75 (9)
H41—N4—H42 120.0 N4—C4—N2 117.53 (9)
C3—N5—H51 120.0 N4—C4—N3 117.05 (8)
C3—N5—H52 120.0 N2—C4—N3 125.42 (10)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1···O2 0.86 1.86 2.7100 (12) 170
N5—H52···O1 0.86 1.94 2.7863 (13) 167
N4—H41···O1i 0.86 2.05 2.8959 (12) 169
N4—H42···O1ii 0.86 2.27 3.0593 (12) 153
N5—H51···N3iii 0.86 2.09 2.9408 (13) 173
N6—H61···N2iv 0.86 2.38 3.2191 (14) 165
N6—H62···O2v 0.86 2.26 2.9690 (13) 140