metal-organic papers
Acta Cryst.(2005). E61, m485–m487 doi:10.1107/S1600536805003764 Duet al. [Mn(NCS)
2(C12H8N4O)2(H2O)2]
m485
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
Diaquabis(2,5-di-3-pyridyl-1,3,4-oxadiazole)dithio-cyanatomanganese(II): a three-dimensional
supra-molecular network formed through O—H
N and
C—H
S interactions
Miao Du,* Xiu-Juan Jiang and Xiao-Jun Zhao
College of Chemistry and Life Science, Tianjin Normal University, Tianjin 300074, People’s Republic of China
Correspondence e-mail: dumiao@public.tpt.tj.cn
Key indicators
Single-crystal X-ray study T= 293 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.035 wRfactor = 0.099
Data-to-parameter ratio = 13.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
The molecular structure of the neutral mononuclear title complex, [Mn(NCS)2(C12H8N4O)2(H2O)2], is
centrosym-metric; the MnIIatom lies on an inversion center and is six-coordinate (MnN4O2), with an octahedral geometry
comprising two trans monodentate 2,5-di-3-pyridyl-1,3,4-oxadiazole ligands, two thiocyanate ligands and two bound water molecules. Intermolecular O—H N hydrogen bonds between these monomeric units result in two-dimensional supramolecular layers with a parallel arrangement, which are stabilized by intralayer aromatic stacking and further extended to a three-dimensional networkviainterlayer weak C—H S interactions.
Comment
Considerable efforts have been devoted to constructing supramolecular architectures using covalent forces together with weaker hydrogen-bonding and/or other intermolecular cooperative interactions (Roesky & Andruh, 2003). Proper selection of the metal centers and organic building blocks is the key issue in the design of these polymeric networks. Recently, one of our research interests has been focused on assemblies based on bent dipyridyl ligands containing the oxadiazole spacer, mainly 2,5-di-4-pyridyl-1,3,4-oxadiazole (4-bpo) and its 3,30-N-donor analog (3-bpo), which could
produce both discrete (Duet al., 2005) and infinite (Duet al., 2004) coordination frameworks under appropriate conditions. Meanwhile, suchN-heterocyclic organic compounds also have a promising ability to generate supramolecular arrays via
hydrogen bonding and aromatic stacking interactions (Du & Zhao, 2003, 2004).
We have reported a series ofMII/4-bpo/NCS isostructural complexes [MII= CoII, MnII, CdIIand FeII; Du & Zhao, 2004;
Du, Liu & Bu, 2002; Fang et al., 2002], which possess the general formula [M(4-bpo)2(NCS)2(H2O)2]. The mononuclear
molecular species (both 4-bpo molecules and NSC anions behave as monodentate terminal ligands) are extended to a three-dimensional supramolecular network via hydrogen-bonding and aromatic stacking interactions. Interestingly, by changing the metal/ligand ratio, in the CdII analog, a new
three-dimensional covalent network with interesting porous properties is formed (Du, Chen & Bu, 2002). As a continua-tion of our investigacontinua-tion, we describe here the molecular/ supramolecular structure of a new MnII/3-bpo/NCS complex, diaquabis(2,5-di-3-pyridyl-1,3,4-oxadiazole)dithiocyanato-manganese(II), (I).
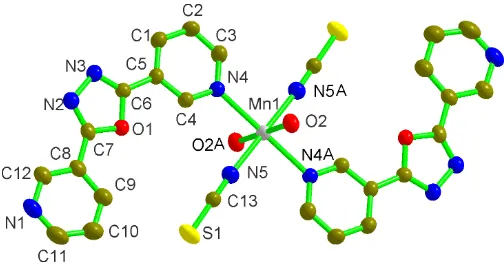
The molecular structure of (I) reveals a neutral octahedral mononuclear centrosymmetric complex, depicted in Fig. 1. The MnII atom, on a crystallographic inversion center, is coordinated by two N-atom donors of two monodentate NSC anions and two water molecules, forming the equatorial plane, and two pyridyl N atoms of a pair of monodentate 3-bpo ligands intransarrangement at the axial sites. The axial Mn1— N4 distances [2.3449 (17) A˚ ] are elongated in comparison with the Mn1—N5 and Mn1—O2 equatorial lengths [2.1431 (19) and 2.2107 (16) A˚ , respectively]. The 3-bpo ligand adopts an unusual transconfiguration with regard to the two terminal pyridyl groups (Du & Zhao, 2003), in order to fulfill the formation of a hydrogen bond between the uncoordinated
3-pyridyl N-atom donor and the aqua ligand. The two terminal pyridyl groups make dihedral angles of 6.40 (14) and 8.80 (16), respectively, with the central oxadiazole ring, and
the dihedral angle between the pyridyl rings is 4.23 (15). The
NSCligands form C13—N5—Mn1 and N5—C13—S1 angles of 173.4 (2) and 179.8 (2), respectively.
Analysis of the crystal packing of the title complex suggests that the monomeric units are linkedvia hydrogen bonds to result in a three-dimensional supramolecular network. The relevant hydrogen-bonding geometries and the symmetry codes are listed in Table 2. As illustrated in Fig. 2, inter-molecular O—H N hydrogen bonds, involving the O atoms of aqua ligands and the N atoms of uncoordinated pyridyl and oxadiazole rings, extend these monomeric units to produce a two-dimensional supramolecular array. The center-to-center and center-to-plane distances between two neighboring almost antiparallel pyridyl rings are 3.87 and 3.5 A˚ , revealing the existence of–stacking interactions, which further stabilize this structure. Furthermore, these two-dimensional hydrogen-bonding arrays are linked via weak interlayer C—H S interactions between SCNanions and 3-bpo ligands, gener-ating a three-dimensional supramolecular network. Exam-ination of this structure withPLATON(Spek, 2003) suggests that there are no solvent-accessible voids in the unit cell.
Experimental
A solution of MnCl24H2O (29 mg, 0.15 mmol) in water (5 ml) was added slowly to a solution of 3-bpo (35 mg, 0.16 mmol) in methanol/ water (1:1v/v, 10 ml) with constant stirring. An excess of NH4SCN (27 mg, 0.35 mmol) was added to the above mixture under reflux for ca30 min. The resulting solution was filtered and left to stand at room temperature. Colorless block crystals suitable for X-ray analysis were obtained by slow evaporation of the solvent over a period of three weeks. Yield: 26 mg (50%). Analysis calculated for C26H20MnN10O4S2: C 47.63, H 3.08, N 21.35%; found: C 47.76, H 2.88, N 20.90%. IR (KBr pellet, cm1): 3258 (b), 3068 (m,) 2070 (vs), 1603 (s), 1547 (m), 1484 (s), 1461 (s), 1435 (s), 1415 (s), 1336 (m), 1274 (w), 1197 (m), 1127 (w), 1088 (s), 1026 (s), 999 (w), 964 (m), 824 (s), 733 (m), 698 (s), 635 (m).
Crystal data
[Mn(NCS)2(C12H8N4O)2(H2O)2]
Mr= 655.58
Triclinic,P1 a= 8.1951 (19) A˚ b= 8.762 (2) A˚ c= 10.619 (3) A˚
= 82.472 (3)
= 77.181 (3)
= 79.873 (3) V= 728.5 (3) A˚3
Z= 1
Dx= 1.494 Mg m
3
MoKradiation Cell parameters from 1513
reflections
= 2.6–26.7
= 0.65 mm1
T= 293 (2) K Block, colorless 0.400.240.11 mm
Data collection
Bruker APEX-II CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.772,Tmax= 0.931
4203 measured reflections
2540 independent reflections 2005 reflections withI> 2(I) Rint= 0.017
max= 25.0
h=9!9 k=10!9 l=10!12
metal-organic papers
m486
Duet al. [Mn(NCS) [image:2.610.44.296.70.209.2]2(C12H8N4O)2(H2O)2] Acta Cryst.(2005). E61, m485–m487 Figure 1
[image:2.610.46.297.277.381.2]ORTEP-3 representation of (I), with the atomic labeling of the asymmetric unit and coordination sphere, shown with 30% probability displacement ellipsoids. H atoms have been omitted. [Symmetry code A: 1x,y, 1z.]
Figure 2
View of the two-dimensional supramolecular array of (I), formed via
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.035
wR(F2) = 0.099 S= 1.04 2540 reflections 196 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0576P)2
+ 0.1207P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.43 e A˚
3
min=0.36 e A˚
3
Table 1
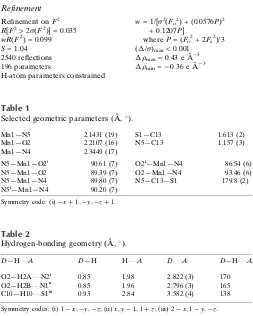
Selected geometric parameters (A˚ ,).
Mn1—N5 2.1431 (19) Mn1—O2 2.2107 (16) Mn1—N4 2.3449 (17)
S1—C13 1.613 (2) N5—C13 1.157 (3)
N5—Mn1—O2i 90.61 (7) N5—Mn1—O2 89.39 (7) N5—Mn1—N4 89.80 (7) N5i—Mn1—N4 90.20 (7)
O2i—Mn1—N4 86.54 (6) O2—Mn1—N4 93.46 (6) N5—C13—S1 179.8 (2)
[image:3.610.44.297.71.387.2]Symmetry code: (i)xþ1;y;zþ1.
Table 2
Hydrogen-bonding geometry (A˚ ,).
D—H A D—H H A D A D—H A
O2—H2A N2i
0.85 1.98 2.822 (3) 170 O2—H2B N1ii
0.85 1.96 2.796 (3) 165 C10—H10 S1iii
0.93 2.84 3.582 (4) 138
Symmetry codes: (i) 1x;y;z; (ii)x;y1;1þz; (iii) 2x;1y;z.
Although all H atoms were visible in difference maps, they were placed in geometrically calculated positions, with C—H distances of 0.93 A˚ and O—H distances of 0.85 A˚, and included in the final
refinement in the riding-model approximation, with displacement parameters derived from their parent atoms [Uiso(H) = 1.2Ueq(C,O)]. Data collection:APEX2(Bruker, 2003); cell refinement:APEX2; data reduction: SAINT (Bruker, 2001); program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: DIAMOND (Brandenburg & Berndt, 1999); software used to prepare material for publication:SHELXTL(Bruker, 2001).
The authors gratefully acknowledge financial support from the National Natural Science Foundation of China (No. 20401012), the Key Project of Tianjin Natural Science Foun-dation and Tianjin Normal University (to MD).
References
Brandenburg, K. & Berndt, M. (1999).DIAMOND. Version 2.1c. Crystal Impact GbR, Bonn, Germany.
Bruker (2001). SAINT and SHELXTL. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (2003).APEX2. Bruker AXS Inc., Madison, Wisconsin, USA. Du, M., Chen, S.-T. & Bu, X.-H. (2002).Cryst. Growth Des.2, 625–629. Du, M., Guo, Y.-M., Chen, S.-T., Bu, X.-H., Batten, S. R., Ribas, J. & Kitagawa,
S. (2004).Inorg. Chem.43, 1287–1293.
Du, M., Liu, H. & Bu, X.-H. (2002).J. Chem. Cryst.32, 57–61. Du, M. & Zhao, X.-J. (2003).J. Mol. Struct.655, 191–197. Du, M. & Zhao, X.-J. (2004).J. Mol. Struct.694, 235–240.
Du, M., Zhao, X.-J. & Guo, J. H. (2005).Inorg. Chem. Commun.8, 1–5. Fang, Y.-Y., Liu, H., Du, M., Guo, Y.-M. & X.-H. Bu. (2002).J. Mol. Struct.608,
229–233.
Roesky, H. W. & Andruh, M. (2003).Coord. Chem. Rev.236, 91–119. Sheldrick, G. M. (1996).SADABS. Version 2.03. University of Go¨ttingen,
Germany.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
metal-organic papers
Acta Cryst.(2005). E61, m485–m487 Duet al. [Mn(NCS)
supporting information
sup-1
Acta Cryst. (2005). E61, m485–m487
supporting information
Acta Cryst. (2005). E61, m485–m487 [https://doi.org/10.1107/S1600536805003764]
Diaquabis(2,5-di-3-pyridyl-1,3,4-oxadiazole)dithiocyanatomanganese(II): a
three-dimensional supramolecular network formed through O
—
H
···
N and C
—
H
···
S interactions
Miao Du, Xiu-Juan Jiang and Xiao-Jun Zhao
Diaquabis(2,5-di-3-pyridyl-1,3,4-oxadiazole)dithiocyanatomanganese(II)
Crystal data
[Mn(NCS)2(C12H8N4O2)2(H2O)2]
Mr = 655.58 Triclinic, P1
a = 8.1951 (19) Å
b = 8.762 (2) Å
c = 10.619 (3) Å
α = 82.472 (3)°
β = 77.181 (3)°
γ = 79.873 (3)°
V = 728.5 (3) Å3
Z = 1
F(000) = 335
Dx = 1.494 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1513 reflections
θ = 2.6–26.7°
µ = 0.65 mm−1
T = 293 K Block, colorless 0.40 × 0.24 × 0.11 mm
Data collection
Bruker APEX-II CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.772, Tmax = 0.931
4203 measured reflections 2540 independent reflections 2005 reflections with I > 2σ(I)
Rint = 0.017
θmax = 25.0°, θmin = 2.0°
h = −9→9
k = −10→9
l = −10→12
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.035
wR(F2) = 0.099
S = 1.04 2540 reflections 196 parameters 3 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0576P)2 + 0.1207P] where P = (Fo2 + 2Fc2)/3
supporting information
sup-2
Acta Cryst. (2005). E61, m485–m487 Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3
Acta Cryst. (2005). E61, m485–m487
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Mn1 0.0430 (3) 0.0531 (3) 0.0307 (3) −0.0230 (2) −0.01120 (19) 0.01186 (19) S1 0.0502 (4) 0.0729 (5) 0.0977 (6) −0.0277 (4) −0.0055 (4) 0.0385 (4) O1 0.0464 (9) 0.0486 (9) 0.0330 (7) −0.0165 (7) −0.0122 (6) 0.0052 (6) O2 0.0680 (11) 0.0561 (10) 0.0333 (8) −0.0192 (8) −0.0134 (7) 0.0070 (7) N1 0.0692 (14) 0.0557 (13) 0.0554 (12) −0.0234 (11) −0.0072 (11) 0.0163 (10) N2 0.0542 (11) 0.0539 (12) 0.0347 (9) −0.0180 (9) −0.0132 (8) 0.0064 (8) N3 0.0515 (11) 0.0574 (12) 0.0324 (9) −0.0198 (9) −0.0130 (8) 0.0066 (8) N4 0.0400 (10) 0.0511 (11) 0.0328 (9) −0.0159 (8) −0.0092 (7) 0.0056 (8) N5 0.0540 (12) 0.0681 (14) 0.0475 (11) −0.0274 (11) −0.0135 (10) 0.0173 (10) C1 0.0425 (12) 0.0562 (14) 0.0406 (12) −0.0144 (11) −0.0142 (10) 0.0032 (10) C2 0.0390 (12) 0.0610 (15) 0.0460 (13) −0.0216 (11) −0.0083 (10) 0.0075 (10) C3 0.0435 (12) 0.0572 (14) 0.0360 (11) −0.0141 (11) −0.0074 (10) 0.0069 (10) C4 0.0381 (11) 0.0486 (13) 0.0352 (11) −0.0148 (10) −0.0089 (9) 0.0041 (9) C5 0.0374 (11) 0.0433 (12) 0.0307 (10) −0.0084 (9) −0.0084 (8) 0.0010 (8) C6 0.0386 (11) 0.0420 (12) 0.0351 (11) −0.0124 (9) −0.0086 (9) 0.0013 (9) C7 0.0482 (12) 0.0390 (12) 0.0310 (11) −0.0058 (10) −0.0080 (9) 0.0027 (9) C8 0.0494 (13) 0.0399 (12) 0.0414 (12) −0.0119 (10) −0.0066 (10) 0.0041 (9) C9 0.0678 (17) 0.0614 (16) 0.0556 (15) −0.0287 (13) −0.0178 (13) 0.0129 (12) C10 0.0761 (19) 0.080 (2) 0.0782 (19) −0.0417 (16) −0.0283 (15) 0.0181 (16) C11 0.0733 (19) 0.0624 (18) 0.0777 (19) −0.0336 (15) −0.0089 (15) 0.0143 (14) C12 0.0562 (15) 0.0490 (14) 0.0496 (13) −0.0158 (11) −0.0088 (11) 0.0084 (11) C13 0.0376 (11) 0.0509 (14) 0.0441 (12) −0.0148 (11) −0.0136 (10) 0.0089 (10)
Geometric parameters (Å, º)
supporting information
sup-4
Acta Cryst. (2005). E61, m485–m487
N5—Mn1—N5i 180.00 (10) N4—C3—C2 123.6 (2) N5—Mn1—O2i 90.61 (7) N4—C3—H3 118.2 N5i—Mn1—O2i 89.39 (7) C2—C3—H3 118.2 N5—Mn1—O2 89.39 (7) N4—C4—C5 123.27 (19) N5i—Mn1—O2 90.61 (7) N4—C4—H4 118.4 O2i—Mn1—O2 180.0 C5—C4—H4 118.4 N5—Mn1—N4 89.80 (7) C1—C5—C4 119.04 (19) N5i—Mn1—N4 90.20 (7) C1—C5—C6 121.52 (19) O2i—Mn1—N4 86.54 (6) C4—C5—C6 119.43 (19) O2—Mn1—N4 93.46 (6) N3—C6—O1 112.63 (17) N5—Mn1—N4i 90.20 (7) N3—C6—C5 129.9 (2) N5i—Mn1—N4i 89.80 (7) O1—C6—C5 117.49 (17) O2i—Mn1—N4i 93.46 (6) N2—C7—O1 112.03 (18) O2—Mn1—N4i 86.54 (6) N2—C7—C8 130.84 (19) N4—Mn1—N4i 180.0 O1—C7—C8 117.09 (19) C7—O1—C6 102.77 (15) C9—C8—C12 118.2 (2) Mn1—O2—H2A 115.9 C9—C8—C7 121.0 (2) Mn1—O2—H2B 116.3 C12—C8—C7 120.8 (2) H2A—O2—H2B 113.7 C10—C9—C8 119.3 (2) C12—N1—C11 117.1 (2) C10—C9—H9 120.4 C7—N2—N3 106.88 (16) C8—C9—H9 120.4 C6—N3—N2 105.70 (17) C9—C10—C11 118.3 (3) C3—N4—C4 116.95 (18) C9—C10—H10 120.9 C3—N4—Mn1 123.21 (13) C11—C10—H10 120.9 C4—N4—Mn1 119.73 (13) N1—C11—C10 124.0 (2) C13—N5—Mn1 173.4 (2) N1—C11—H11 118.0 C5—C1—C2 117.98 (19) C10—C11—H11 118.0 C5—C1—H1 121.0 N1—C12—C8 123.1 (2) C2—C1—H1 121.0 N1—C12—H12 118.5 C1—C2—C3 119.2 (2) C8—C12—H12 118.5 C1—C2—H2 120.4 N5—C13—S1 179.8 (2) C3—C2—H2 120.4
supporting information
sup-5
Acta Cryst. (2005). E61, m485–m487
C2—C1—C5—C6 −179.2 (2) C8—C9—C10—C11 0.6 (4) N4—C4—C5—C1 0.7 (3) C12—N1—C11—C10 −0.4 (4) N4—C4—C5—C6 179.64 (19) C9—C10—C11—N1 0.1 (5) N2—N3—C6—O1 0.0 (2) C11—N1—C12—C8 0.0 (4) N2—N3—C6—C5 −179.1 (2) C9—C8—C12—N1 0.7 (4) C7—O1—C6—N3 0.0 (2) C7—C8—C12—N1 −178.3 (2)
Symmetry code: (i) −x+1, −y, −z+1.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O2—H2A···N2ii 0.85 1.98 2.822 (3) 170 O2—H2B···N1iii 0.85 1.96 2.796 (3) 165 C10—H10···S1iv 0.93 2.84 3.582 (4) 138