First-Principles Calculation of Point Defects in Uranium Dioxide
Misako Iwasawa
1, Ying Chen
2, Yasunori Kaneta
2, Toshiharu Ohnuma
1,
Hua-Yun Geng
2and Motoyasu Kinoshita
3;41
Materials Science Research Laboratory, Central Research Institute of Electric Power Industry, Tokyo 201-8511, Japan
2Department of Quantum Engineering and Systems Science, School of Engineering,
The University of Tokyo, Tokyo 113-8656, Japan
3Nuclear Technology Research Laboratory, Central Research Institute of Electric Power Industry, Tokyo 201-8511, Japan 4Japan Atomic Energy Agency, Ibaraki 319-1195, Japan
A first-principles calculation for uranium dioxide (UO2) in an antiferromagnetic structure with four types of point defects, uranium
vacancy, oxygen vacancy, uranium interstitial, and oxygen interstitial, has been performed by the projector-augmented-wave method with generalized gradient approximation combined with the Hubbard U correction. Defect formation energies are estimated under lattice relaxation for supercells containing 1, 2, and 8 unit cells of UO2. The electronic structure, the atomic displacement and the stability of defected systems are
obtained, and the effects of cell sizes on these properties are discussed. The results form a self-consistent dataset of formation energies and atomic distance variations of various point defects in UO2with relatively high precision. We show that a supercell with 8 UO2unit cells or larger
is necessary to investigate the defect behavior with reliable precision, since point defects have a wide-ranging effect, not only on the first nearest neighbor atoms of the defect, but on the second neighbors and on more distant atoms. [doi:10.2320/matertrans.47.2651]
(Received August 3, 2006; Accepted October 6, 2006; Published November 15, 2006)
Keywords: first-principles method, density functional theory, generalized gradient approximation, projector-augmented-wave method, Hubbard U correction, electronic structure, uranium dioxide, point defect, formation energy, lattice relaxation
1. Introduction
Uranium dioxide, UO2, is widely used as fuel in nuclear
power generation. In the high burn-up of the fuel, the radial periphery of UO2 pellets forms a characteristic fine grain,
which is known as a rim structure.1) To clarify the
micro-scopic mechanism of this structuring behavior, a basic understanding of the thermodynamic, structural, and kinetic properties of UO2 is very important for both the practical
operation of nuclear reactors and the theoretical interest in this metal oxide with strongly correlated electrons. In particular, the rim structure is believed to be formed by accumulated irradiation damage and the effects of high energy electronic excitation. Since it is difficult to analyze these complicated dynamical processes using current theo-retical tools, the first step of this research is to provide by information on the point defect behavior in UO2, which is the
most fundamental information on the elemental processes in the formation of the complex defect structure.
Petitet al.have carried out a series of electronic structure calculations on UO2 with point defects by the linear
muffin-tin orbital method with the atomic-sphere approximation (LMTO-ASA) and by the plane-wave pseudopotential method based on density functional theory (DFT) with the local density approximation (LDA) or the generalized gradient approximation (GGA).2–5) They investigated the
electronic structure of the defect systems, and estimated the formation energies of several types of point defects in UO2.
Their results qualitatively provided the correct trend of defect formation in UO2. However, their studies have two
limi-tations. One is the relatively small 211 supercell (U8O16) used in each calculation, which may lead to an
overestimation of the defect formation energies. The other is the LDA or GGA scheme adopted in all calculations, which gave a metallic ground state. UO2 is well known to be a
conventional Mott insulator,6) but in the framework of the LDA or GGA, UO2is predicted to be a metal.2–5)This failure
of the conventional LDA or GGA has been attributed to the absence of strong correlations for the 5f electrons in the uranium atom in UO2. The LDA+U7,8) method has been
developed to describe such a correlation by introducing a strong intra-atomic interaction. This approach has been used for uranium 5f electrons,6,9,10)and has yielded the electronic
structure as a Mott insulator for UO2 in the ground state.
In the present study, we perform a series of comprehensive calculations on UO2with four types of point defects to obtain
point defect properties with high accuracy. We have focused on the following two aspects: to construct a suitable theoretical framework for UO2 that reflects the correct
antiferromagnetic insulator ground state of UO2 crystal by
incorporating the effect of strongly correlated 5f electrons in uranium atoms through the Hubbard U correction, and to calculate point defect properties such as formation energies and lattice deformations by taking the currently plausible maximal supercell (222 unit cells of UO2, U32O64) as
the model system.
In the next section, the calculation method and the calculation systems are briefly described. In Section 3, results obtained for various cases are presented and dis-cussed. The results are then summarized in Section 4.
2. Calculation Method
The present work focuses on four types of point defects in UO2: uranium vacancy (U-vacancy), oxygen vacancy
(O-vacancy), uranium interstitial (U-interstitial) and oxygen interstitial (O-interstitial). The ideal UO2crystal has a CaF2
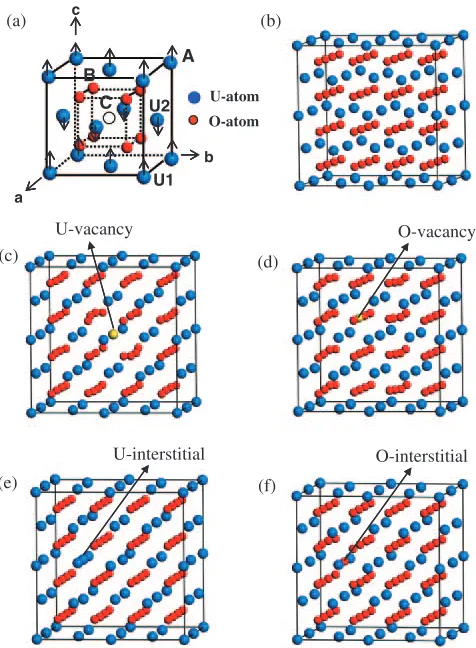
-type structure (cF12, Fm33m), and its unit cell is shown in Fig. 1(a). Systems containing point defects are modeled using supercell technique. A specific supercell has been
constructed to introduce each point defect. Examples of an U-vacancy and an O-U-vacancy are illustrated as site ‘‘A’’ and site ‘‘B’’, respectively, in Fig. 1(a). For the interstitial defects, only the octahedral site (the center of the oxygen cube) is taken into account for both U- and O-interstitials, as marked by ‘‘C’’ in Fig. 1(a). To examine the effect of the cell size, calculations are performed for three types of supercells with containing 1, 2, and 8 unit cells of UO2. We use the following
notation to show the number of atoms in each unit cell, U4O8
(111), U8O16 (211) and U32O64 (222) for
each type of defect. Figures 1(b)–(f) show five U32O64
(222) supercells corresponding to an ideal UO2crystal
and the four types of point defects. Note that these defected structures show the situations after relaxation, but the initial regular structures can be imaged easily.
To discuss the ground state properties of UO2with defects,
we assume an antiferromagnetic structure for electron spins in our calculations in accordance with experimental re-sults.11) The magnetic moments of uranium ions lie in the
(001) plane ferromagnetically, and this plane is stacked along the [001] direction antiferromagnetically, as illustrated in Fig. 1(a).
An antiferromagnetic configuration remains when defects are introduced. All lattice constants and cell shapes are relaxed by minimizing the stress tensor, and all atomic positions are relaxed by minimizing the Hellman-Feynman forces using the RMM-DIIS algorithm.12) In the
paramag-netic state, UO2 crystal belongs to the CaF2-type structure
with cubic symmetry. However, the antiferromagnetic structure causes UO2 to have a tetragonal lattice. The initial
atomic positions and lattice constants are set corresponding to the cubic lattice, while during calculation, the lattice symmetries turn to tetragonal for U4O8 and U32O64, and
orthorhombic for U8O16 as a natural result of introducing
antiferromagnetism.
Calculations in the present work are carried out by the projector-augmented-wave (PAW) method,13,14)using
PBE-GGA exchange-correlation functional,15)as implemented in the Vienna ab initio Simulation Package (VASP).16)The spin polarization calculations are performed, whereas the spin-orbit interactions are neglected as a simplification of the complex magnetism in UO2. The Hubbard U correction is
introduced to describe strongly correlated uranium 5f electrons in accordance with a simplified version of GGA+U proposed by Dudarevet al.8)They determined the LSDA+U
parameters to be U¼4:5eV and J¼0:51eV by a careful estimation using LMTO method.9)It may be reasonable to
select these values when we use the simplified version of GGA+U for Hubbard U correction. We undertook electronic structure calculation for U4O8by the PAW-GGA+U method
using these values in the antiferromagnetic configuration whose lattice constant was experimental value, 0.547 nm. The electronic structure of U4O8obtained was that of a Mott
insulator, so we decided to adopt these values in our PAW-GGA+U calculations.
Regarding to other calculation parameters, the cut-off energy of plane-waves is set to 400 eV. The Wigner-Seitz radii of uranium and oxygen atoms are 0.1588 and 0.0820 nm, respectively. The Monkhorst-Pack17)(4,4,4) set is used
for k-space sampling in U4O8, whereas a (3,3,3) set is used
in large cells to reduce the amount of calculation. The tetrahedron method with Blo¨chl’s corrections18) is used in
k-mesh integration. The bulk modulus is estimated by fitting to the Birch-Murnaghan equation of state19)based on the total
energy vs unit cell volume plot.
3. Results and Discussion
3.1 Structural properties of pure UO2
First, the calculation for an ideal UO2 crystal was carried
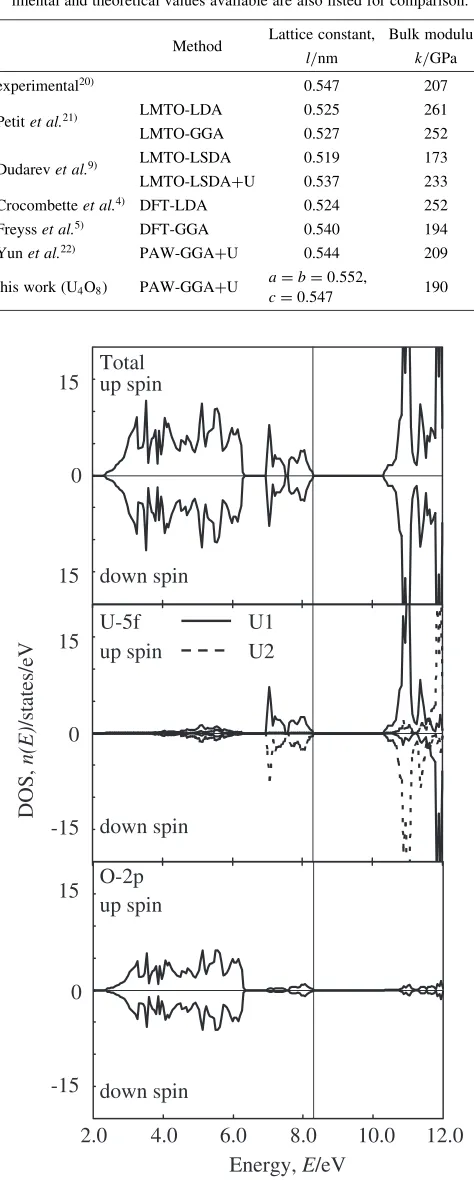
out to test the framework with a combination of several theoretical treatments so as to obtain an optimized calcu-lation condition. The calculated equilibrium properties are listed in Table 1, as well as the available values from experiments20) and other calculations21,22) for comparison.
A good agreement among these data can be found. The tetragonal symmetry (a¼b6¼c) in the present result is due to thec-axis-direction antiferromagnetic structure.
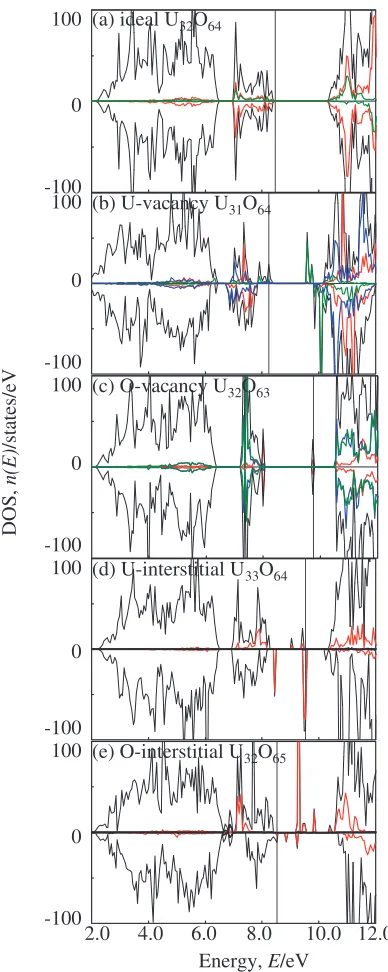
Figure 2 shows the total density of states (DOS) of ideal UO2 and the site projected density of states (PDOS) for
uranium (U) and oxygen (O) atoms. Since U 7s, 6p and 6d components are very small, only the U 5f components are illustrated for the U atom. Because of the antiferromagnetic configuration, there are two types of U sites: U1 indicates the U site in thec¼0plane with up spin, and U2, the U site in thec¼1=2 plane with down spin, as shown in Fig. 1. It can be seen that the strong correlation for 5f electrons of U
(f) a
A B
C
(a)
(c)
(e)
(d) (b)
b c
U-vacancy U1 U2
O-atom U-atom
U-interstitial O-interstitial O-vacancy
Fig. 1 Calculation systems. (a) Unit cell of ideal UO2crystal, and relaxed
defect systems in U32O64 supercell: (b) Ideal, (c) U-vacancy, (d)
[image:2.595.50.288.73.397.2]atoms results in the removal of the degeneracy of 5f bands near the Fermi level. The 5f bands split into two energy regions, one is mainly the top of the valence states and the
other is the bottom of the conduction states, which correctly reproduces an insulator characteristic. It is also found that the 2p orbitals of O predominate the lower valence states and partially hybridize with 5f orbitals of U at the top of the valence band. These calculated results are consistent with the experimental and theoretical values reported previous-ly,6)which confirms that the method we employed can be
taken as a suitable platform for further investigation of various defect structures.
3.2 Electronic structures of defect systems
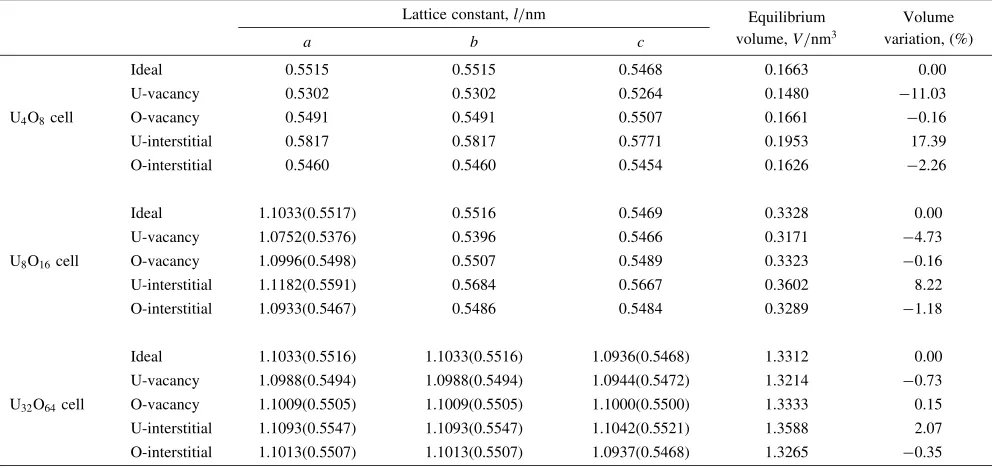
Figure 3 shows the total DOS of ideal and four defect structures in U32O64, as well the PDOS of defect-related sites
in various types of defects. The components of the first nearest neighbor (‘‘n.n.’’ hereafter), 2nd n.n. and 3rd n.n. U atoms are plotted in red, green and blue, respectively. The ‘‘reference sites’’ to which the neighboring atoms are referred in different situations are, in Fig. 3(a), the U atom site; in Figs. 3(b) and (c), the U vacancy and O vacancy sites, respectively; and in Figs. 3(d) and (e), the U interstitial and O interstitial sites, respectively. For the ideal crystal, only the contributions of the 1st n.n. and 2nd n.n. atoms are drawn, because, with respect to one U atom, the 3rd n.n. U atoms are fully geometrically equivalent to the 1st n.n. U atoms but only at a different distance from the U atom, their contributions are the same as those of the 1st n.n. U atoms (red). Comparing the DOS of defect structures with the ideal situation, the effect of introducing point defects and of the introduced defects themselves on the electronic structures can be explained as following.
The electronic structure of the system with the U-vacancy, U31O64 (Fig. 3(b)), shows p-type semiconductor behavior
with an acceptor level at 0.5 eV higher than the top of the valence band. The missing U atom results in an O 2p hole state; at same time, the antiferromagnetism is broken and a non zero magnetic moment in the U 5f bands occurs. It is interesting to see the effect of the 1st n.n. U atoms on the vacancy (red) most appears at the top of the valence band, whereas the acceptor level that newly appeared in the energy gap is mainly created by the 2nd n.n. U atoms (green). It is also observed that the PDOS distribution of the 3rd n.n. U atoms (blue) is significantly different from that of 1st n.n. U atoms (red) although they are the same in the ideal crystal. Such a redistribution of the band structure is due to the fact that the interatomic interactions among atoms are amended according to the vacancy introduced.
The situation for the O-vacancy, U32O63(Fig. 3(c)), seems
to be that of an n-type semiconductor with a donor level at 1.7 eV higher than the top of the valence band, near the bottom of the conduction band. This impurity state is mainly formed from the 5f states of U (which almost cannot be seen in the figure due to its small value) hybridized with plane-wave-like states that are attributed to an interstitial compo-nent of about 45% electrons in that state. It is intriguing to note the deviation of the PDOS of the 1st and 3rd n.n. U atoms, and the remaining antiferromagnetism with the existing O vacancy.
The electronic structure of the U-interstitial, U33O64
[image:3.595.49.286.95.685.2](Fig. 3(d)), is as that of an n-type semiconductor. The component of the U interstitial atom steadily crosses the
Table 1 Calculated equilibrium properties of relaxed structure with point defects in UO2: lattice constants (nm) and bulk moduli (GPa).
Exper-imental and theoretical values available are also listed for comparison.
Method Lattice constant, Bulk modulus, l/nm k/GPa
experimental20Þ 0.547 207
Petitet al.21Þ LMTO-LDA 0.525 261
LMTO-GGA 0.527 252
Dudarevet al.9Þ LMTO-LSDA 0.519 173
LMTO-LSDA+U 0.537 233
Crocombetteet al.4Þ DFT-LDA 0.524 252
Freysset al.5Þ DFT-GGA 0.540 194
Yunet al.22Þ PAW-GGA+U 0.544 209
this work (U4O8) PAW-GGA+U
a¼b¼0:552,
c¼0:547 190
DOS,
n(E)
/states/eV
up spin
down spin
up spin
down spin
up spin
down spin
Energy,
E
/eV
0
0
15
15
15
15
-15
-15
Total
O-2p
U-5f
U1
U2
2.0
4.0
6.0
8.0
10.0
12.0
0
Fig. 2 Total DOS and PDOS of ideal UO2crystal with antiferromagnetic
range from the bottom of the valence band to the top of the conduction band, although the portion of the single atom is too small to be seen in the figure. It can be seen that the donor level is formed mainly from 5f electrons of the 1st n.n. U atoms (red) and is located at about 1.3 eV higher than the top of the valence band. The defect state is similar to the U32O63
case, but the extra U 5f electrons impose a larger magnetic effect to form an obvious asymmetric distribution of majority and minority spins.
The O-interstitial system, U32O65 (Fig. 3(e)), acts as a
p-type semiconductor; the acceptor level, which consists of mainly 1st n.n. U 5f (red) states slightly hybridized by the 2p of the interstitial O atom (which almost cannot be seen), spans the energy gap while showing complicated features. The effect of the single O interstitial atom (bold black line) can be observed at the joint place of two parts of the valence band. The defect state is similar to the U31O64case, but since
some of the occupied 5f electrons on the U site are absorbed into the interstitial O site, the unoccupied 5f states appear above the Fermi level. This also leads to a large magnetic-state redistribution in the DOS that creates a remarkable shift of the majority and minority spin states in the energy space. Figure 4 shows the partial charge density distributions projected onto the (011) plane in the lattice corresponding to the 5f band at the top of the valence band spanned from the Fermi level down to the lower energy states by2:0eV, for ideal and defect systems in U32O64supercells. The localized (b) U-vacancy U31O64
(c) O-vacancy U32O63
(d) U-interstitial U33O64
(e) O-interstitial U32O65 (a) ideal U32O64
2.0 4.0 6.0 8.0 10.0 12.0 100
-100 0 100
-100 0 100
-100 0
100
-100 0
100
-100 0
DOS,
n(E)
/states/eV
Energy, E/eV
Fig. 3 Total DOS of ideal UO2crystal and four types of point defects in
U32O64supercell (thin line), and PDOS of defect relevant sites. (a) Ideal
UO2crystal: total DOS and PDOS of 1st n.n. (red), 2nd n.n. (green) and
3rd n.n. (blue) U atom sites with respect to U atom. (b) U-vacancy case: total DOS and PDOS of 1st n.n. (red), 2nd n.n. (green) and 3rd n.n. (blue) U atom sites with respect to U vacancy site. (c) O-vacancy case: total DOS and PDOS of 1st n.n. (red), 2nd n.n. (green) and 3rd n.n. (blue) U atom sites with respect to O vacancy site. (d) U-interstitial case: total DOS and PDOS of 1st n.n. (red) U atom sites with respect to interstitial U site and U interstitial site (bold). (e) O-interstitial case; total DOS and PDOS of 1st n.n. (red) U atom sites with respect to interstitial O site and O interstitial site (bold). (Vertical bar indicates Fermi level.)
(a)
(b)
(e)
(c)
(d)
U-atom
O-atom
U-vacancy
O-vacancy
O-interstitial
U-interstitial
Fig. 4 Partial charge density distributions projected onto (011) plane corresponding to U 5f band at the top of valence band spanned from Fermi level down to lower energy states by2:0eV. (a) Ideal UO2crystal, and
relaxed defect systems in U32O64supercell: (b) U-vacancy, (c) O-vacancy,
[image:4.595.71.265.70.556.2] [image:4.595.305.543.73.449.2]f-orbital feature is clearly displayed. Note that a high spherical charge density exists in UO2, particularly around
O atoms. The non spherical charge density of occupied U 5f electrons, atomic displacement and change in bonding state in various defect structures can be observed clearly, particularly in the O-vacancy case.
3.3 Lattice relaxations surrounding defects
A summary of the lattice parameters and the volume variations of relaxed structures in the ideal crystal and the four types of point defects in the UO2 system are shown in
Table 2. These four types of point defects in the U32O64
supercell after relaxation are displayed in Figs. 1(c)–(f), and the ideal U32O64is shown in Fig. 1(b).
We found that the lattice parametercis less thana(orb) in all cases. This can be assigned to the asymmetry of charge densities in theða;bÞplane and along thec-axis, which results from the magnetic interaction among partially occupied 5f electrons.
The volume variations with respect to different sizes of supercells listed in Table 2 are depicted in Fig. 5. One can see that the volume variations are large in the case of the U defects; the U-interstitial causes a large swelling, whereas the U-vacancy induces a decrease in volume shrink. On the other hand, O defects do not cause significant variations in volume. Figure 5 also shows the effective ranges of various point defects. We found rather large volume variations in the U8O16 cell; even in our largest cell, U32O64, the volume
variations still do not reach zero. This clearly indicates the insufficiency of the U8O16 cell as a non interacting point
defect model.
It is of high interest to analyze the details of changes in atomic distances among the defect site and its neighboring atoms in our largest supercell U32O64 (Fig. 6). It is
[image:5.595.52.548.105.339.2]note-worthy that all distances of the 1st n.n. atoms with respect to
Table 2 Calculated lattice parameters and volume variations of relaxed structures with point defects in UO2with respect to the ideal crystal in three types of
supercells: U4O8, U8O16and U32O64. The volume variation is calculated as the difference from the ideal crystal in each system. (Values in parentheses are
converted to one unit cell of UO2: U4O8.)
Lattice constant,l/nm Equilibrium Volume
a b c volume,V/nm3 variation, (%)
Ideal 0.5515 0.5515 0.5468 0.1663 0.00
U-vacancy 0.5302 0.5302 0.5264 0.1480 11.03
U4O8cell O-vacancy 0.5491 0.5491 0.5507 0.1661 0.16
U-interstitial 0.5817 0.5817 0.5771 0.1953 17.39
O-interstitial 0.5460 0.5460 0.5454 0.1626 2.26
Ideal 1.1033(0.5517) 0.5516 0.5469 0.3328 0.00
U-vacancy 1.0752(0.5376) 0.5396 0.5466 0.3171 4.73
U8O16cell O-vacancy 1.0996(0.5498) 0.5507 0.5489 0.3323 0.16
U-interstitial 1.1182(0.5591) 0.5684 0.5667 0.3602 8.22
O-interstitial 1.0933(0.5467) 0.5486 0.5484 0.3289 1.18
Ideal 1.1033(0.5516) 1.1033(0.5516) 1.0936(0.5468) 1.3312 0.00
U-vacancy 1.0988(0.5494) 1.0988(0.5494) 1.0944(0.5472) 1.3214 0.73
U32O64cell O-vacancy 1.1009(0.5505) 1.1009(0.5505) 1.1000(0.5500) 1.3333 0.15
U-interstitial 1.1093(0.5547) 1.1093(0.5547) 1.1042(0.5521) 1.3588 2.07
O-interstitial 1.1013(0.5507) 1.1013(0.5507) 1.0937(0.5468) 1.3265 0.35
Volume Variation (%)
Number of unit cells of UO2 -15.0
-10.0 -5.0 0.0 5.0 10.0 15.0 20.0
1 2 4 6 8 U-vacancy O-vacancy U-interstitial O-interstitial
Fig. 5 Volume variations of relaxed defect structures in UO2(in %).
Distance Variation,
d
/nm
Distance to Defect, d/nm
U32O64supercell U-vacancy
O-vacancy U-interstitial O-interstitial
-0.02 -0.01 0.00 0.01 0.02 0.03
0.20 0.25 0.30 0.35 0.400.45 0.50 0.55 0.60
Fig. 6 Bond length variations in relaxed defect structures in UO2
[image:5.595.312.540.378.543.2] [image:5.595.312.541.604.758.2]defect sites in the four defect structures are increased, whereas for further distant atoms, the corresponding distance varies complicatedly according to the types of defect. In Fig. 6, several pairs of points at about 0.27, 0.39 and 0.46 nm from the defect site are the same neighboring atoms with slight distance differences due to the tetragonal symmetry of the lattice.
In the case of the U-vacancy (solid line in Fig. 6), the eight 1st n.n. O atoms move outward from the vacancy site to form a large vacancy hole, although the total volume change is negative, as shown in Table 2. This large vacancy hole manifests the feature of the ionic crystal, and is caused by effective Coulomb repulsion between the 1st n.n. O atoms due to the missing cation. The further n.n. atoms adjust themselves in an oscillatory manner,i.e., the twelve 2nd n.n. U atoms turn to be closer to the vacancy due to the removal of the Coulomb interactions among the cations; then the next nearest atoms adjust themselves slightly further from the defect site, and so on.
In the case of the O-vacancy (dashed line in Fig. 6), the displacement of n.n. atoms is similar to the behavior in the previous case of the U-vacancy; a vacancy hole is created, originating from repulsive forces among the four 1st n.n. U atoms. However, the displacements of the six 2nd n.n. O atoms depend on their direction with respect to the vacancy site. Though the two O atoms along the [001] direction in the upper or lower (001) plane from the vacancy site approach the vacancy site very slightly, the four O atoms in the same (001) plane as the vacancy site become rather closer to the vacancy site. These complex phenomena result from the asymmetric occupied 5f electrons at the U site, which are released from the removed O. The O removal corresponds to an additional two electrons in the crystal, and then these electrons mainly occupy the unoccupied U 5f orbitals. This situation can be seen in Fig. 4(c), as well as the asymmetry of charge density. The two additional electrons spread to U sites from the vacancy site, and the charge density around the U atoms near the vacancy is modified markedly from the cubic symmetry.
In the case of interstitials at the octahedral site, the displacements of the 1st n.n. atoms may be attributed to the atomic size effect of interstitial atoms. However, displace-ments of further n.n. atoms also affect ionic interactions among atoms. In fact, a larger variation in 2nd n.n. than in 1st n.n. atoms is observed in both U-interstitial and O-interstitial defects; this cannot be explained simply by the size effect.
Figure 6 also gives information on the convergence of the supercell size to the model of noninteracting defects. It can
be seen that the changes in the volumes and bond lengths of the four defect structures become stable towards an asymptotic limit with increasing of the supercell size to U32O64. The most important point manifested in Fig. 6 is
that the long-range interactions exist among the defect and the surrounding atoms. In the U32O64 supercell, there are
still large distance variations of 2% at about 0.4 nm from the defect site, which is beyond the range of the U8O16 cell.
This suggests that a large U32O64 supercell is necessary to
estimate the precise defect properties. This will be discussed again in the next section.
3.4 Defect formation energies
The vacancy formation energy can be defined by
EFV
X ¼E
N1
VX E
N
þEX; ð1Þ
where EVFX is the vacancy formation energy of atom X (X¼U, O),ENVX1is the calculated free energy of a cell with defect X,EN is the free energy of an ideal crystal without a
defect, andEX is the internal energy of the pure substance
formed from an X-atom in the reference state. -Uranium (oC4, Cmcm) is used as the reference state of U, and an oxygen molecule in a 1.0 nm cubic cell is taken as the reference state of O. For-uranium, the free energy was also calculated by the GGA+U method with the same U and J values as used by Dudarev9)in order to obtain a consistent
reference state for all UO2calculations in the present work.
The interstitial formation energy is defined in a similar way to eq. (1):
EIF
X ¼E
Nþ1
X E
NEX; ð2Þ
whereEFIXis the interstitial formation energy of atom X, and
EXNþ1is the calculated free energy of the cell when an X atom is placed at the interstitial site.
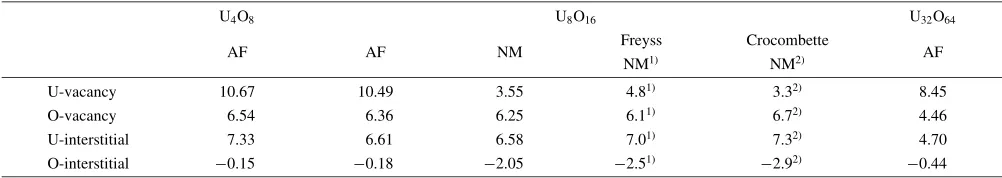
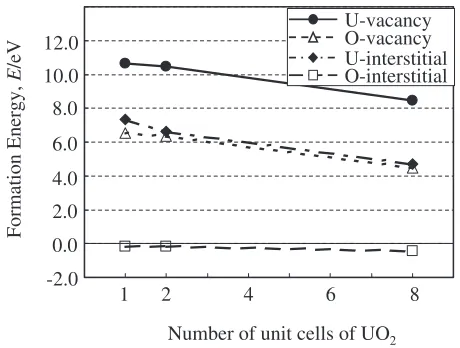
The calculated formation energies of the four types of defects, U- and O-vacancies, and U- and O-interstitials, in the three kinds of supercells are shown in Table 3 and plotted in Fig. 7. A gradual decrease in the values of formation energies with increasing size of the supercell can be observed. It can be seen that the U-vacancy has a large positive formation energy, whereas the U-interstitial and O-vacancy have roughly half the formation energy of the U-vacancy defect. The most noteworthy result is the negative formation energy of the O-interstitial in the octahedral site, which implies that the O-interstitial has lower energy than the O2molecule, and
that UO2becomes oxidized in the presence of O, which is in
agreement with the well-known fact that UO2 is inclined to
[image:6.595.46.547.94.183.2]be hyperstoichiometric.
Table 3 Formation energies (eV) of point defects in UO2in three types of supercells: U4O8, U8O16and U32O64. Other calculated results
available are also listed for comparison.
U4O8 U8O16 U32O64
AF AF NM Freyss Crocombette AF
NM1Þ NM2Þ
U-vacancy 10.67 10.49 3.55 4.81Þ 3.32Þ 8.45
O-vacancy 6.54 6.36 6.25 6.11Þ 6.72Þ 4.46
U-interstitial 7.33 6.61 6.58 7.01Þ 7.32Þ 4.70
O-interstitial 0.15 0.18 2.05 2.51Þ 2.92Þ 0.44
Table 3 also gives the values for U8O16obtained by Freyss
et al.5)and Crocombetteet al.4)as a comparison. Note that
there is a large discrepancy in the values of formation energy between the two calculations, particularly for the U-vacancy; our value is about double that of Freyss et al.’s value, and moreover, Freyss et al.’s formation energy of the O-interstitial is more than 10 times our result. In addition to the difference in values arising from the different methods, we expect that the difference in magnetic states also resulted in this discrepancy, since our calculations treated all systems as antiferromagnetic (AF) structures, whereas Freyss et al. used nonmagnetic (NM) ground states. To clarify this point, we performed the NM calculation on U8O16. The results are
also listed in Table 3 under ‘‘NM’’ next to the column of our AF values for U8O16. One can see that our NM calculation
gives very similar formation energies to Freyss et al.’s values, and we can confirm the necessity of using AF configuration to obtain accurate values of formation energy. Although it is difficult to see the full convergence from Fig. 7 even in the 8 unit cells of the U32O64 supercell, we
believe that the defect formation energies in a large supercell would be more accurate than in small cells.
4. Conclusion
We have performed first-principles electronic structure calculations of point defects in UO2, in which we included
antiferromagnetism, strong correlations, and the relaxation of lattice and atomic positions. The results form for the first time, a self-consistent dataset of formation energies and atomic distance variations of various point defects in UO2
with relatively high precision, which provides important information on the elementary processes of complex struc-ture formation, and provides a basis for further investigations of the thermodynamic, stability and kinetic properties of UO2. The change in the electronic structures in UO2
introduced by point defects can be explained in the picture of the ionic interactions among atoms. The important effect of the magnetism in the UO2 system is revealed through the
present calculations. We believe that our results for the supercell of 222 UO2 unit cells (U32O64) provide
reliable formation energies and volume relaxation values and also suggest that a larger supercell is necessary for investigating the impurity behavior with higher precision, since point defects have a wide-ranging effect, not only on the first nearest neighbor atoms of the defect but on the second nearest neighbor and on more distant atoms.
Acknowledgement
This study was financially supported by the Budget for Nuclear Research of the Ministry of Education, Culture, Sports, Science and Technology, based on the screening and counseling by the Atomic Energy Commission.
REFERENCES
1) T. Sonoda, M. Kinoshita, I. L. F. Ray, T. Wissb, H. Thiele, D. Pellottiero, V. V. Rondinella and Hj. Matzke: Nucl. Instr. and Meth. B
191(2002) 622–628.
2) T. Petit, C. Lemaignan, F. Jollet, B. Bigot and A. Pasturel: Philos. Mag. B77(1998) 779–786.
3) T. Petit, G. Jomard, C. Lemaignan, B. Bigot and A. Pasturel: J. Nucl. Mater.275(1999) 119–123.
4) J. P. Crocombette, F. Jollet, L. Thien Nga and T. Petit: Phys. Rev. B64
(2001) 104107.
5) M. Freyss, T. Petit and J. P. Crocombette: J. Nucl. Mater.347(2005) 44–51.
6) F. Jollet, T. Petit, S. Gota, N. Thromat, M. Gautier-Soyer and A. Pasturel: J. Phys.: Condens. Matter9(1997) 9393–9401.
7) A. I. Liechtenstein, V. I. Anisimov and J. Zaanen: Phys. Rev. B52
(1995) R5467–R5470.
8) S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton: Phys. Rev. B57(1998) 1505–1509.
9) S. L. Dudarev, G. A. Botton, S. Y. Savrasov, Z. Szotek, W. M. Temmerman and A. P. Sutton: phs. stat. sol. (a)166(1998) 429–443. 10) R. Laskowski, G. K. H. Madsen, P. Blaha and K. Schwarz: Phys. Rev.
B69(2004) 140408(R).
11) B. C. Frazer, G. Shirane, D. E. Cox and C. E. Olsen: Phys. Rev.140
(1965) A1448–A1452.
12) P. Pulay: Chem. Phys. Lett.73(1980) 393. 13) P. E. Blo¨chl: Phys. Rev. B50(1994) 17953–17979. 14) G. Kresse and J. Joubert: Phys. Rev. B59(1999) 1758–1775. 15) J. P. Perdew, K. Burke and M. Ernzerhof: Phys. Rev. Lett.77(1996)
3865–3868.
16) G. Kresse and J. Furthmu¨ller: Phys. Rev. B54(1996) 11169–11186. 17) H. J. Monkhorst and J. D. Pack: Phys. Rev. B13(1976) 5188–5192. 18) P. E. Blo¨chl, O. Jepsen and O. K. Andersen: Phys. Rev. B49(1994)
16223–16233.
19) F. D. Murnaghan: Proc. Natl. Acad. Sci. U.S.A.30(1944) 244. 20) M. Idiri, T. Le Bihan, S. Heathman and J. Rebizant: Phys. Rev. B70
(2004) 014113.
21) T. Petit, B. Morel, C. Lemaignan, A. Pasturel and B. Bigot: Philos. Mag. B73(1996) 893–904.
22) Y. Yun, H. Kim, H. Kim and K. Park: Int. J. Korean Nucl. Soc. Nucl. Eng. and Tech.37(2005) 293–298.
U-vacancy O-vacancy U-interstitial O-interstitial
1 2 4 6 8 -2.0
0.0 2.0 4.0 6.0 8.0 10.0 12.0
Number of unit cells of UO2
Formation Energy,
E
/eV
Fig. 7 Formation energies (eV) of point defects in UO2in three types of
[image:7.595.54.284.71.245.2]