organic papers
Acta Cryst.(2005). E61, o1093–o1094 doi:10.1107/S160053680500663X Hapipah M. Aliet al. C
16H16N2O3S
o1093
Acta Crystallographica Section EStructure Reports
Online
ISSN 1600-5368
N
-(4-Methoxybenzoyl)-
N
000-(4-methoxyphenyl)-thiourea
Hapipah M. Ali, Siti Nadiah Abdul Halim, Wan Jefri Basirun and Seik Weng Ng*
Department of Chemistry, University of Malaya, 50603 Kuala Lumpur, Malaysia
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 295 K
Mean(C–C) = 0.003 A˚
Rfactor = 0.042
wRfactor = 0.132
Data-to-parameter ratio = 16.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
Molecules of the title compound, C16H16N2O3S, are linked by
N—H S hydrogen bonds to form centrosymmetric dimers
[N S = 3.4501 (13) A˚ ]. TheN0-phenyl andN-phenyl rings are
twisted by 52.7 (1) and 23.3 (1), respectively, from the
essentially planar —NHC( S)NC( O)— moiety.
Comment
The crystal structures of a number of aromatic thioureas have
been determined; the parent compound N-benzoyl-N0
-phenylthiourea exists as a weakly held dimer arising from N— H S interactions [N S = 3.654 (1) A˚ ; Yamin & Yusuf, 2003]. A non-planar conformation is adopted by the homolog having an electron-donating methoxy substituent in the N0
-phenyl ring [N S = 3.507 (3) A˚ ; Caoet al., 1996].
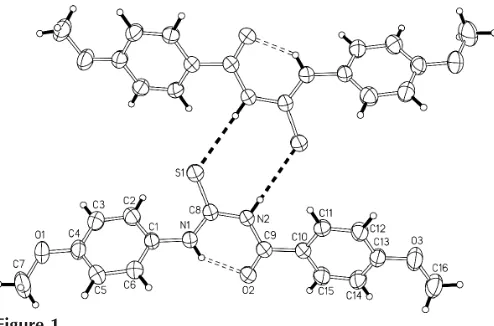
The title compound, (I), has a methoxy substituent in the 4-position of both aromatic rings (Fig. 1); the central
—NHC( S)NC( O)— moiety is flat, being held in such a
conformation owing to the strong intramolecular hydrogen bond [N1 O2 = 2.626 (2) A˚ , H1n O2 = 1.90 (2) A˚ and N1—H1n O2 = 141 (2)]. TheN0-phenyl group is twisted by
52.7 (1) with respect to this moiety, whereas the N-phenyl
group is twisted by only 23.3 (1). In the crystal structure,
[image:1.610.209.456.535.698.2]Received 25 February 2005 Accepted 3 March 2005 Online 25 March 2005
Figure 1
molecules are linked through N—H S hydrogen bonds to form centrosymmetric dimers [N2 S1i = 3.4501 (13) A˚ , H2n S1i = 2.629 (16) A˚ and N2—H2n S1i = 164 (2);
symmetry code: (i) 1x, 1y, 1z]. The bond distances in the title compound are similar to those found in related systems (Yamin & Yusuf, 2003).
Experimental
An acetone solution of ammonium thiocyanate (0.50 g, 6.57 mmol) and 4-anisoyl chloride (1.12 g, 6.57 mmol) was vigorously stirred. To the solution was added 4-anisidine (0.80 g, 6.57 mmol) and the mixture was heated for 2 h. The solution when cooled yielded a brown precipitate; the crude compound was purified by recrystal-lization from ethyl acetate to give colorless crystals.
Crystal data
C16H16N2O3S
Mr= 316.37 Monoclinic,P21=c a= 11.4198 (7) A˚
b= 11.0121 (7) A˚
c= 12.2262 (8) A˚ = 93.319 (1) V= 1534.94 (17) A˚3
Z= 4
Dx= 1.369 Mg m
3
MoKradiation Cell parameters from 3590
reflections = 2.5–26.8
= 0.23 mm1
T= 295 (2) K Block, colorless 0.440.400.24 mm
Data collection
Bruker SMART area-detector diffractometer
’and!scans
Absorption correction: none 9010 measured reflections 3338 independent reflections
2369 reflections withI> 2(I)
Rint= 0.019 max= 27.1
h=14!14
k=14!13
l=15!7
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.042
wR(F2) = 0.132
S= 1.03 3338 reflections 209 parameters
H atoms treated by a mixture of independent and constrained refinement
w= 1/[2
(Fo2) + (0.0728P)2 + 0.3087P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.23 e A˚
3
min=0.19 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
S1—C8 1.655 (2)
N1—C1 1.426 (2)
N1—C8 1.330 (2)
N2—C8 1.390 (2)
N2—C9 1.385 (2)
C1—N1—C8 126.3 (2)
C8—N2—C8 128.0 (2)
N1—C8—N2 115.7 (2)
N1—C8—S1 124.8 (1)
N2—C8—S1 119.4 (1)
Carbon-bound H atoms were placed at calculated positions (C— H = 0.93 A˚ for the aromatic H atoms and C—H = 0.96 A˚ for the methyl H atoms) and were included in the refinement in the riding-model approximation, withUiso= 1.2Ueq(C) for the aromatic H atoms
andUiso= 1.5Ueq(C) for the methyl H atoms. The torsion angle of the
methyl groups was refined. Nitrogen-bound H atoms were located in a difference Fourier map and were refined with an N—H = 0.86 (1) A˚ distance restraint.
Data collection:SMART(Bruker, 2001); cell refinement:SAINT (Bruker, 2001); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEPII (Johnson, 1976); software used to prepare material for publication:SHELXL97.
The authors thank the Ministry of Science, Technology and the Environment for supporting this study (grant No. IPRA 33-02-03-3055). We acknowledge Mr Xiao-Long Feng of Sun Yat-Sen University for the diffraction measurements.
References
Bruker (2001).SAINTandSMART. Bruker AXS Inc., Madison, Wisconsin, USA.
Cao, Y., Zhao, B., Zhang, Y.-Q. & Zhang, D.-C. (1996).Acta Cryst.C52, 1772– 1774.
Johnson, C. K. (1976).ORTEPII. Report ORNL-5138. Oak Ridge National Laboratory, Tennessee, USA.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of Go¨ttingen, Germany.
supporting information
sup-1 Acta Cryst. (2005). E61, o1093–o1094
supporting information
Acta Cryst. (2005). E61, o1093–o1094 [https://doi.org/10.1107/S160053680500663X]
N
-(4-Methoxybenzoyl)-
N
′
-(4-methoxyphenyl)thiourea
Hapipah M. Ali, Siti Nadiah Abdul Halim, Wan Jefri Basirun and Seik Weng Ng
N-(4-Methoxybenzoyl)-N′-(4-methoxyphenyl)thiourea
Crystal data
C16H16N2O3S
Mr = 316.37
Monoclinic, P21/c
Hall symbol: -P 2ybc
a = 11.4198 (7) Å
b = 11.0121 (7) Å
c = 12.2262 (8) Å
β = 93.319 (1)°
V = 1534.94 (17) Å3
Z = 4
F(000) = 664
Dx = 1.369 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 3590 reflections
θ = 2.5–26.8°
µ = 0.23 mm−1
T = 295 K Block, colorless 0.44 × 0.40 × 0.24 mm
Data collection
Bruker SMART area-detector diffractometer
Radiation source: medium-focus sealed tube Graphite monochromator
φ and ω scans
9010 measured reflections 3338 independent reflections
2369 reflections with I > 2σ(I)
Rint = 0.019
θmax = 27.1°, θmin = 1.8°
h = −14→14
k = −14→13
l = −15→7
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.042
wR(F2) = 0.132
S = 1.03 3338 reflections 209 parameters 2 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H atoms treated by a mixture of independent and constrained refinement
w = 1/[σ2(F
o2) + (0.0728P)2 + 0.3087P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.23 e Å−3
Δρmin = −0.19 e Å−3
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.9838 (1) 0.9425 (2) 0.1794 (1) 0.0700 (5) O2 0.4909 (1) 0.8975 (1) 0.5931 (1) 0.0569 (4) O3 0.1999 (2) 0.6879 (1) 0.9740 (1) 0.0686 (5) C1 0.7323 (2) 0.8331 (2) 0.3887 (2) 0.0413 (4) C2 0.7308 (2) 0.7934 (2) 0.2809 (2) 0.0510 (5) C3 0.8174 (2) 0.8304 (2) 0.2148 (2) 0.0537 (5) C4 0.9057 (2) 0.9065 (2) 0.2542 (2) 0.0491 (5) C5 0.9088 (2) 0.9442 (2) 0.3612 (2) 0.0567 (5) C6 0.8208 (2) 0.9069 (2) 0.4280 (2) 0.0523 (5) C7 1.0673 (2) 1.0323 (3) 0.2130 (3) 0.0847 (8) C8 0.5989 (2) 0.6983 (2) 0.4826 (2) 0.0414 (4) C9 0.4681 (2) 0.7923 (2) 0.6154 (2) 0.0419 (4) C10 0.3960 (2) 0.7609 (2) 0.7076 (2) 0.0403 (4) C11 0.4006 (2) 0.6493 (2) 0.7611 (2) 0.0443 (4) C12 0.3347 (2) 0.6288 (2) 0.8487 (2) 0.0500 (5) C13 0.2616 (2) 0.7186 (2) 0.8862 (2) 0.0466 (4) C14 0.2568 (2) 0.8303 (2) 0.8351 (2) 0.0522 (5) C15 0.3240 (2) 0.8505 (2) 0.7471 (2) 0.0503 (5) C16 0.1289 (3) 0.7778 (2) 1.0208 (2) 0.0767 (8) H1n 0.603 (2) 0.867 (2) 0.488 (2) 0.067 (7)* H2n 0.482 (2) 0.626 (1) 0.568 (2) 0.044 (5)* H2 0.6715 0.7419 0.2535 0.061* H3 0.8163 0.8037 0.1426 0.064* H5 0.9691 0.9941 0.3890 0.068* H6 0.8225 0.9327 0.5005 0.063* H7a 1.1122 1.0545 0.1520 0.127* H7b 1.1189 1.0007 0.2709 0.127* H7c 1.0274 1.1026 0.2387 0.127* H11 0.4491 0.5883 0.7369 0.053* H12 0.3387 0.5538 0.8838 0.060* H14 0.2087 0.8913 0.8599 0.063* H15 0.3212 0.9262 0.7132 0.060* H16a 0.0942 0.7449 1.0841 0.115* H16b 0.0681 0.8025 0.9680 0.115* H16c 0.1764 0.8468 1.0420 0.115*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2005). E61, o1093–o1094
C4 0.042 (1) 0.047 (1) 0.061 (1) 0.005 (1) 0.020 (1) 0.005 (1) C5 0.048 (1) 0.059 (1) 0.065 (1) −0.01 (1) 0.014 (1) −0.007 (1) C6 0.054 (1) 0.053 (1) 0.051 (1) −0.010 (1) 0.014 (1) −0.007 (1) C7 0.057 (1) 0.090 (2) 0.110 (2) −0.019 (1) 0.036 (1) 0.009 (2) C8 0.039 (1) 0.040 (1) 0.046 (1) −0.003 (1) 0.009 (1) 0.001 (1) C9 0.042 (1) 0.038 (1) 0.046 (1) −0.003 (1) 0.008 (1) −0.001 (1) C10 0.042 (1) 0.036 (1) 0.044 (1) −0.002 (1) 0.008 (1) 0.000 (1) C11 0.049 (1) 0.035 (1) 0.049 (1) 0.005 (1) 0.012 (1) 0.000 (1) C12 0.062 (1) 0.038 (1) 0.052 (1) 0.008 (1) 0.017 (1) 0.010 (1) C13 0.053 (1) 0.045 (1) 0.044 (1) 0.002 (1) 0.014 (1) 0.002 (1) C14 0.059 (1) 0.041 (1) 0.059 (1) 0.012 (1) 0.021 (1) 0.004 (1) C15 0.058 (1) 0.035 (1) 0.059 (1) 0.007 (1) 0.019 (1) 0.008 (1) C16 0.090 (2) 0.069 (2) 0.076 (2) 0.016 (1) 0.046 (1) 0.005 (1)
Geometric parameters (Å, º)
S1—C8 1.655 (2) C12—C13 1.389 (3) N1—C1 1.426 (2) C13—C14 1.379 (3) N1—C8 1.330 (2) C14—C15 1.376 (3) N2—C8 1.390 (2) N1—H1n 0.86 (1) N2—C9 1.385 (2) N2—H2n 0.86 (1) O1—C4 1.372 (2) C2—H2 0.93 O1—C7 1.419 (3) C3—H3 0.93 O2—C9 1.222 (2) C5—H5 0.93 O3—C13 1.360 (2) C6—H6 0.93 O3—C16 1.421 (3) C7—H7a 0.96 C1—C6 1.363 (3) C7—H7b 0.96 C1—C2 1.388 (3) C7—H7c 0.96 C2—C3 1.374 (3) C11—H11 0.93 C3—C4 1.377 (3) C12—H12 0.93 C4—C5 1.371 (3) C14—H14 0.93 C5—C6 1.392 (3) C15—H15 0.93 C9—C10 1.475 (2) C16—H16a 0.96 C10—C15 1.388 (2) C16—H16b 0.96 C10—C11 1.391 (3) C16—H16c 0.96 C11—C12 1.364 (3)
O1—C4—C3 115.7 (2) C5—C6—H6 119.5 C4—C5—C6 119.4 (2) O1—C7—H7a 109.5 C1—C6—C5 121.0 (2) O1—C7—H7b 109.5 N1—C8—N2 115.7 (2) H7a—C7—H7b 109.5 N1—C8—S1 124.8 (1) O1—C7—H7c 109.5 N2—C8—S1 119.4 (1) H7a—C7—H7c 109.5 O2—C9—N2 121.5 (2) H7b—C7—H7c 109.5 O2—C9—C10 121.9 (2) C12—C11—H11 119.7 N2—C9—C10 116.6 (2) C10—C11—H11 119.7 C15—C10—C11 118.1 (2) C11—C12—H12 119.6 C15—C10—C9 117.8 (2) C13—C12—H12 119.6 C11—C10—C9 123.9 (2) C15—C14—H14 120.3 C12—C11—C10 120.5 (2) C13—C14—H14 120.3 C11—C12—C13 120.7 (2) C10—C15—H15 119.2 O3—C13—C14 124.8 (2) O3—C16—H16a 109.5 O3—C13—C12 115.6 (2) O3—C16—H16b 109.5 C14—C13—C12 119.6 (2) H16a—C16—H16b 109.5 C15—C14—C13 119.3 (2) O3—C16—H16c 109.5 C14—C15—C10 121.7 (2) H16a—C16—H16c 109.5 C14—C15—H15 119.2 H16b—C16—H16c 109.5
C8—N1—C1—C6 −127.9 (2) C8—N2—C9—O2 13.4 (3) C8—N1—C1—C2 57.4 (3) C8—N2—C9—C10 −165.3 (2) C6—C1—C2—C3 −1.1 (3) O2—C9—C10—C15 20.1 (3) N1—C1—C2—C3 173.6 (2) N2—C9—C10—C15 −161.2 (2) C1—C2—C3—C4 0.0 (3) O2—C9—C10—C11 −156.1 (2) C7—O1—C4—C5 −5.5 (3) N2—C9—C10—C11 22.5 (3) C7—O1—C4—C3 172.5 (2) C15—C10—C11—C12 1.1 (3) C2—C3—C4—C5 1.3 (3) C9—C10—C11—C12 177.3 (2) C2—C3—C4—O1 −176.8 (2) C10—C11—C12—C13 0.0 (3) O1—C4—C5—C6 176.4 (2) C16—O3—C13—C14 −3.0 (3) C3—C4—C5—C6 −1.4 (3) C16—O3—C13—C12 176.2 (2) C2—C1—C6—C5 0.9 (3) C11—C12—C13—O3 180.0 (2) N1—C1—C6—C5 −174.0 (2) C11—C12—C13—C14 −0.8 (3) C4—C5—C6—C1 0.4 (3) O3—C13—C14—C15 179.7 (2) C1—N1—C8—N2 176.7 (2) C12—C13—C14—C15 0.5 (3) C1—N1—C8—S1 −0.8 (3) C13—C14—C15—C10 0.6 (3) C9—N2—C8—N1 −7.1 (3) C11—C10—C15—C14 −1.4 (3) C9—N2—C8—S1 170.5 (2) C9—C10—C15—C14 −177.8 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
N1—H1n···O2 0.86 (1) 1.90 (2) 2.626 (2) 141 (2) N2—H2n···S1i 0.86 (1) 2.62 (1) 3.450 (2) 165 (2)